I would also like to thank all my professional collaborators who contributed to the thesis: Research Center of Dr.
INTRODUCTION
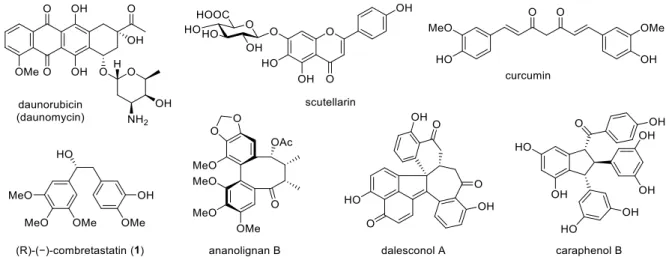
Natural polyphenols
3 alone is a precursor of coumarins, the combination of 3 with malonyl-CoA (4) results in the formation of flavonoids, 10 curcuminoids and stilbenoids. Oxidation of p -coumaric acid ( 2a ) or 3 , followed by O -methylation, leads to an important series of substituted cinnamic acids 2b–d ( Scheme 1 ).
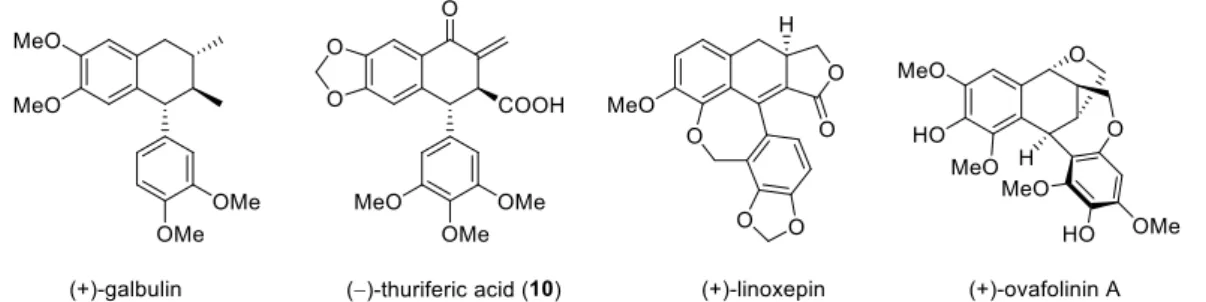
Lignans
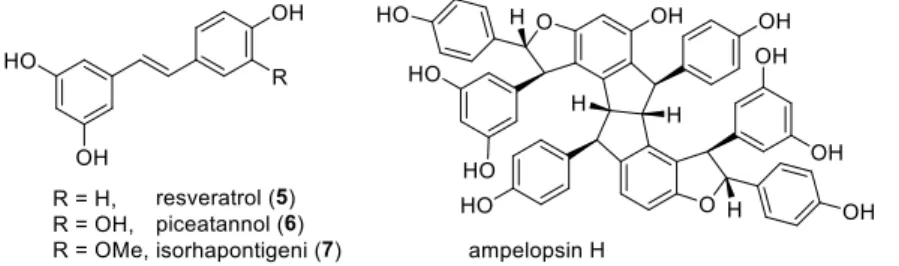
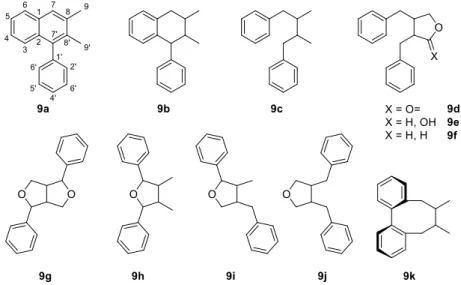
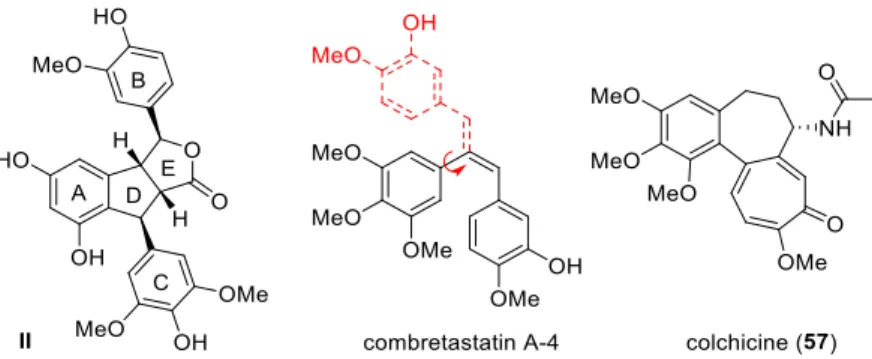
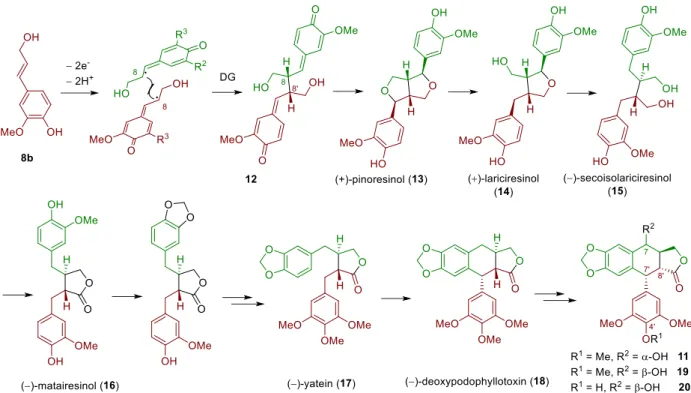
Stilbenoids include simple stilbenes such as 5-7 (Figure 2), dihydrostilbenes such as combretastain 1 (Figure 1) and dimeric or oligomeric stilbenoids, which are formed in a wide range of structural types by the coupling of free radicals formed by stilbenes during the oxidation of laccases and other enzymes. Cyclolignans from the aryltetralin subgroup are characterized by the additional carbon-carbon bond between the atoms C-2 and C-7'.
Podophyllotoxin
- Isolation and biosynthesis of podophyllotoxin
- Medicinal relevance of podophyllotoxin
- Selected total syntheses of podophyllotoxin
