I would like to express my sincere gratitude to my supervisor Annette Meyer, who gave me the opportunity to carry out my master thesis at Eurofins WEJ Contaminants GmbH in Hamburg. In this work, liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) was used for the detection of bacitracin A and B, colistin A and B as well as polymyxin B1 and B2 in milk and animal tissue samples. Grenzwerte for die Summe von Bacitracin A, B and C sowie für die Summe and Colistin A and B festgelegt.
CHAPTER I: INTRODUCTION AND LITERATURE REVIEW
- B ACKGROUND TO THE PROBLEM
- A IMS
- O BJECTIVES
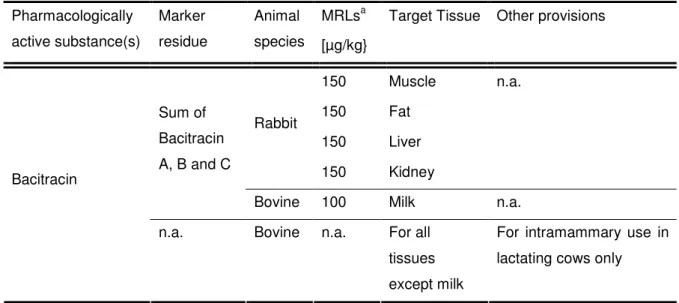
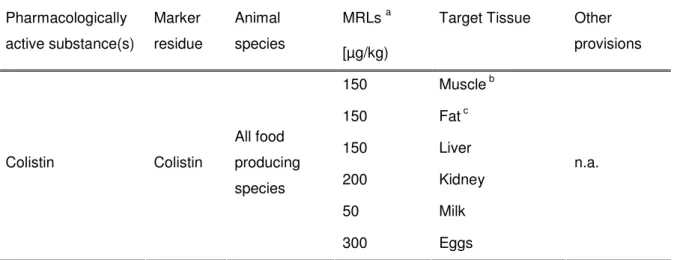
- L EGAL REQUIREMENTS
- P EPTIDE ANTIBACTERIAL COMPOUNDS
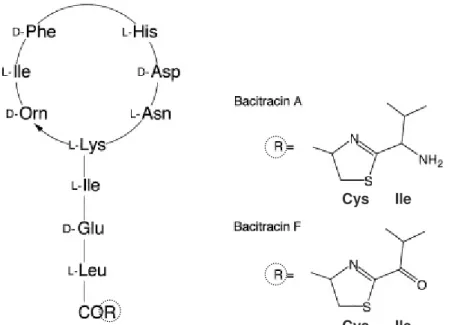
- Bacitracin
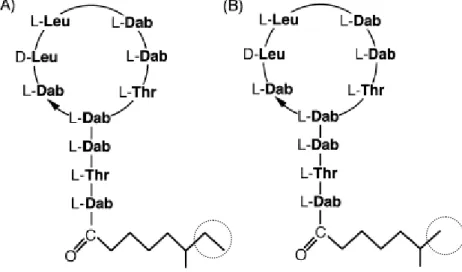
- Colistin (Polymyxin E)
- Polymyxin B
- A NALYTICS OF POLYPEPTIDE ANTIBIOTICS – STATE OF THE ART
- Sample preparation
- Chromatography
- Detection
- P ROJECT OVERVIEW
In Europe, the most important polypeptide antibiotics used in veterinary medicine are bacitracin and colistin (Brabander et al., 2009), which together with polymyxin B will be described shortly. Polymyxin B is again a mixture of closely related components produced by Bacillus polymyxa and is active against gram-negative bacteria (Orwa et al., 2001b). However, UV detection in general lacks selectivity and sensitivity in complex matrices (Decolin et al., 1997).
