"JULIO DE MESQUITA FILHO"
FACULDADE DE CIÊNCIAS FARMACÊUTICAS
CÂMPUS DE ARARAQUARA
“AVALIAÇÃO DE PROCESSOS PARA OBTENÇÃO DE COMPRIMIDOS
DE
ȕ
-CICLODEXTRINA-PARACETAMOL”
NELSON PEREIRA DA SILVA JÚNIOR
Dissertação apresentada ao programa de Pós-Graduação
em Ciências Farmacêuticas, Área de Pesquisa e
Desenvolvimento de Fármacos e Medicamentos, da
Faculdade de Ciências Farmacêuticas, UNESP, como
parte dos requisitos para a obtenção do Título de Mestre
em Ciências Farmacêuticas.
ORIENTADOR: Profa. Dra.MARIA PALMIRA DAFLON GREMIÃO
CO-ORIENTADOR: Profa. Dra. ANA DÓRIS DE CASTRO
Faculdade de Ciências Farmacêuticas UNESP – Campus de Araraquara
Silva Júnior, Nelson Pereira da
S586a Avaliação de processos para obtenção de comprimidos de
β-ciclodextrina--paracetamol / Nelson Pereira da Silva Júnior. – Araraquara, 2006.
77 f.
Dissertação (Mestrado) – Universidade Estadual Paulista. “Júlio de Mesquita Filho”. Faculdade de Ciências Farmacêuticas. Programa de Pós Graduação em Ciências Farmacêuticas
Orientador: Maria Palmira Daflon Gremião Co-orientador: Ana Dóris de Castro
. 1.Beta Ciclodextrina. 2.Comprimidos. 3.Tecnologia farmacêutica. I.Gremião, Maria Palmira Daflon , orient. .II.Castro, Ana Dóris, co-orient. III. Título.
CDD: 615.4
Avaliação de processos para obtenção de comprimidos de beta-ciclodextrina-paracetamol
A comissão julgadora dos trabalhos de defesa da dissertação de mestrado, em sessão
pública em 28/07/2006 considera o candidato:
( ) REPROVADO ( x ) APROVADO
1) Examinador (Profa. Dra. Leila Aparecida Chiavacci): __________________________
2) Examinador (Prof. Dr. Osvaldo de Frietas): __________________________________
3) Examinador (Profa. Dra. Maria Palmira Daflon Gremião):_______________________
À Deus, por me dar a oportunidade da vida, por me dar uma família especial, por me
conceder obstáculos para que eu pudesse crescer, por me mostrar o caminho, pelo amor
incondicional.
À minha família por ter acreditado que meu sonho era possível, por estarem ao meu lado nos momentos mais importantes, por dividirem comigo este momento tão especial.
À Profa Dra Maria Daflon Gremião e Profa Dra Ana Dóris de Castro pela oportunidade a mim concedida, pela amizade, pelos ensinamentos e dedicação colaborando dessa forma para que eu pudesse ser um profissional melhor.
Ao Prof. Dr. Luís Alexandre de Pedro de Freitas e Prof. Dr. Marcos Vinícius Chaud pela contribuição concedida no exame geral de qualificação.
À todos os professores que me deram a oportunidade da convivência nesses anos de pesquisa.
Às secretárias do programa de pós graduação, Cláudia, Laura e Sônia pelo auxílio, disposição e atenção constantes.
À Keila , secretária do departamento de fármacos e medicamentos.
Aos técnicos da Faculdade de Ciências Farmacêuticas- USP- Ribeirão Preto- Franklin e Paulo.
À Professora Dra. Leila Aparecida Chiavacci do departamento de físico-química do Instituto de Química da UNESP-Araraquara pelos ensinamentos, colaboração e discussão em análises.
À Profa Dra Beatriz S. Cury, pela paciência, dedicação e amizade que dispensou a mim.
Aos estagiários e amigos Renan, Amanda, Fernanda, Daniel, Denis e Alexandre, pelo auxílio no desenvolvimento desse trabalho.
Aos amigos que proporcionaram momentos especiais e tornaram minha vida mais feliz em especial: Arnóbio, Cristina, Luana, Daniela Longo, Helen, Andréia, Daniele Michelin, Rubiana, Tina, Marlus, Gustavo, Thalita, Kelly, Thiago, Priscila, Fernando, Mara, Gisele, Maria Carolina, Vanessa, Ednir, Thais, Karen e Traudi.
