In this work, we develop a program to solve the protein design problem using Answer Set Programming. The protein design problem consists of determining the best components of a protein for a given protein structure.
Contributions
The function of a protein is determined by its structure. Therefore, it is important to determine the best protein structure for a specific function. The study of protein structure and the best structure of a protein for a particular function can be useful in many areas.
Outline
In this chapter, the components of proteins will be described, and then the structure of amino acids and the relationship between them will be described in particular.
Protein Structure
Note that Figure 2.4 illustrates the Ψ angle for Amino Acid 1 and the Φ angle for Amino Acid 2, but each amino acid has the two dihedral angles. In Figure 2.5, position 7 of the protein has the amino acid Valine (V), which has only one dihedral angle (χ1), position 8 has Arginine (R), which has four dihedral angles (χ1, χ2, χ3, χ4), and position 10 has glutamic acid ( E), which has three dihedral angles (χ1,χ2,χ3).

Protein Design
Protein Design Problem
The purpose of protein design is to determine the set of amino acids that fold into a defined backbone structure, minimizing the energy of the protein. In protein design, it is common to keep the protein backbone fixed and determine the best side chain conformation of each amino acid.
Tools
On the website of the Protein Data Bank it is possible to see an image with the model of a protein and in most cases it is also possible to interact with a three-dimensional model. This library consists of rotational frequencies, mean dihedral angles, and variance as a function of backbone dihedral angles Φ and Ψ.

Related Work
The purpose of the dead-end elimination is to eliminate rotamers that cannot be in the GMEC. In the left branch of the tree, Alanine (A) is chosen to occupy the position of the protein.
Answer Set Programming Concepts
It is possible to optimize by the number of instances of specific predicates; for example, it is possible to minimize the number of instances of the predicate x. It is also possible to optimize according to weights, that is, it is possible to assign a weight to each predicate and then optimize the sum of the weights of the instances of the predicates3.

Answer Set Programming Language
The weak negation is negation as failure, that is, a literal is considered false if nothing says it is true. This rule represents that we wear a hat if we can assume that it is not raining. The second rule represents strong (or "classical") negation and states that we wear a hat if we know for sure that it will not rain.
Note that in this rule, the ability to assume that it is not raining is not enough to use a hat.
Tools
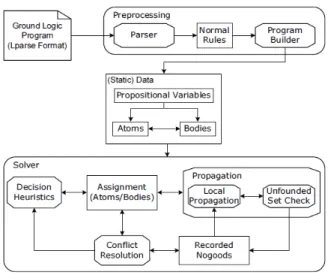
So if we have {eatCake, eatChocolate}1, this means that the answer sets must contain at most 1 atom of the set {eatCake, eatChocolate}, i.e. a response set can contain oneatCakeoreatChocolate or none, but cannot contain both. If we have 1{eatCake, eatChocolate}, this means that the answer must contain at least 1 atom of the set {eatCake, eatChocolate}, i.e. a response set can contain eatCake and/oreatChocolate, and must therefore have at least one of them. The square brackets in the rule mean that the sum of the weights of the occurrences of the predicate must be between 1 and 7.
Our goal is to solve a simplified version of the protein design optimization problem using answer set programming.
Problem Codification
Simple Codification
To represent the energy scores of the interaction between two rotamers at different positions, the predicate interEnergy(4.1.1.3) is used. For example, if we want to represent the interaction between the first rotamer at position 1 of the protein (e.g. it is also necessary to indicate that a rotamer is part of the solution, that is, a specific rotamer belongs to the designed protein).
To solve our problem, it is now necessary to present the rules of the problem.
Double Optimization
The rule that defines that if a rotamer belongs to the solution and has an energy score, then this score is The rule (4.1.2.7) means that if a rotamer with a non-negative energy score belongs to the solution, then the non-negative energy score also belongs to the solution. Analogously, the rule (4.1.2.8) means that if a rotamer with a negative energy score belongs to the solution, then the negative score also belongs to the solution.
The rule (4.1.2.9) means that if two rotamers belong to the solution and they have a non-negative interaction energy point, then that point also belongs to the solution and the rule means the same thing, but for negative interaction energies .
Alternative Codifications
Both codifications run out of memory for 20 positions very quickly due to the large number of possible combinations. Therefore, some entries that do not contribute to the best solution should be removed. The index predicate (4.1.3.3) represents the index for protein positions, where the index argument refers to the index value and the post-predicate refers to the position of the protein.
Note that this only applies to the second approach to the problem, where not only the rotamers but also the amino acids are also designed.
Dead End Elimination
Dead-End Elimination with ASP
The first (4.2.1.2) considers positions (Pos2) greater than the position (Pos) of the rotamer. The second rule (4.2.1.3) considers positions (Pos2) smaller than the position (Pos) of the rotamer. The above two rules summarize all the best and worst cases, respectively, and the result of the interaction energy of the considered rotamer with the protein backbone.
A rotamer is impossible if at the same position there is another rotamer with a worst situation that is worse than the best situation of the first rotamer.
Dead-End Elimination with Java
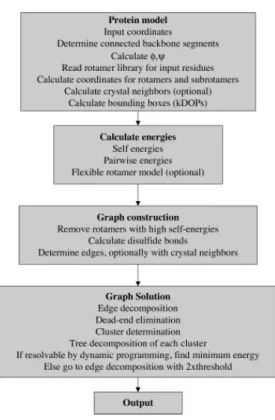
Therefore, a rotamer is eliminated if there is another rotamer that can replace the first one and reduce the total energy of the protein. If all the rotamers of the cleavage position are labeled, the original rotamer can be eliminated. The algorithm 3 [37] is the pseudo-code of the simple split dead-end elimination algorithm implemented.
Furthermore, we will describe the results of simple coding and dual optimization implementations.
Instances
Additionally, for the second approach described in Section 4.1, where the amino acids in the protein are not fixed, we considered two sets of amino acids. One set of amino acids considered contains all 20 essential amino acids described in Table 2.1. The second set of amino acids discussed contains only the 9 hydrophobic amino acids mentioned earlier (A, C, F, I, L, M, V, W, and Y).
Furthermore, for each set of positions we considered three sets of amino acids: assuming that the amino acids are fixed (first approach described in section 4.1); the set of hydrophobic amino acids; and all 20 essential amino acids (second approach).

