A ce jour, tous les gènes responsables identifiés dans l'épilepsie monogénique codent pour des canaux ioniques, à l'exception du gène récemment découvert dans l'épilepsie partielle avec hallucinations auditives. Les principaux syndromes d'épilepsie idiopathique généralisée (IGG) (épilepsie-absence de l'enfant, épilepsie myoclonique juvénile (EMJ), épilepsie grand mal éveillée) ont un mode de transmission complexe et multigénique. Le gène CLCN2 correspondrait à un gène majeur à l'origine de l'épilepsie dans ces 3 familles, mais d'autres gènes non identifiés ou un fond génétique différent peuvent expliquer les phénotypes distincts observés au sein d'une même famille.
Le CF+ peut être associé à d'autres types de crises : i) les absences, qui sont rares, les distinguant de l'épilepsie-absence de l'enfant, ii) les myoclonies, iii) les crises atoniques voire l'épilepsie myoclonique-astatique. Cependant, une bilinéarité a été notée chez les sujets atteints d'EAE (antécédents d'épilepsie dans les branches paternelle et maternelle), suggérant que d'autres gènes mutés participent à ce phénotype. Des mutations de ce gène ont même été identifiées dans des cas d'épilepsie myoclonique sévère de l'enfant (SMEI) [10].
Des variations interfamiliales et intrafamiliales considérables sont observées, notamment en ce qui concerne la sévérité de l'épilepsie ou la réponse aux médicaments antiépileptiques. La recherche de mutation du récepteur nicotinique a également été réalisée dans des cas a priori non familiaux d'épilepsie frontale avec crises nocturnes et a permis d'identifier une mutation dans un seul cas. Cela montre que l'origine de l'épilepsie partielle peut être génétique même en l'absence d'antécédents familiaux d'épilepsie.
Épilepsie partielle familiale à foyer variable L'épilepsie partielle familiale à foyer variable est une épilepsie partielle autosomique dominante avec des crises partielles provenant de différents lobes du cerveau chez différents membres affectés d'une famille [27].
Chromosomenaberrationen und Epilepsie
Patau-Syndrom) und Trisomie 18 (Ed- wards Syndrom)
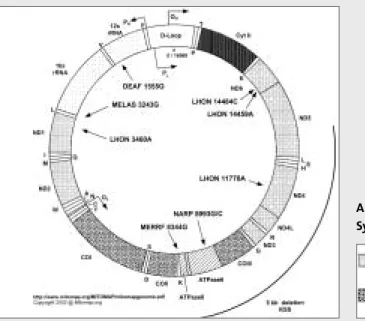
Die Diagnose einer Trisomie 13 oder 18 kann heute sehr oft pränatal gestellt werden (Fehlbildungen durch Ultraschall, pränatale Chromosomenuntersuchung) und die Schwangerschaft auf Wunsch der Eltern abgebrochen oder bei der Geburt wird das Neugeborene intensivmedizinisch betreut, lebenslang auf verlängernde Maßnahmen wird verzichtet. Bei der Bildung der Ringchromosomen geht in der Regel ein Teil der beteiligten Subtelomere verloren, manchmal liegt aber auch eine größere Deletion vor, wie im Fall des Ringchromosoms 13. Die meisten Patienten mit einer De-novo-Deletion des distalen Segments von 1p (dh keine familiäre unausgeglichene Translokation) wurden durch subtelomeres Screening entdeckt.
Epilepsie beim Wolf-Hirschhorn-Syndrom manifestiert sich meist innerhalb des ersten Lebensjahres und ist ein häufiger Grund für die kurze Überlebenszeit: Etwa 80 % der Patienten sterben innerhalb der ersten 24 Monate nach der Geburt. Ein Kandidatengen für Epilepsie beim Wolf-Hirschhorn-Syndrom war das für eine GABAA-Rezeptor-Untereinheit. Die gelöschte Region ist Bande 17p13.3, die das Lissenzephalie-Typ-1-Gen (LIS1) enthält und durch Haploinsuffizienz Lissenzephalie Typ 1 (vollständige Agyrie) verursacht [35].
Ein unspezifischer dysmorphologischer Phänotyp, Entwicklungsverzögerung, Mikrozephalie und Epilepsie sind charakteristische Befunde vieler Patienten mit autosomalen Ringchromosomen. Epilepsie tritt bei einem extrem hohen Anteil der Patienten mit den Ringchromosomen 14, 17 und 20 auf (Tabelle 2). Bei der Diagnose dieser Patienten besteht möglicherweise eine Tendenz zur Epilepsie, da Epilepsie praktisch bei jedem Menschen diagnostiziert wurde, der das dritte Lebensjahr erreicht hat.