Very very thanks à Wagner e Sandro pelas correções de inglês e amizade
Não Sei... Cora Coralina
Não sei...se a vida é curta ou longa demais para nós,
Mas sei que nada do que vivemos tem sentido,
Se não tocamos o coração das pessoas.
Muitas vezes basta ser:
Colo que acolhe,
Braço que envolve,
Palavra que conforta,
Silêncio que respeita,
Alegria que contagia,
Lágrima que corre,
Olhar que acaricia,
Desejo que sacia,
Amor que promove.
E isso não é coisa de outro mundo, é o que dá sentido à vida.
É o que faz com que ela não seja nem curta,
nem longa demais,mas que seja intensa,
SUMÁRIO
LISTA DE FIGURAS i
LISTA DE TABELAS iv
RESUMO v
ABSTRACT vi
ABREVIATURAS vii
1. INTRODUÇÃO 01
1.1. Ciclodextrinas 01
1.1.1. Formação de complexos 05
1.1.2. Diagrama de solubilidade 07
1.2. Comprimidos 08
1.2.1. Características físicas dos comprimidos 13
1.3. Técnicas termoanalíticas 15
1.4. Difração de raios-X 15
1.5. Processos de obtenção de comprimidos 17
2. OBJETIVOS 24
3. MATERIAIS 25
3.1. Matérias-primas 25
3.2. Equipamentos 25
4. MÉTODOS 27
4.1. Metodologia analítica 27
4.2. Determinação da solubilidade do paracetamol em solução aquosa de ȕ-CD 27
4.3. Obtenção do granulado em estufa (Processo I) 28
4.5. Avaliação dos granulados obtidos pelos processos I e II 33
4.5.1. Umidade 33
4.5.2. Análise morfológica 34
4.5.3. Distribuição Granulométrica 34
4.5.4. Escoamento 35
4.5.5. Densidades aparentes bruta e compactada, índice de compressibilidade
percentual e fator de Hausner 36
4.5.6. Difração de raios X do granulado obtido pelo processo II 38
4.5.7. Obtenção das curvas de DSC do granulado obtido pelo processo II 38
4.6. Avaliação dos comprimidos obtidos a partir dos processos I e II 39
4.6.1. Aspecto 39
4.6.2. Variação do peso 39
4.6.3. Espessura 39
4.6.4. Resistência mecânica 39
4.6.5. Tempo de desintegração 40
4.7. Análise estatística 40
5. RESULTADOS E DISCUSSÃO 41
5.1. Metodologia analítica 41
5.2. Determinação da solublidade do paracetamol em solução aquosa de β-CD 42
5.3. Avaliação dos granulados obtidos pelo processo I 44
5.4. Compressão dos granulados obtidos pelo processo I 53
5.5. Avaliação do granulado obtido pelo processo II 55
5.6. Compressão do granulado obtido pelo processo II 64
5.8. Análise estatística 70
6. CONCLUSÕES 72
LISTA DE FIGURAS
FIGURA 1: Detalhe da ligação α-1,4 entre duas unidades de glicose de uma
molécula de CD 01
FIGURA 2: Representação esquemática da estrutura funcional das CDs 02 FIGURA 3: Formação de dextrinas cíclicas e acíclicas a partir do amido 02
FIGURA 4: Estrutura de α,β e β-CDs 04
FIGURA 5: Esquema da associação da molécula hospedeira (CD) e da molécula
hóspede (p-xileno); 06
FIGURA 6: Isotermas de solubilidade 07
FIGURA 7: Etapas da compressão 11
FIGURA 8: Deformações (a) tênsil (b) por compressão (c) por cisalhamento 12 FIGURA 9 Esquema do processo usual de obtenção de comprimidos contendo
CDs 20
FIGURA 10: Diagrama esquemático do regime de jorro 23
FIGURA 11: Esquema do primeiro processo para obtenção de grânulos secos em
leito estático (estufa). 28
FIGURA 12: Esquema do segundo processo para obtenção de grânulos secos em
leito fluidizado (leito de jorro). 30
FIGURA 13: Diagrama esquemático do leito de jorro e periféricos 31
FIGURA 14: Espectro de absorção do paracetamol em solução aquosa no UV 41
FIGURA 15: Curva analítica do paracetamol em água destilada 42
FIGURA 16: Isoterma de solubilidade 43
FIGURA 17: Fotomicrografia do GLE-amido 45
FIGURA 18: Fotomicrografia do GLE-celulose 45
FIGURA 20: Distribuição do tamanho de partículas do GLE- amido GLE-
celulose GLE- lactose 47
FIGURA 21: Cotangente do ângulo de escoamento: GLE- amido, GLE- celulose e GLE-lactose. Abertura do funil de escoamento: 12mm 48
FIGURA 22: Velocidade de escoamento: GLE- amido, GLE- celulose e GLE-
lactose. Abertura do funil de escoamento:12mm 49
FIGURA 23: Cotangente do ângulo de escoamento: GLE- amido, GLE- celulose e GLE-lactose. Abertura do funil de escoamento: 9mm 50