CHAPTER II: MATERIAL AND METHODS
M ATERIAL
- Chemicals and Reagents
- Biological samples
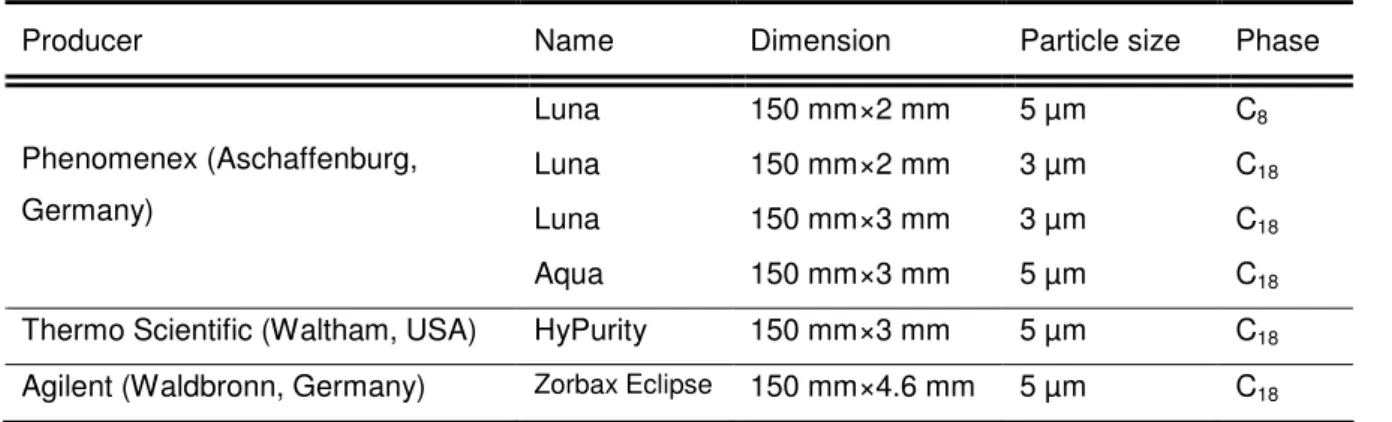
- Apparatus
All samples were stored at -20°C prior to analysis and thawed in small aliquots overnight in the refrigerator at 4°C. Thawed samples were stored in the refrigerator at 4°C for several days, if sample preparation and analysis occurred during consecutive days. The optimized gradient program shown in Table 2-3 was developed in the course of this work.
M ETHODS
- Preparation of solutions
- Preparation of analyte and spiking solutions
- Mass spectrometry
- Liquid chromatography
- Sample preparation
- Calculations used for results
- Method validation
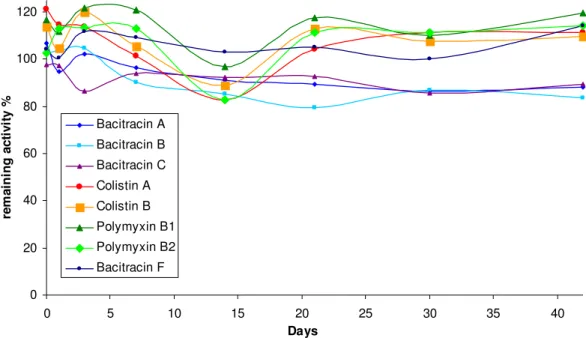
Initial tuning of the reference substances in the mass spectrometer was performed to determine the dominant precursor ions of the substances. To estimate the proportion of components in the mixture, doubly and triply charged precursor ions were measured in a pseudo-MRM experiment (the same ions were chosen as product ions). The remaining analyte concentration was calculated according to equation 2-4, where At represents the area measured at the corresponding time point and Af.
The validation of the method is carried out according to the sample preparation described in section 2.2.4, method 1. For milk validation, 1 ml HCl was used during sample preparation instead of the 0.5 ml mentioned. Each of the sample series included a non-enriched matrix blank, as well as four levels of enrichment in the case of meat and five in the case of milk.
To determine the repeatability of the method, seven to eight samples, with the same volumes applied, were processed on the same day under identical conditions (1 sample, 1 person, 1 instrument, same chemicals). The repeatability is then calculated according to Equation 2-5, whereby s is the standard deviation of the parallel determinations and x is the mean value. The corrected µg/kg values used for the repeatability of the method were also used to determine the truth of the method.
Corrected values for yield and corrected values for yield and internal standard were expressed as a percentage of the calculated expected increased values.
CHAPTER III: RESULTS AND DISCUSSION
LC-MS/MS METHOD
- Mass spectrometry
- Liquid Chromatography
More detailed analysis of the standard assembly will follow later in this chapter. Attempts to direct the formation of ions in either direction were not made. This can either indicate the presence of several single amino acids in the reference substance, as well as their formation in the injection chamber of the mass spectrometer.
For general purposes only the product scans of the triply charged precursor ions for each analyte will be shown and discussed. In the initial phase of the project, several product ions were measured for precursors during the tuning process and initial experiments. Consequently these are excluded from the measurement method and for reasons of clarity are not included in the figures below.