This reduction demonstrates the weak nature of the benzyl ether bond with respect to heterolysis (Section 4.2.10). Another approach to ring C formation was published in 2017 by Harja et al.67 An intramolecular Heck reaction of bromide 40 is used to form a bond between C-2 and C-7', yielding the unsaturated lactone 41 (Scheme 9).
Stereoselective coupling of phenoxyl radicals
- Stereoselective coupling by laccases and monooxygenases
- Dirigent proteins and the stereoselective coupling of lignols
- Diastereoselective coupling of persistent radicals in free solution
The fungal monooxygenase KtnC from Aspergillus niger was found to catalyze the regio- and stereoselective dimerization of coumarin demethylsiderine (42) to give P-(+)-orlandine (43) (Scheme 10),73 which is then O-methylated to give P-ine > 9%-cotane. However, the biggest paradigm-changing discovery came with the description of so-called conducting proteins (DIRs).
Non-classical lignans - furoindane stilbenolignans (FIS)
- Biological activity of FIS
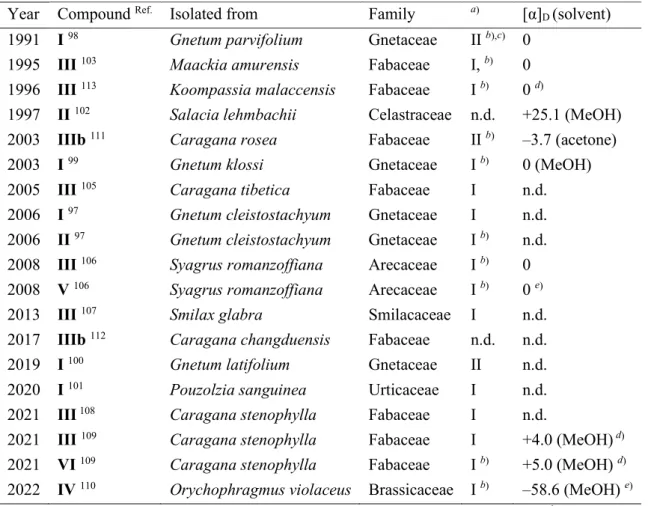
Later, based on the NOE study of the free phenol, the relative configuration was reassigned as rel-(7S,8R,7'R,8'S), type-I.99. I 100 Anti-inflammatory: neuroinflammation reduction, suppresses the upregulation of NO release by Aβ1-42 (amyloid beta1-42) transfection in BV2 cells (microglial cells).