FIGURA 24: Velocidade de escoamento: GLE- amido, GLE- celulose e GLE-
lactose. Abertura do funil de escoamento:9mm 50
FIGURA 25: Fotomicrografia do paracetamol 56
FIGURA 26: Fotomicrografia da ȕ-CD 56
FIGURA 27: Fotomicrografia do GLJ- amido (750x) 57
FIGURA 28: Fotomicrografia do GLJ-amido (aumento 3500x) 57
FIGURA 29: Distribuição do tamanho de partículas do GLJ- amido 58
FIGURA 30: Difratograma do paracetamol, ȕ-CD e do GLJ- amido 61
FIGURA 31: Difratograma da mistura física e do GLJ- amido 62
FIGURA 32: Curvas DSC: amido, paracetamol, ȕ-CD, mistura física e GLJ-
amido 63
FIGURA 33: Comprimidos obtidos do GLE-amido 64
FIGURA 34: Comprimidos obtidos do GLE-celulose 65
FIGURA 35: Comprimidos obtidos do GLJ-amido 65
FIGURA 36: Comprimidos obtidos do GLE-lactose 65
FIGURA 37: Variação de peso dos comprimidos: GLE- amido, GLE- celulose e
GLJ- amido 67
FIGURA 38: Espessura dos comprimidos: GLE- amido, GLE- celulose e GLJ-
FIGURA 39: Dureza dos comprimidos: GLE- amido, GLE- celulose e GLJ-
amido 68
FIGURA 40: Tempo de desintegração dos comprimidos: GLE- amido, GLE-
ÍNDICE DE TABELAS
TABELA 1: Limites de variação de peso 13
TABELA 2: Composição das amostras 29
TABELA 3: Condições operacionais empregadas na secagem do material 32
TABELA 4:Número dos tamises e abertura da malha 35
TABELA 5: Valores das absorvâncias das soluções aquosa do paracetamol
obtidas por espectrofotometria no Ȝ = 243 nm 42
TABELA 6:Influência da ȕ-CD na solubilidade do paracetamol 44
TABELA 7:Densidade aparente bruta do GLE- amido, GLE- celulose e
GLE- lactose 51
TABELA 8: Densidade aparente compactada do GLE- amido, GLE-
celulose e GLE- lactose 51
TABELA 9:Índice de compressibilidade percentual e fator de Hausner do
GLE- amido, GLE- celulose e GLE- lactose 53
TABELA 10: Densidade aparente bruta do GLJ- amido 59
TABELA 11: Densidade aparente compactada do GLJ- amido 60
TABELA 12: Índice de compressibilidade percentual e fator de Hausner
do GLJ- amido. 60
TABELA 13:Medidas da largura em determinados ângulos do
difratograma (mistura física) 63
TABELA 14:Medidas da largura em determinados ângulos do
difratograma (GLJ-amido) 63
TABELA 15: Análise de variância para peso 70
TABELA 16: Análise de variância para espessura 70
TABELA 17: Análise de variância para dureza 71
TABELA 18: Análise de variância para tempo de desintegração 71
RESUMO
Ciclodextrinas (CD) têm sido relatadas em inúmeros estudos por interagir com
muitos fármacos para a formação de complexos de inclusão com o objetivo de aumentar a
solubilidade, estabilidade e biodisponibilidade. No processo usual para obtenção de
comprimidos contendo β-CD, as dispersões líquidas de fármaco/β-CD são submetidas a
processos de secagem por liofilização, evaporação ou spray-drying e o material seco é
incorporado a vários excipientes. O objetivo principal deste trabalho foi avaliar processos
de obtenção de comprimidos de β-CD/paracetamol. O paracetamol foi utilizado como
fármaco modelo por ser pouco solúvel em água. No primeiro processo, a dispersão líquida
de ȕ-CD/paracetamol foi incorporada ao amido de milho, celulose microcristalina ou
lactose monoidratada e o material foi granulado e submetido à secagem em leito estático
(estufa). No segundo processo, a dispersão líquida de ȕ-CD/paracetamol foi incorporada ao
amido de milho e o material submetido à secagem em leito fluidizado (leito de jorro). Os
materiais obtidos em ambos os processos foram comprimidos. Comparando os três
excipientes utilizados no primeiro processo, tanto o amido quanto celulose são os
excipientes que possibilitariam a incorporação de quantidade maior de fármaco. Como
resultados, os granulados obtidos a partir dos excipientes amido e celulose apresentaram
boas características de escoamento e compressibilidade. O segundo processo, originou um
material que apresentou boas características de compressibilidade e comprimidos que
apresentaram as melhores características físicas durante o processo de compactação.