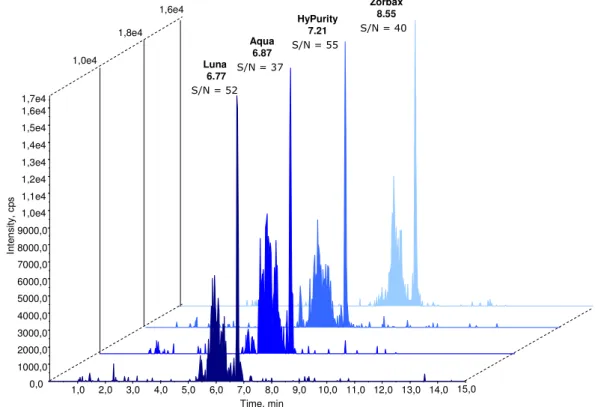
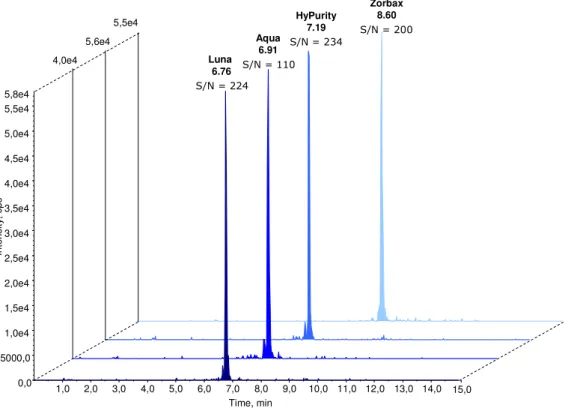
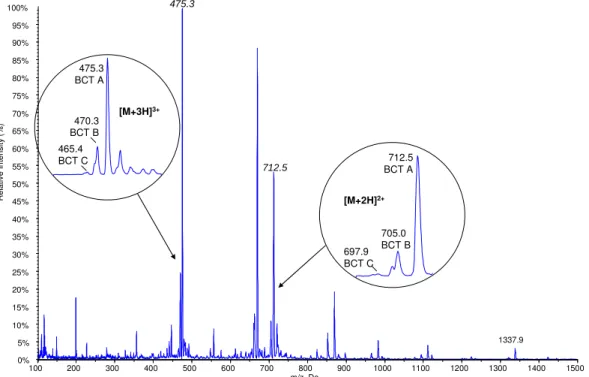
Spectra of triple-charged precursor ions are illustrated along with the product ions used for quantification. In the present work, the length of the isocratic phase at high organic mobile phase B was shortened from 4 min. The figure shows that the retention time, the signal-to-noise ratio, as well as the peak intensity change according to the selected column.
All the peaks have a small shoulder before the main peak, which probably depends on the substance. Ideally, the retention time of the analytes should be the same in standard solutions and prepared samples. Therefore, this solution was used for further experiments and to dilute the calibration standards.
S AMPLE PREPARATION
- Liquid-Liquid sample extraction (method 3)
- Liquid-Liquid extraction and solid-phase clean-up (method 1 and 2)
- Extraction solutions
- Sample quantity
- Solid-phase extraction cartridges
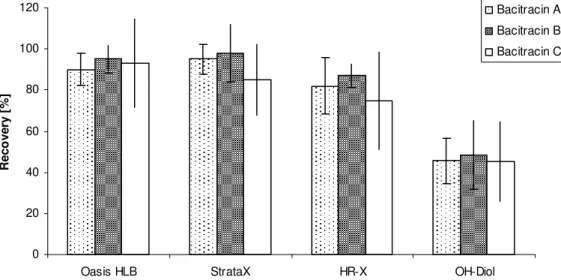
- Recovery values
- Robustness of the method
- Internal standard
During the course of method development, experiments were performed with amounts of meat ranging from 0.5 to 2.5 g (spiked at the same µg/kg level, adjusting the peak volume) to assess whether the sample amount could reduced for meat. A 10% solution was used and tested during the development of the method, in order to wash away more of the interferences of the carrier matrix. Therefore the loss of analyte in the third milliliter was accepted in the development of the protocol.
The results show that the highest recovery values were obtained when the eluents were evaporated to dryness in the presence of ethylene glycol. Depending on the matrix (meat, milk or water samples), the extraction loss was between 20-40% for bacitracin, while it was in the range of 10-20% for colistin and polymyxin. Final experiments were performed to assess the robustness of the method with respect to the extraction time and the amount of HCl used.