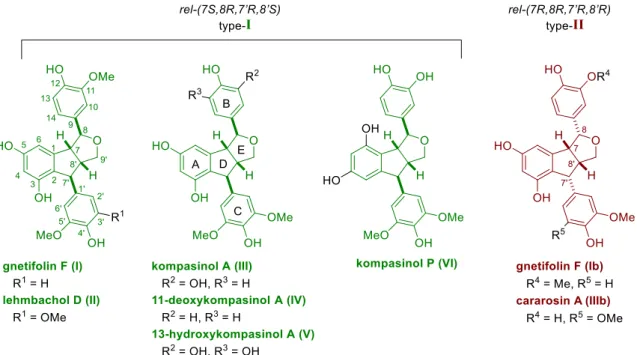
Synthetic approaches to arylindanes
Interestingly, compared to the reported exclusive 5-exo-tet cyclization of epoxymalonate 67 (Scheme 21), both products of the 5-exo-tet (72) and 6-exo-tet (73) cyclizations were isolated, with the ratio depending on the nature of the base used. The difficult predictability of the cyclization mode during nucleophilic opening of epoxides is further demonstrated by the 6- exo -selective cyclization of Fischer carbene complex-stabilized anion 76 derived from lithiostilbene 75 , previously reported by Florio et al.
- Transition metal catalysed asymmetric conjugate addition
- Direct asymmetric conjugate addition of organolithium compounds
The synthesis began with conjugate addition of PhB(OH)2 to cinnamate 78, yielding adduct 79 in 88% yield and 96% ee. Two catalytic systems for the conjugate addition to cinnamates were developed by the Miyaura group, both based on the bidentate phosphine ligand chiraphos.
HYPOTHESIS AND RETROSYNTHETIC ANALYSIS
Bioinspired approach to natural arylindanes and aryltetralins
The bond between atoms C-2 and C-7' in 92 can be formed by conjugate addition, the second bond by addition of radical at C-8' to C-7 followed by SET oxidation, and the third bond between C-8 and the oxygen atom at C-9' can be formed via lactonization of the benzyl carbocation at C-8. Similarly in 11, the dissonant relationship between nucleophilic atoms C-8 and C-8' can be resolved by successive SET oxidations with an "in-between" radical cyclization.
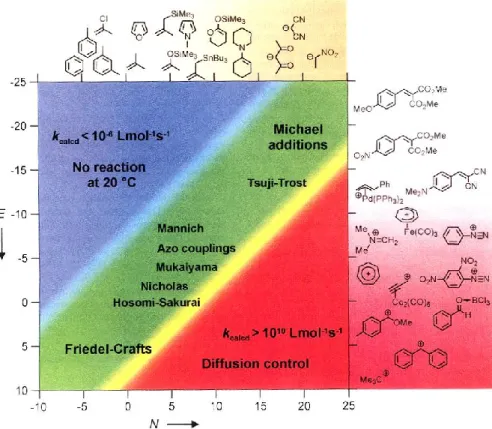
Dirigent protein hypothesis and optical purity of natural FIS
AIMS OF THE WORK
RESULTS AND DISCUSSION
Part A. Cu-catalysed conjugate addition/oxidative cyclisation
- Design of the key annulation combining conjugate addition/oxidation
- Initial exploration of in situ oxidative cyclisation
- Cu-catalysed conjugate addition of lithiated stilbenes to ylidene malonates
- Radical v. polar annulation of model stilbenes
- Racemic synthesis of kompasinol A pentamethyl ether
With the optimized conditions for lithiation and conjugate addition established, we proceeded to in situ oxidation of the enolate. Because successful one-pot oxidative annulation requires full conversion to the Michael adduct, re-optimization of the conjugate addition was necessary.