Concluiu-se que ambos processos representam uma estratégia tecnologicamente viável para
obtenção de comprimidos contendo ȕ-CD.
ABSTRACT
Cyclodextrins (CDs) have been reported in a number of studies in the
pharmaceutical field since it interact with many drugs to form water soluble inclusion
complexes, thus improving not only the solubility but also the stability and biovailability of
various drugs. In order to obtain ȕ-CD tablets, liquid dispersions of drug/ȕ-CD are usually
submitted to different drying process, like spray drying, freeze-drying or slow evaporation
and further added to several excipients. In this work we evaluated different process for the
preparation of ȕ-CD/ acetaminophen tablets. Due to its low solubility, we have used
acetaminophen as a drug model. In the first process, an aqueous ȕ-CD/ acetaminophen
dispersion were added to corn starch, microcrystalline cellulose or monohydrated lactose
and the material was dried in a static bed. In the second process, an aqueous ȕ-CD/
acetaminophen dispersion were added to corn starch and further dried in a fluidized bed
(spouted bed). As a result, in the first process the use of starch or cellulose led to mixtures
with higher amount of drug with good flowability and compressibility, whereas the second
process led to a mixture of good compressibility and to tablets that presented the best
physical proprieties. In conclusion, both process represent viable technological approaches
to obtain ȕ-CD tablets.
KEYWORDS: β-cyclodextrins, tablets, spouted bed, static bed.
ABREVIATURAS
Å Angstron
Abs Absorvâncias
CD Ciclodextrina
Į- CD Alfa ciclodextrina
ȕ- CD Beta ciclodextrina
Ȗ- CD Gama ciclodextrina
CGTase Ciclodextrina-glicosil-transferase
CV Coeficiente de variação
DSC Calorimetria exploratória diferencial
Fc Distribuição probabilidade calculada
FH Fator de Hausner
FV Fator de variação
GL Grau de liberdade
m/m Massa/massa
GLE Granulado obtido em leito estático
GLJ Granulado obtido em leito de jorro
IC Índice de compressibilidade
QM Quadrado médio
SQ Soma de quadrado
x Média
σ Desvio padrão
1. INTRODUÇÃO 1.1. Ciclodextrinas
As ciclodextrinas (CDs) são oligossacarídeos cíclicos formados por 6 a 12 unidades
de glicose. As unidades de glicose que constituem as CDs encontram-se unidas por ligações
do tipo α-1,4 como mostra a Figura 1.
Figura 1: Detalhe da ligação α-1,4 entre duas unidades de glicose de uma molécula
de CD (Adaptado por FERNANDES e VEIGA, 1999).
As CDs possuem uma forma tronco-cônica com uma cavidade hidrófoba (Figura 2)
na qual pode ser incluída uma grande variedade de moléculas-hóspedes de tamanho e forma
adequados, resultando em uma associação estável sem a formação de ligações covalentes,
formando complexos de inclusão. As CDs têm sido largamente empregadas em tecnologia
farmacêutica para aumentar a solubilidade, velocidade de dissolução, estabilidade e
biodisponibilidade de várias substâncias ativas em diferentes formas farmacêuticas como,
Figura 2: Representação esquemática da estrutura funcional das CDs (SZEJTLI,
1990).
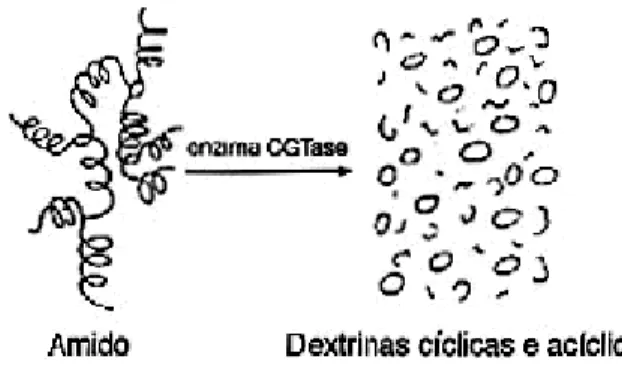
As CDs são obtidas a partir do amido por ação da enzima
ciclodextrina-glicosil-transferase (CGTase) que atua no substrato a partir das extremidades redutoras e, ao mesmo
tempo, catalisa as ligações α-1,4, promovendo a ciclização do amido (Figura 3) (VETTER
e THORN, 1992).
