Simulação molecular do equilíbrio de fases de hidrocarbonetos e suas misturas com CO2 e H2O utilizando o método de Monte Carlo. Simulação molecular de fases de equilíbrio de hidrocarbonetos e suas misturas com CO2 e H2O utilizando o método de Monte Carlo.
REVISÃO BIBLIOGRÁFICA
Métodos de simulação
A simulação computacional utilizando Monte Carlo é bastante difundida e já foi utilizada com sucesso para prever o equilíbrio de fases de hidrocarbonetos puros e misturas de hidrocarbonetos em diferentes faixas de pressão e temperatura, obtendo resultados em muitos casos concordantes com dados experimentais. O objetivo do trabalho é investigar o equilíbrio entre as fases de hidrocarbonetos e suas misturas com dióxido de carbono e água através de simulação molecular pelo método de Monte Carlo.
Superfície de Energia potencial e Campo de
Esses campos de força superestimam algumas propriedades dos alcanos de cadeia curta usando o potencial de Lennard-Jones e a regra de mistura de Lorentz-Berthelot. Os campos de força SPC e TIP4P (pertencentes ao grupo OPLS) são frequentemente usados para descrever as propriedades termodinâmicas da água líquida em condições ambientais, mas não são necessariamente adequados para prever as propriedades de equilíbrio de fase em temperaturas mais altas (Vorholz et al. al. , 2000). ).Esses campos.

O Método de Monte Carlo
É importante destacar que no método Monte Carlo a aceitação do próximo movimento proposto é influenciada pelo último movimento aceito. Ulam e Metropolis (1949 apud Ulam; Smith, Haymet, 1993) publicaram seu primeiro trabalho utilizando o método Monte Carlo em 1949.
Função de distribuição de probabilidades e
Em 1953, Metropolis, Rosenbluth e Teller (Metropolis et al.) apresentaram o método de Monte Carlo envolvendo o critério de equilíbrio detalhado. O desenvolvimento da simulação ocorre através de uma série de ciclos, conhecidos como ciclos de Monte Carlo.
Aplicação da metodologia
Existem vários métodos e todos visam gerar ajustes de acordo com a distribuição de equilíbrio de um conjunto. Apenas os arranjos criados na fase de equilíbrio são levados em consideração nos cálculos das propriedades termodinâmicas.
Condição de contorno periódica
Em resumo, iniciando a simulação com uma randomização configuracional, cada molécula 'i' é selecionada e um movimento aleatório é realizado nesta molécula. Portanto, outra molécula é então selecionada e o mesmo procedimento adotado com a molécula “i” é repetido até que um ciclo com “N” moléculas seja completado.

A Função de autocorrelação temporal
No final da simulação há uma cadeia de arranjos que descrevem a evolução da simulação (Coutinho. Uma função de autocorrelação próxima de +1 significa que os dados estão diretamente correlacionados, próxima de -1 significa que os dados estão inversamente correlacionados, e próxima de zero significa que os dados não estão correlacionados.
A Função de distribuição radial de pares
Integrando a função G(r) é possível estimar o número de átomos que estão espalhados em torno de outro átomo. Em sistemas com átomos do tipo i e j, a equação (13) representa o número de átomos i que estão distribuídos em torno de um átomo j.