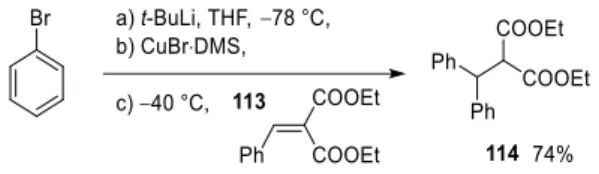
Part B: Total synthesis of FIS via direct ArLi addition/oxidative cyclisation
- Direct conjugate addition of ArLi to cinnamates – model study
- Retrosynthetic analysis of gnetifolin F
- Synthesis of tert-butyl protected isorhapontigenin 151 and attempted conjugate
- Synthesis of benzyl-protected isorhapontigenin 157
- Selection and synthesis of Li-specific ligands
- Conjugate addition of stilbene 157 to cinnamate 150
- Asymmetric conjugate addition of 157 to malonates
- Enantioselectivity rationale – transition state
- Total synthesis of (+)-gnetifolin F (I)
- Total synthesis of (+)-11-deoxykompasinol A (IV)
- Towards kompasinol A (III)
Addition of conjugate can be decoupled from the oxidative cyclization, and instead capture of the enolate of 150 by ethyl chloroformate would generate malonate 149 required for cyclization. By running the reaction under controlled vacuum, the by-products can be continuously removed during addition of Boc2O, preserving the high reactivity of the catalyst. We reasoned that the high steric demand of the t-Bu group adjacent to the lithiated center was probably incompatible with the conjugate addition.
Based on the unsuccessful addition of 151 , the strategy for protecting the isorhapontigenin moiety of gnetifolin F (I) was reconsidered. Finally, the ligands must be sufficiently soluble in aromatic hydrocarbons to be compatible with the cryogenic conditions of the conjugate addition. Next, we turned our attention to the last component of the direct conjugate addition, which is the Michael acceptor.
Coordination of the malonate can occur either from its Re side (Scheme 54, upper path) leading to the 174-I transition state, or from its Si side (lower path) leading to TS 174-II.
Part C: Approaches to podophyllotoxin via direct conjugate addition to cinnamate
- First generation radical 6-exo-tet approach
- Second generation approach based on RCM/conjugate addition
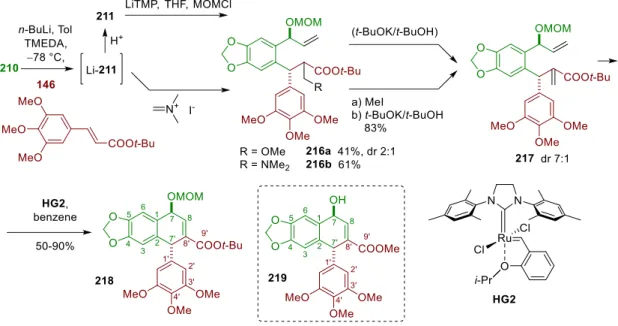
Due to the lower boiling point of the MOMCl and its contaminants, this should circumvent the above problem. In this revised approach, we decided to install one additional carbon atom at C-8' via one of the standard methyleneation procedures. The chemical shifts of the minor diastereomer 218' on the other hand differed significantly from 219. d) Not detected. e) Detected by HMBC.
Since toluene is a strictly non-coordinating solvent with respect to Li, the four coordination sites of the Li atom must be occupied by substrates and/or TMEDA. The basis of the model is a 5-membered metallacycle, formed by chelation of the Li atom by the ether oxygen of the MOM group (scheme 68). After establishing the shortcut to 218 , we planned to continue the synthesis by adding the remaining C-9 carbon via another conjugate addition ( Scheme 69a ).
In order to make the synthesis asymmetric, enantioselective version of the initial vinylation of 6-bromopiperonal was investigated.
Part D: Approaches to neopodophyllotoxin polar bicyclisation of epoxystilbenes
- Synthesis of neopodophyllotoxin analog
- Rapid synthesis of neopodophyllotoxin core – racemic approach
- Initial optimization of organocatalytic Corey-Chaykovsky epoxidation
- Asymmetric Corey-Chaykovsky epoxidation with activated alkyl halides
Since 231b is derived from the minor cis isomer of epoxide 230 , the next round of efforts focused on optimizing the Corey–Chaykovsky epoxidation (Section 4.4.3). A series of parallel experiments was performed with increased loading of different bases (5 equivalents) to speed up the reaction. The increased reactivity of cinnamyl bromide probably results from the faster rate of alkylation of ITC as well as the higher stabilization of the ylide form.