Figura 3: Formação de dextrinas cíclicas e acíclicas a partir do amido
Diversos tipos de CDs são conhecidos, a α-CD, que contém 6 unidades de
glicopiranose, a β-CD com 7 unidades de glicopiranose e a β-CD com 8 unidades de
glicopiranose (Figura 4). Todas são produzidas industrialmente como substâncias
cristalinas homogêneas (SZEJTLI, 1982). As quantidades relativas das α-CD, β-CD e β-CD
dependem do tipo de microorganismo que produz as enzimas envolvidas no processo de
obtenção e das condições de reação (SAENGER, 1980). A enzima CGTase produzida pelo
Bacillus macerans e Klebsiella pneumoniae produz preferencialmente a α-CD, enquanto
que, a proveniente do Bacillus alcalofílico produz principalmente β-CD. Quando a
conversão enzimática do amido se processa na presença de tolueno e em determinadas
condições de pH, temperatura e o tempo de reação, o principal produto obtido é a β-CD. A
adição de decanol tem revelado que este é o agente mais indicado para aumentar a
produção de α-CD, enquanto que a presença de metiletilcetona conduz a um maior
rendimento na obtenção de β-CD (SZEJTLI, 1994). A β-CD é a mais hidrossolúvel a 25ºC
(23g se dissolvem em 100ml de água), enquanto a β-CD é a menos hidrossolúvel (1,85g se
dissolvem em 100ml de água). A solubilidade da α-CD é 15g em 100 ml de água
Figura 4: Estrutura de α,β e β-CDs (PSZCZOLA, 1988).
As modificações químicas das CDs podem possibilitar o aumento da capacidade de
inclusão (UEKAMA, 1982).
A substituição dos grupos hidroxila por grupos metílicos no C2, C3 ou C6 (Figura
1), originam as CDs metiladas. Essas substituições, impedem a formação de ligações de
hidrogênio intramoleculares, o que torna a estrutura molecular mais flexível, originando
alterações relacionadas com a solubilidade. As hidrossolubilidades a 25ºC da dimetil-β-CD
(57g/100ml de água) e a trimetil-β-CD (31 g/100ml de água) são muito superiores à da α
-CD, β-CD e β-CD (DUCHENE e WOUESSIDJEWE, 1990).
Como exemplos das CDs hidroxialquiladas temos a 2-hidroxietil-ȕ-CD, a
2-hidroxipropil-ȕ-CD, a 3-hidroxipropil-ȕ-CD e a 2,3-dihidroxipropil-ȕ-CD cujas
solubilidades em água a 25ºC são superiores a 50g em 100ml. A preparação destes
derivados ocorre de forma não seletiva e processa-se por condensação com os referidos
As CDs ramificadas podem ser obtidas por síntese química, mas na maioria dos
casos são preparadas por via enzimática. Podem ser subdivididas em duas categorias. As
CDs ramificadas homogêneas, cujas cadeias laterais são constituídas por unidades de
glicose, tais como, a glicosil-ȕ-CD, a maltosil-ȕ-CD, a maltotriosil-ȕ-CD ou a dimaltosil-ȕ
-CD e as -CDs ramificadas heterogêneas, que possuem uma ou mais unidades de galactosil
ou unidades similares na cadeia lateral da CD. Todas apresentam solubilidade em água a
25º C superiores a 50g em 100 ml (DUCHENE e WOUESSIDJEWE, 1990).
As CDs etiladas têm como principal função controlar a velocidade de dissolução de
fármacos solúveis em água. São exemplos deste tipo a heptakis (2,6-di-O-etil)-ȕ-CD e a
heptakis (2,3,6-tri-O-etil)- ȕ-CD que apresentam solubilidade em água a 25º C inferiores à
0,005g em 100 ml (DUCHENE e WOUESSIDJEWE, 1990).