Patienten mit Ring 17 können in zwei Gruppen eingeteilt werden: solche mit Lissenzephalie (und damit verbundenen Hirnfehlbildungen) und solche ohne diese Hirnbefunde. Bei Patienten mit Lissenzephalie liegt der Bruchpunkt auf dem kurzen Arm von Chromosom 17 proximal zur Miller-Dieker-Deletion (17p13.3), was zu schweren neurologischen Anomalien führt. Abhängig von den Auswahlkriterien können Screening-Untersuchungen auf submikroskopische Chromosomenanomalien im Bereich des Subtelomers bei 4–8 % der untersuchten Patienten unausgeglichene Chromosomenanomalien zeigen (meist bei nicht klassifizierbaren multiplen Fehlbildungen mit Entwicklungsverzögerung).
Die folgenden Punkte können dem Arzt bei der Entscheidung helfen, ob bei einer entwicklungsverzögerten Person ein subtelomeres Screening durchgeführt werden soll oder nicht: 1. Aus diesem Grund ist das durchschnittliche Alter der Mutter bei der Empfängnis erhöht, ähnlich wie bei anderen Trisomien. Epilepsie ist nicht leicht zu kontrollieren und manchmal kommt es zu einer Beeinträchtigung der geistigen Fähigkeiten.
Klinische und genetische Aspekte familiärer Kavernome
In ihrer bahnbrechenden Studie postulierten Rigamonti et al., dass bis zu 50 % aller Kavernomträger die familiäre Form haben [19]. Der erste genetische Durchbruch gelang 1995 Dubovsky et al., die einen Zusammenhang zwischen Kavernomen und Markern auf Chromosom 7q (7q11-22) fanden. De-novo-Läsionen bei einer familiären Form zerebraler kavernöser Fehlbildungen: klinische und MR-Merkmale in 29 nicht-hispanischen Familien.
Linkage of the locus for cerebral cavernous hemangiomas to human chromosome 7q in four families of Mexican-American descent. Multilocus linkage identifies two new loci for a Mendelian stroke form, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Linkage to the CCM 2 locus and evidence of genetic heterogeneity in familial cerebral cavernous malformations.
Mutations in the gene encoding Krit 1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations. Interaction between Krit 1 and icap1alpha implicates integrin beta1-mediated disruption of angiogenesis in the pathogenesis of cerebral cavernous malformation. Analysis of the CCM 1 gene in families segregating cerebral cavernous malformations: identification of novel mutations and identification of extracranial manifestations.
Mutations in a gene encoding a novel phosphotyrosine-binding domain protein cause cerebral cavernous malformations type 2 (CCM 2).
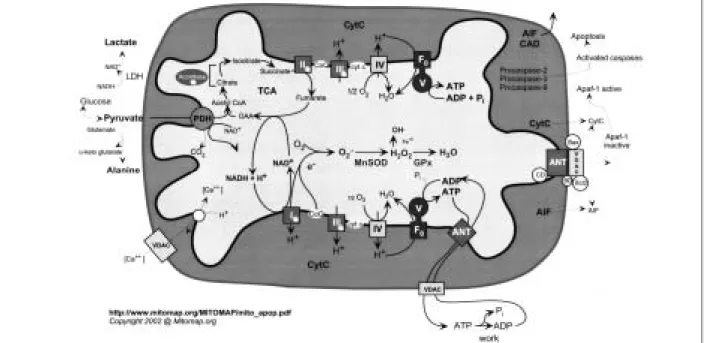
Epilepsie als Symptom mitochondrialer Zytopathien
Die Bedeutung der molekulargenetischen Diagnostik in der Differenzialdiagnose der progressiven myoklonischen Epilepsie. Differenzialdiagnose der progressiven myoklonischen Epilepsie. Sabina Gallati, Molekulare Humangenetik, Inselspital Bern, für die Durchführung der mtDNA-Analyse bei Patient 1 und Dr. Michael Morris, Medizinische Genetik an der Universität Genf, für den konstruktiven Dialog und die Analysen der mtDNA bei Patient 1 und des Cystatin-B-Gens bei Patient 2, Dr.
Hinsichtlich der klinischen Symptome waren die wesentlichen Unterschiede zum Zeitpunkt der endgültigen Diagnose wie folgt: Patient 1 hatte die ersten Myoklonien und generalisierten tonisch-klonischen Anfälle im Alter von 14 Jahren, Patient 2 im Alter von 8 Jahren. Bei Patient 1 traten 1999 Im Alter von 25 Jahren fiel im neurologischen Status erstmals eine Hypakusis auf, bei der Fingerreiben auf beiden Seiten nicht mehr zu hören war, was in der Audiometrie objektivierbar war (Tabelle 4), während bei Patient 2 das Hörvermögen vorhanden ist blieb bis heute erhalten. Patient 2 beschrieb im Alter von 28–29 Jahren auch nicht-neurologische Symptome, nämlich Anfälle von Atemnot, Angstzuständen und Gesichtsrötung.