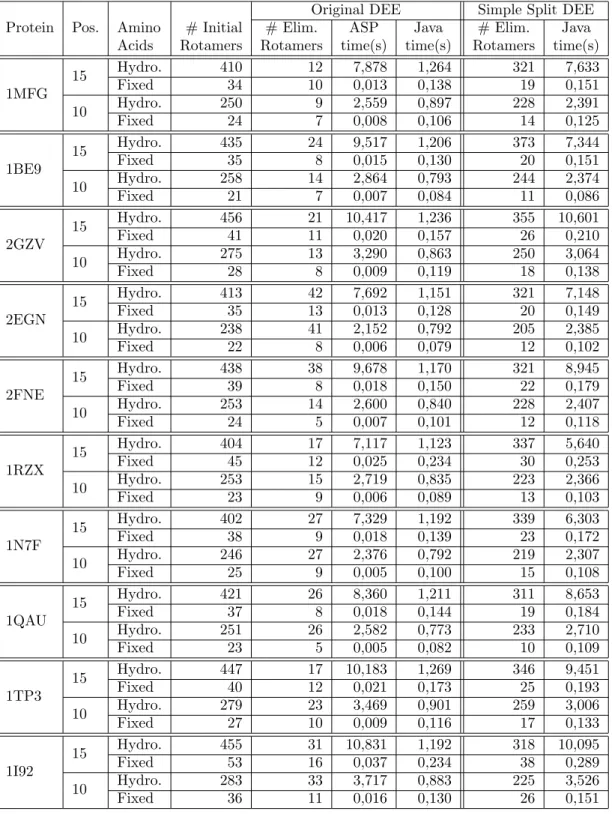
Dead-End Elimination Results
Problem Codifications Results
Table 5.3 shows that both codifications, the simple and the double optimization, do not differ much from each other. It is also possible to see that using the DEE algorithm first, because it reduces the number of rotamers, allows a larger number of cases to be solved. Of these 120 cases, 40 consider the 20 essential amino acids, the other 40 consider only hydrophobic amino acids, and the remaining 40 consider the amino acids specified as input.
In Appendix D it is possible to verify that the majority of unresolved cases were out of memory.
Discussion
It is possible to verify that time increases exponentially with the number of rotamers given as input. The goal of DEE is to find rotamers that cannot belong to the optimal solution of the protein design problem. In Figure 5.1(e), it is possible to verify that the dissolution time of ASP increases exponentially with the increase in the number of rotamers.
Moreover, it is possible to see that the implementation of the original DEE algorithm in ASP is worse than the implementation in Java of the same algorithm for a larger number of rotamers.

Future Work
A hint in a left diagonal means that the sum of the consecutive white boxes vertically above the hint must add up to the number in the hint. A hint in a straight diagonal means that the sum of the horizontally consecutive white cells must be the number given in the hint. To define a group of consecutive white cells (horizontal or vertical), we define the predicate group (Id,X,Y), which means that the cell with coordinates (X,Y) belongs to group Id.
The third rule in kakuro states that a consecutive group of white blood cells (horizontally or vertically) must add up the value of the respective hint.

Simple codification
Code
This section will show the complete code for simple codification and for double optimization. For better understanding, an example of input and corresponding output for each codification will be given.
Example
The optimization value of the optimal solution in this case is 12 because the conversions of negative values have been made. In fact, the value comes from the sum of the positive energies 3 and 0, from the (1,1, ala) rotamer and from the interaction between the two solvation rotamers respectively. The energy of the rotamer(2,2,ala), since it is a negative value, after the transformation has the absolute value of the negative interactions that do not belong to the solution.
Double Optimization Codification
Code
Example
This section shows the complete implementation of the original dead-end elimination algorithm using ASP. This section will show the complete results tables for the implemented dead-end elimination algorithms and for the two codifications performed. Tables D.4, D.5, and D.6 contain results for the Original, Simple Goldstein, and Simple Split DEE algorithms implemented in Java.
Tables D.10, D.11, and D.12 show the results of the simple coding where the Simple Split DEE algorithm was applied to each instance. The first three tables consider cases where no DEE algorithms have been applied. The last three tables show the results of the double optimization considering cases where the Simple Split DEE algorithm was applied. 12] Martin Gebser, Benjamin Kaufmann, Roland Kaminski, Max Ostrowski, Torsten Schaub and Marius Thomas Schneider.