This facilitates the use of the method in a routine laboratory with respect to time and instrumental requirements. At the end of the work, after the validation, experiments were carried out with different amounts of HCl, as listed in Table 3-8 together with the appropriate pH values in milk and meat. Measurement of pH values was done in the diluted solution (aliquot and water) which was then loaded onto the SPE column.
However, the results of these experiments indicate that pH is an important factor in sample preparation and that further experiments may be necessary.
M ETHOD VALIDATION
Ideally, the retention time of the analytes should be identical in the standard solutions and the prepared samples, indicating the selectivity of the method, as well as its robustness. For all substances, the resulting surface area was substantially lower compared to the surface area of the lowest standard used. They were calculated by combining the results of duplicate sample preparation from all three days.
For the determination of reproducibility in this work, the analysis day and operators were varied. If certified reference material according to Directive 2002/657/EC is not available to determine the accuracy of the method, an spiked blank sample may be used instead. Validation results for recovery and correctness of the method (milk samples) Analyte recovery (corrected) [%] Correctness.
Method recovery and correctness validation results (meat samples) Analyte recovery (corrected) [%] Trueness. It is associated with the CCα value as well as the dispersion of the measured results. The tables indicate that for milk samples which have good repeatability and reproducibility as described above, CCβ values indicate a good performance of the method.
For this purpose, guidelines for the implementation of decision 2002/657/EC have been issued by the Directorate for Health and Consumer Protection of the EC (SANCO.
CHAPTER IV: CONCLUSION
C ONCLUSION
Furthermore, a multi-residue method for confirmatory determination of antibiotics in milk was presented by Samanidou and Nisyriou Multiclass, multi-residue method for the detection of antibiotic residues in distilled grains by liquid chromatography and ion trap tandem mass spectrometry”, Journal of Chromatography A, Full. 2003), "Ion suppression in mass spectrometry", Clinical Chemistry, Vol. 1992), "Preparative high-performance liquid chromatographic separation and isolation of bacitracin components and their relationship to microbiological activity", Journal of Chromatography A, Vol. 1963), “Bacitracin biosynthesis and spore formation:. Physiological role of an antibiotic", Archives of Biochemistry and Biophysics, Vol. 2010), "Development of a multi-class method for the quantification of veterinary drug residues in animal feed by liquid chromatography-tandem mass spectrometry", Journal of Chromatography A, Vol. Future trends from a historical perspective”, Journal of Chromatography A, Vol. 2008), “Development and validation of a reversed-phase high-performance liquid chromatography assay for polymyxin B in human plasma”, Journal of Antimicrobial Chemotherapy, Vol. 2001), “Determination of the Antibiotic Zinc Bacitracin in Animal Foods by High Performance Liquid Chromatography with Ultraviolet Detection”, Chromatographia, Vol. MRS.
/MS method development and validation for the determination of polymyxins and vancomycin in rat plasma”, Journal of Chromatography B, Vol. 2337/90 establishing a Community procedure for the establishment of maximum residue limits for veterinary medicinal products in foodstuffs of animal origin", Official Journal of the European Union, L of 17 December 1998 amending, as regards the withdrawal of authorization for certain antibiotics, Directive 70 /524/EEC on feed additives", L351. 1997), "Hyphenated Liquid Chromatography Method for the Determination of Colistin Residues in Bovine Tissues", Journal of Chromatography Science, Vol. 2011), “Validation of a new LC-MS/MS method for the quantification of colistin A and B in human plasma”, Journal of Separation Science, Vol. 1986), "Absorption, metabolism and excretion of zinc 14C-bacitracin fed to young pigs", Journal of Animal Physiology and Animal Nutrition, Vol. 2012), "Mass spectrometric analysis of muscle samples to detect potential misuse of antibiotic growth promoters in broilers", Food Additives &.