Obwohl die visuell evozierten Reaktionen von Patient 1 im Laufe der Zeit ebenfalls pathologisch wurden, blieben die visuell evozierten Fähigkeiten von Patient 2 normal. Die Ergebnisse der MRT-Untersuchungen spiegelten den Ernst der klinischen Situation wider, nämlich dass Patient 1 im Jahr 1999 nur eine leichte Kleinhirnatrophie aufwies, Patient 2 jedoch im Jahr 2000 eine generalisierte, kleinhirnbetonte Hirnatrophie. Ein für den weiteren Verlauf der Diagnose bedeutsamer Unterschied zwischen beiden zeigte sich in der Audiometrie, wobei Patient 1 entsprechend der im Neurostatus 1999 festgestellten Hypakusis eine Schallempfindungsschwerhörigkeit aufwies, während Patient 2 erwartungsgemäß einen normalen Befund aufwies.
Bei Patient 1 war jedoch bereits 1995 für Prof. eine Analyse der mtDNA aus Blutzellen durchgeführt worden. Untersuchung des Cystatin-B-Gens in Genf durch Dr. Neben der mütterlichen Migräneanamnese weisen vor allem leicht erhöhte Liquorlaktatwerte auf eine MZ hin.
Ungewöhnlich ist auch die bei Patient 1 beobachtete und bisher nur für ULE beschriebene Verschlechterung unter Phenytoin [1]. Der Patient leidet an progressiver Myoklonus-Epilepsie ohne gerissene rote Fasern, Laktatazidose und Hypokusis, und zwar zweimal. Die bei Patient 1 festgestellte Mutation wurde bisher hauptsächlich bei Patienten mit MELAS beschrieben[17].
Patient 1 erläutert konkret die Bedeutung subtiler klinischer Befunde und Aufarbeitung einerseits und des Dialogs mit dem Molekulargenetiker andererseits. Zusammenfassung und Schlussfolgerungen Ein Überblick über den aktuellen Wissensstand zur molekularen Genetik von PME legt nahe, dass die molekulargenetische Diagnostik in Zukunft eine immer wichtigere Rolle bei der Aufklärung der PME zugrunde liegenden Entität spielen wird.
Pharmakogenetik von Antiepileptika
Oktober fand in Bern der von Epilepsie-Liga und epi-suisse gemeinsam organisierte Hauptanlass
Die Gespräche, die Ellinor von Kauffungen mit zwei betroffenen Jugendlichen und ihren Eltern führte, bildeten die Grundlage für eine von Regina Henggeler-Dimmler, epi-Suisse, moderierte Diskussion. Basierend auf dem MOSES-Bildungsprogramm für Erwachsene soll der derzeit in Vorbereitung befindliche FAMOSES-Kurs Eltern, Geschwistern und Betroffenen helfen, sich der Krankheit zu stellen, sie zu akzeptieren und ihre Auswirkungen besser zu verstehen. Geplant sind drei separate Kurse, einer für Schüler, einer für Jugendliche und einer für Eltern, die sich alle mit der Stellung des Betroffenen in der Familie und seinem Verhältnis zu Geschwistern und Eltern befassen.
Ziel der Bewegung ist die Stärkung des Selbstvertrauens, der Alltag soll selbständiger bewältigt werden und Eltern versuchen, Überfürsorglichkeit zu vermeiden. Diesbezüglich wurden relevante Erfahrungen mit dem bereits bestehenden MOSES-Trainingsprogramm für Erwachsene gesammelt. Neben einer optimalen medizinischen Betreuung durch einen Facharzt ist die soziale Integration betroffener Kinder und Jugendlicher äußerst wichtig.
Dazu gehört die Gewährung von Freiheit, Unabhängigkeit und Teilhabe an altersgerechten sozialen Aktivitäten.
Tag der Epilepsie
Bewerbungen sind aus allen Fachgebieten und Berufsgruppen möglich und willkommen, sowohl aus den Kernfächern als auch aus den klinischen Fächern. Die Jury besteht aus drei Mitgliedern des SLgE-Vorstands, die bei Bedarf weitere externe Experten hinzuziehen können. Die neuen Broschüren der Epilepsie-Liga liegen zum Ausdrucken in Arztpraxen oder Krankenhäusern auf.
Ausschreibung - Forschungsförderung
April
Die Schweizerische Liga gegen Epilepsie (SLgE) vergibt neu einen jährlichen Preis von
Jahrestagung der Deutschen Sektion der Internatio- nalen Liga gegen Epilepsie
Arbeitstagung des Deutsch-Österreichischen- Schweizer Arbeitskreises (Dach-AK) Epilepsie
Kongresskalender
Impressum
Autorenverzeichnis Jahrgang 20 | 2003
April 2003
August 2003
Dezember 2003
Inhaltsverzeichnis Jahrgang 20 | 2003


![Tabelle 2: Klinik der progressiven Myoklonus-Epilepsien nach „Clinical Synopsis“ aus OMIM [5], Stand 8/03](https://thumb-eu.123doks.com/thumbv2/pubpdfco/325573.41366/34.892.104.817.108.367/tabelle-klinik-progressiven-myoklonus-epilepsien-clinical-synopsis-stand.webp)