Na2CO3 failed to promote the reaction completely, while NaOH caused excessive degradation of the product even at low conversion. In addition, the concentration of the catalyst decreased more significantly over time in MeCN compared to t-BuOH. This is probably due to the heterogeneous nature of the base and the high viscosity of the solvent.
The planned final steps of the total synthesis of 8'-fluoropodophyllotoxin (25) via neopodophyllotoxin (222) are outlined in Scheme 81.
Part E: Regioselective C-C scission of ketone enolates mediated by nitrosation
- Cleavage of phenones and symmetrical ketones
- Cleavage of unsymmetrically substituted and complex ketones
- Mechanistic considerations and conclusions
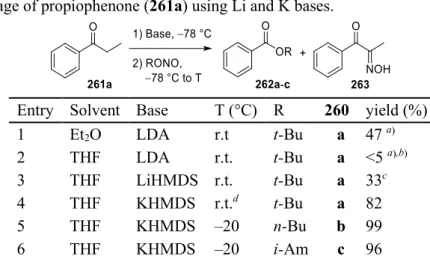
When considered all together, these reports reveal an interesting dichotomy in the reactivity of enolates toward nitrosating reagents, which can be explained by a simple model (Scheme 83, C) in which the reaction pathways diverge in the nitrosoketone phase 258. The rest of the mass balance consisted of one of the aldoxime materials (2, unselected reactants) resulting from aldoximes (entries 3, 5) or α-hydroxyiminone. unbroken tone (entries 4, 7-9). Nitrosation of the sodium enolate of O -methylestrone ( 286b ) by i -AmONO resulted in opening of the D ring, giving hydroxyiminoester 287b in 78–89% yield (entries 4, 5).
This finding is consistent with the incomplete cleavage of the conformationally locked cyclopropanated cyclohexanone 278b (Table 28, entry 9). Conjugate addition to R-(−)-carvone (294b) proceeds with a high degree of diastereoselectivity, affording TMS enol ether 295b after silylation of the magnesium enolate. By carefully controlling the loading of base and oxidant, the reaction can be terminated at the hydroxyamino ester product(s), making nitrosative cleavage a potentially useful method for cleavage of the α-C-C bond in ketones.
Both steric hindrance in the vicinity of the carbonyl group and the conformational rigidity of some cyclic substrates can prevent cleavage (Scheme 85).
CONCLUSIONS AND PERSPECTIVES
In addition to existing ligands, some new ligands, such as the dianhydrosugar-derived diethers, have been shown to be competent promoters of conjugate addition. The organocatalytic Corey-Chaykovsky epoxidation used in the neopodophyllotoxin approach was inspired by an earlier dual-catalytic method. The accidental discovery of nitrosative cleavage of ketonolates via retro-aza-Claisen fragmentation turned out to be a common reactivity pattern of most simple and more complex ketones.
The reaction is site-specific and allows the anti-Beckmann cleavage of other unsymmetrically substituted phenones and ketones via their kinetic or thermodynamic enolates. Stereocomplex Ω-hydroxyiminoesters can be achieved by Cu-catalyzed conjugate addition followed by cleavage of the resulting enolates, potentially opening the way to optically pure complex unnatural ω-amino acids. Conductor and conductor-like proteins may play a more important role in the biosynthesis of secondary metabolites of fungi, bacteria and plants than previously believed.
In the future, the discovered or newly engineered DIRs may find important applications in the biotechnological production of pharmaceuticals such as etoposide and teniposide.
EXPERIMENTAL PART
Part A
- Initial exploration of in situ oxidative cyclisation
- Cu-catalysed conjugate addition of ArLi to ylidene malonates
- Annulation of model stilbenes
- Racemic synthesis of kompasinol A pentamethylether (140)
The combined organic extracts were dried over Na2SO4, concentrated and purified by flash chromatography (hexane/EA 15:1 to pure EA) affording 125 mg (74%) of 114 as a colorless amorphous solid. The aqueous phase was extracted with additional n-hexane (100 mL) and the combined organic layers were dried over Na2SO4. The combined organic extracts were dried over anhydrous Na2SO4, concentrated under vacuum and purified by flash chromatography (hexane/EA 10:1 to pure EA) to afford 153 mg of 123a (70%) as a colorless amorphous solid.