As pressões de bolha para dez misturas ternárias foram previstas usando regras de mistura e correlações desenvolvidas a partir de dados experimentais para apenas vinte e uma misturas binárias. Devido à falta de dados experimentais sobre a densidade da fase líquida na literatura, foi realizada uma estimativa qualitativa da densidade com base no comportamento da pressão de bolha descrito pela equação de estado. Os campos de força TraPPE foram aplicados a substâncias puras de CO2 e N2 e conseguiram representar os dados experimentais com boa precisão.
No estudo, simulações de Monte Carlo usando dinâmica molecular são realizadas no conjunto de Gibbs (NVT e NPT) para descrever o equilíbrio líquido-vapor da água em temperaturas entre 323k e 573K, do dióxido de carbono em temperaturas entre 230k e 290K, e do binário mistura CO2 – H2O em temperaturas entre 348k e 393K. O objetivo do artigo foi prever o equilíbrio de fases do sistema binário CO2 – H2O por simulação molecular e comparar os resultados obtidos com dados experimentais. As previsões do equilíbrio de fases de substâncias puras mostraram boa concordância com os dados experimentais. Modelos termodinâmicos foram desenvolvidos para calcular a solubilidade mútua ou unilateral desta mistura (Hu et al, 2007). A baixas temperaturas, geralmente é alcançada concordância com os dados experimentais (Spycher, Pruess, 2010; Spycher, Pruess, 2003;
O melhor campo de força para prever a solubilidade do CO2 em água a 323,15 K e 423,15 K é o TraPPE/TIP4P2005, pois geralmente concorda com os dados experimentais para a faixa de pressão investigada. Os dados da fase gasosa são mais difíceis de obter devido à falta de concordância entre os dados experimentais de diferentes laboratórios. Todos os modelos testados capturam qualitativamente a dependência da solubilidade com pressão e temperatura, mas ainda não são capazes de reproduzir os dados experimentais em toda a faixa de pressão e temperatura investigada.
METODOLOGIA
- Pentano
- Pentano + CO 2
- Metano + CO 2 + H 2 O
- O simulador MCCCS Towhee
A função de distribuição radial foi calculada em cada fase a uma temperatura de 343k com o campo de força TraPPE para fornecer evidências da estrutura das moléculas nas caixas no estado de equilíbrio. O conjunto a ser utilizado (comando 'ensemble', seguido do conjunto selecionado na linha seguinte, no caso deste trabalho, NVT); O número de componentes do sistema (comando 'nmolty', seguido do número de componentes selecionados na linha seguinte);
O número de partículas de cada componente (comando 'nmolectyp', seguido do número de partículas de cada componente na linha seguinte); O número de movimentos que as partículas irão realizar (comando 'nstep', seguido do número de ciclos selecionado na próxima linha); Em quantas partes deseja dividir o sistema, ou seja, em quantos subblocos (comando 'tamanho do bloco', seguido do número de divisões escolhido na próxima linha);
Número inicial de partículas em cada caixa de simulação (comando 'initmol', seguido do número de moléculas selecionadas na linha seguinte); Campo de força potencial (o comando 'ffnumber', seguido do caminho para a pasta onde o campo de força está armazenado); O potencial de interação intermolecular (comando 'potencial-clássico', seguido do potencial selecionado na linha seguinte, foi utilizado no caso deste trabalho.
RESULTADOS E DISCUSSÕES
Pentano
Foram realizadas simulações com os campos de força SKS, Amber e MM2, na temperatura de 343K, para fazer comparação com os dados obtidos com o campo de força TraPPE. No campo de força SKS, as variações observadas entre as entalpias de vaporização estimadas foram maiores que no campo de força TraPPE. O tempo computacional para completar as simulações com o campo de força SKS foi de 2 a 4 dias, semelhante ao realizado com o campo de força TraPPE.
No campo de força Amber, a diferença nas estimativas de entalpia de vaporização obtidas a partir de termalizações e equilíbrios teóricos foi pequena, mas maior que a observada no campo de força TraPPE. Nas simulações do campo de força Amber, a mudança observada na entalpia teórica dos dados de vaporização foi maior que os dados experimentais. No campo de forças MM2, a diferença nas estimativas de entalpia de vaporização obtidas nas etapas de termalização e nas etapas de equilíbrio foi significativa, representando um erro relativo superior a 10%.
O expoente não pôde ser ajustado para a caixa de fluido (caixa 1) no campo de força SKS. Para a caixa fluida, no campo de força TraPPE os dados apresentaram grande dispersão e também a função de autocorrelação (ρk) ficou muito próxima de zero desde os primeiros ciclos e durante todo o intervalo de simulação (entre 0,002 e -0,005), indicando que o sistema está em equilíbrio e que os dados de energia não estão correlacionados. Para a caixa de vapor pode ser feito um ajuste exponencial no campo de força do TraPPE nos primeiros 4000 ciclos.