Determination of polymyxin E1 in rat plasma by high-performance liquid chromatography", Journal of Chromatography B, Vol. 2010), "Assay of colistin and colistin methanesulfonate in plasma and urine by liquid chromatography-tandem mass spectrometry", Antimicrobial Agents and Chemotherapy, Vol. Structural Mass Spectrometry : Rapid Methods for Separation and Analysis of Natural Peptide Products", Journal of Natural Products, Vol. 2003a), "Hyphenation of liquid chromatography to ion trap mass spectrometry to identify minor components in polypeptide antibiotics", Analytical and Bioanalytical Chemistry, Vol. 2003b), "Sequencing of Bacitracin A and Related Minor Components by Liquid Chromatography/Electrospray Ionization Ion Trap Tandem Mass Spectrometry", Rapid Communications in Mass Spectrometry, Vol. 1999), "Peptide Antibiotics", Antimicrobials and Chemotherapy, Vol. 1995), "Total structures and antimicrobial activity of small components of bacitracin", The Journal of Antibiotics, Vol. class, multi-residue liquid chromatography/tandem mass spectrometry screening and confirmation methods for drug residues in milk”, Rapid Communications in Mass Spectrometry, Vol. 2011), “Identification of impurities in polymyxin B and colistin bulk sample using liquid chromatography coupled to mass spectrometry”, Talanta, Vol.
Calculation of decision limit (CCα) and detection capability (CCβ) for banned substances: The imperfect marriage between quantitative and qualitative criteria,” Analytica Chimica Acta , vol. 2003), "Liquid chromatography-tandem mass spectrometric detection of inhibited antibacterial growth promoters in animal feed", Analytica Chimica Acta, Vol. 2006), "Detection of residual bacitracin A, colistin A and colistin B in milk and animal tissues by mass spectrometry coupled to liquid chromatography", Analytical and Bioanalytical Chemistry, Vol. 2012), "Analysis of Colistin A and B in Fishery Products by Ultra Performance Liquid Chromatography with Electrospray Positive Ionization Mass Spectrometry", Journal of Chromatography B, Vol.
Bacitracin
Colistin and Polymyxin
Tuning results (double-charged ions)
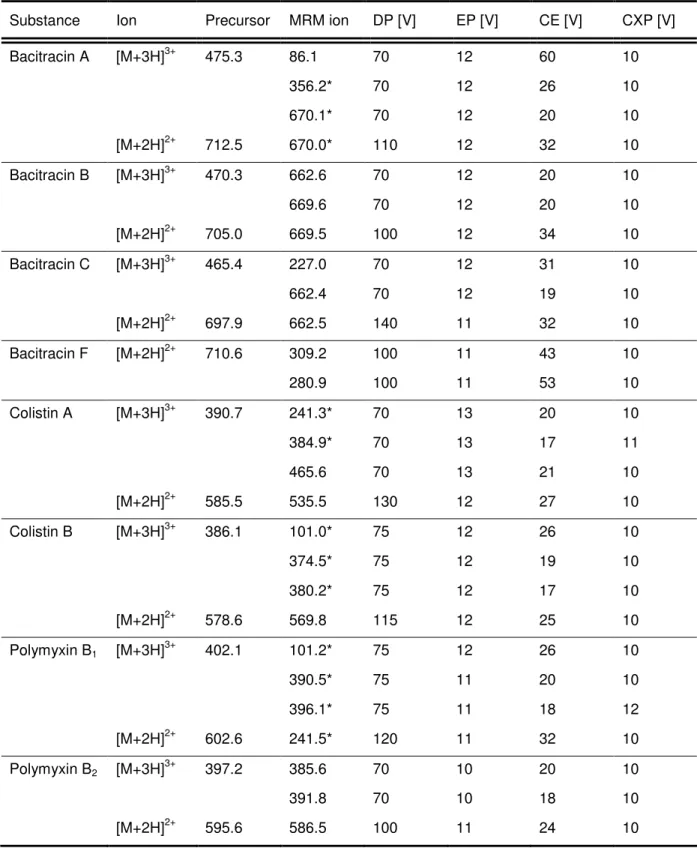
MRM transitions
![Table 2-6. Mass spectrometric parameters for standard composition determination Substance DP [V] EP [V] CE [V] CXP [V]](https://thumb-eu.123doks.com/thumbv2/pubdocorg/274339.42876/31.892.194.699.712.1170/table-mass-spectrometric-parameters-standard-composition-determination-substance.webp)
![Table 3-2. Initial HPLC gradient elution program (adapted from from Wan et al., 2006) Time [min.] Mobile phase A [%] Mobile phase B [%]](https://thumb-eu.123doks.com/thumbv2/pubdocorg/274339.42876/52.892.213.683.863.1034/table-initial-gradient-elution-program-adapted-mobile-mobile.webp)