The combined organic extracts were dried over Na 2 SO 4 , concentrated and dried under high vacuum to give 600 mg of a pale yellow oil. The combined organic extracts were dried over Na 2 SO 4 , concentrated and dried under high vacuum to give 666 mg of yellow thick oil. The combined organic layers were dried over Na 2 SO 4 , concentrated in vacuo and purified by flash chromatography (hexane/EA 9:1 for pure EA) to give 74 mg (98%) of 138b as a colorless sticky solid.
The reaction was quenched by the addition of water (15 mL), transferred to a separatory funnel and extracted twice with a 1:1 mixture of THF and Et 2 O (2 x 25 mL).
Part B
- Direct conjugate addition of 136 to cinnamate 146
- Synthesis of tert-butyl protected isorhapontigenin 151
- Synthesis of 157
- Synthesis of Li-specific ligands
- Addition of stilbene 157 to cinnamate 150
- Synthesis of malonates 171a-d, 188, 195
- Optimization of asymmetric conjugate addition of 157 to malonates 171a-d
- Tandem addition/oxidation of S4M3 (isolation of alcohol)
- Total synthesis of gnetifolin F (I)
- Total synthesis of (+) and (–)-11-deoxykompasinol A (IV)
- Towards kompasinol A
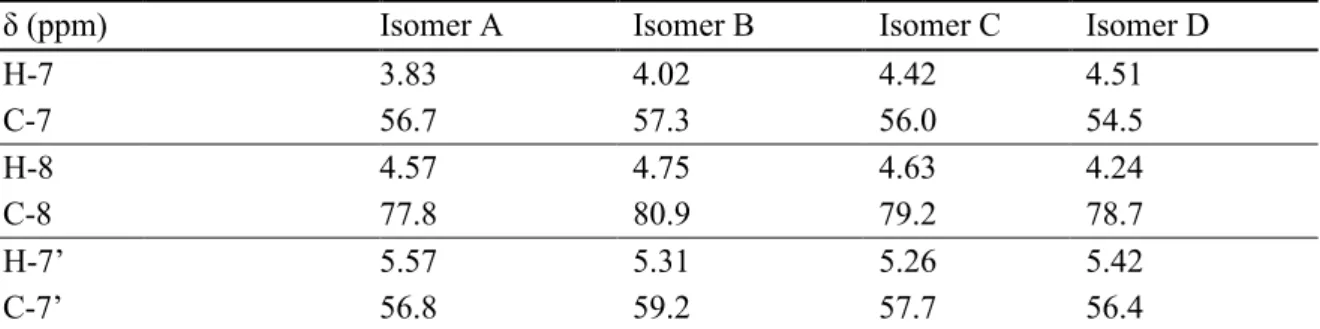
- Comparison of 1 H and 13 C NMR spectra of natural and synthetic FIS
The mixture was stirred for 10 min, transferred to a separatory funnel and extracted with PE (350 mL). The mixture was stirred under dry N 2 at ambient temperature overnight and heated to reflux for 1 hour. After the azeotropic distillation of water had ceased, the mixture was cooled to room temperature, concentrated and dried under high vacuum.
The solvent was removed under vacuum, THF (20 mL), water (4 mL) and acetic acid (4 mL) were added and the mixture was stirred for 30 minutes. After stopping the azeotropic distillation of water, the mixture was cooled, concentrated and dried under high vacuum. After stopping the azeotropic distillation of water, the mixture was cooled and concentrated and dried under high vacuum.
After the azeotropic distillation of water had ceased, the mixture was cooled and concentrated in vacuo to -1/2 volume. Under a N 2 atmosphere, a stir bar was added and the mixture redissolved in dry toluene (0.4 mL). After cooling, the reaction mixture was partitioned between 30 wt% aqueous citric acid (20 mL) and DCM (30 mL).
Part C
- First generation radical 6-exo-tet approach to 11
- Second generation approach based on RCM/conjugate addition
Part D
- Synthesis of neopodophyllotoxin analogue by polar bicyclization
- Rapid synthesis of neopodophyllotoxin core – racemic approach
- Towards asymmetric synthesis of (2-fluoro)neopodophyllotoxin
Part E
- General experimental procedures
- Synthesis and characterization of starting materials
- Characterization data of cleavage products
X-ray crystallography
AUTHOR’S PUBLICATIONS AND SCIENTIFIC PRESENTATIONS