A mistura binária Pentano – CO 2
A função de distribuição radial foi calculada para cada fase da mistura de pentano e dióxido de carbono a uma temperatura de 408,15 K em relação ao átomo central de cada uma. Foi possível observar no gráfico 19 (ANEXO Z) e no gráfico 20 (ANEXO AA) que o comportamento da função distribuição radial no quadro 1 indica que ela descreve a mistura na fase líquida. Além disso, é possível observar que as moléculas de pentano são mais solvatadas pelas moléculas de CO2 do que seriam num sistema de distribuição aleatória, e as moléculas de CO2 também são mais solvatadas nas moléculas de pentano em comparação com o mesmo sistema.
O sistema atingiu o equilíbrio com 506 moléculas na fase líquida, 456 moléculas de pentano e 50 moléculas de CO2. A curva de dissolução de CO2 para moléculas de CO2 na caixa líquida pode ter sido comprometida devido à baixa quantidade de moléculas de CO2 na fase líquida. O comportamento da função distribuição radial no quadro 2 pode ser observado no gráfico 21 (BILAG AB) e gráfico 22 (BILAG AC), e indica a descrição das moléculas na fase vapor.
As moléculas de pentano são mais solúveis que as moléculas de CO2 em comparação com um sistema de distribuição aleatória, assim como as moléculas de CO2 também são mais solúveis que as moléculas de pentano em comparação com o mesmo sistema. O sistema atingiu o equilíbrio com 494 moléculas na fase de vapor, 344 moléculas de pentano e 150 moléculas de CO2. A curva G(r) para solvatação de CO2 a partir de moléculas de CO2 na caixa de vapor foi praticamente zero, o que não era esperado.

As moléculas de metano são dissolvidas principalmente por moléculas de CO2 e igualmente por moléculas de CH4 e H2O. As funções G(r) da solvatação de H2O por moléculas de H2O e de CO2 por moléculas de H2O podem ter sido insignificantes devido à baixa concentração global de água no sistema. As funções de solvatação de H2O das moléculas de CO2 e CO2 das moléculas de CO2 foram negativas.
O sistema atingiu o equilíbrio com 173 moléculas na fase líquida, 20 moléculas de metano, 77 moléculas de CO2 e 80 moléculas de água. As moléculas de metano são mais solvatadas por moléculas de CH4, as moléculas de CO2 e H2O são solvatadas por moléculas de metano. O sistema atingiu o equilíbrio com 823 moléculas na fase líquida, 280 moléculas de metano, 523 moléculas de CO2 e 20 moléculas de água.
D'Souza R.; Patrick JR; Teja AS; 1988, High-pressure phase equilibria in the carbon dioxide – n-hexadecane and carbon dioxide – water systems, The Canadian Journal of Chemical Engineering. Jimenez, R.; Galicia, LA; Solis, O.E.; Experimental vapor-liquid equilibria for the carbon dioxide + octane and carbon dioxide + decane systems, Journal of Chemical Engineering and Data. Potoff, J.J.; Siepmann, J.I.; Vapor – Liquid Equilibria of Mixtures Containing Alkanes, Carbon Dioxide and Nitrogen, AIChE Journal, Vol.
J.; Lin H.; Chao K.; Vapor-liquid equilibrium in binary mixtures of carbon dioxide + n-decane and carbon dioxide + n-hexadecane, J. Su, C.S.; Prediction of volumetric properties of carbon dioxide expanded organic solvents using the Predictive Soave–Redlich–Kwong (PSRK) equation of state, J .

Tabela 4
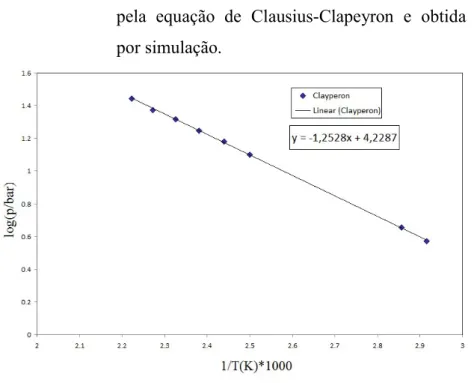
Gráfico 17
Gráfico 18