A substituição dos grupos hidroxila das CDs por grupos ionizáveis confere a CDs
ionizáveis uma hidrofilia e uma capacidade de complexação dependentes do pH. A
carboximetiletil- ȕ-CD pode ser utilizada como agente complexante de fármacos solúveis
que sejam instáveis em pH baixo e/ou irritantes para a mucosa gástrica e cuja absorção se
verifique no intestino (HORIUCHI, 1991).
1.1.1. Formação de complexos
Devido à estrutura das CDs, estas apresentam a habilidade de formar complexos
com compostos sólidos, líquidos e gasosos. Na Figura 5, é apresentado o esquema de
formação de complexo de inclusão de CD com uma substância líquida, sendo o p-xileno a
Figura 5: Esquema da associação da molécula hospedeira (CD) e da
molécula hóspede (p-xileno); os pequenos círculos representam as
moléculas de água (SZEJTLI, 1990).
Uma das condições para que ocorra a complexação é que o tamanho da molécula
a ser complexada seja compatível com a cavidade da CD em questão. A α-CD é utilizada
essencialmente para complexar moléculas pequenas ou cadeias laterais de moléculas
grandes (por exemplo, prostaglandinas), enquanto a β-CD é muito utilizada para complexar
moléculas contendo grupamento fenila ou com massa molecular entre 200 a 800 e a β-CD é
mais utilizada na complexação de moléculas grandes, como antibióticos macrolídeos e
esteróides (LOFTSSON e BREWSER, 1996).
Outra condição a ser observada é a polaridade da molécula hóspede e sua
competição com os demais compostos no meio. As moléculas muito hidrofílicas, muito
pequenas ou muito grandes ou, ainda, que estejam ionizadas, dificilmente se complexam
(LOFTSSON e BREWSER, 1997). Os compotos inorgânicos não são, na sua maioria,
adequados para a complexação com CDs, embora existam algumas exceções como ácidos
não dissociados, halogênios e outros gases, tais como o dióxido de carbono e o propano. As
moléculas apolares, cujas dimensões sejam inferiores às da cavidade da CD, podem ser
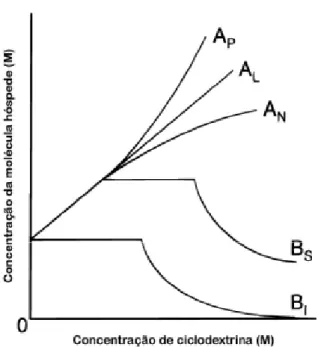
1.1.2. Diagrama de solubilidade
Os valores de concentração da molécula-hóspede podem ser representados
graficamente em função das concentrações de CD. Podem ser obtidos dois grandes tipos de
isotermas de solubilidade: o tipo A e o tipo B (Figura 6).
Figura 6: Tipos de isotermas de solubilidade, de acordo com Fromming e Szejtli, (1994).
A isoterma do tipo A indica que a solubilidade da molécula-hóspede aumenta com
as concentrações crescentes da CD, enquanto que o diagrama tipo B indica a formação de
um complexo de solubilidade limitada e inferior à da CD. No tipo A vários
comportamentos podem ser observados. O tipo AL, que se caracteriza por um aumento
linear da solubilidade, indica que se formam complexos solúveis de composição constante.
O tipo AP com um desvio positivo de linearidade reflete a formação de complexos de maior
uma molécula-hóspede. O tipo AN é idêntico ao anterior, mas com desvio negativo, que
corresponde ao estabelecimento de interações soluto/soluto e soluto/solvente. No tipo B
pode-se observar dois tipos de comportamento: tipo BS e tipo BI. No primeiro, onde
inicialmente há um aumento da solubilidade do soluto, seguido de um platô e posterior
diminuição da solubilidade em altas concentrações de CD, geralmente é ocasionado por
uma precipitação do complexo. No tipo BI, o complexo de inclusão é praticamente
insolúvel, o que justifica a ausência da parte inicial da curva de solubilidade (SZETJTI,
1994).
1.2. Comprimidos
Dentre as formas farmacêuticas destinadas à administração pela via oral, a forma
farmacêutica sólida comprimido tem sido o sistema de liberação de fármacos preferido para
obtenção de efeito sistêmico e é a forma farmacêutica mais difundida por apresentar as
maiores vantagens em relação ao seu uso. Dentre estas vantagens, podem ser citadas a
grande precisão de dosagem, a baixa variabilidade do conteúdo e a facilidade de
administração (BANKER e ANDERSON, 2001).
De modo geral, as formulações de comprimidos são compostas por um ou mais
fármacos além dos excipientes que devem exercer diversas funções (BANKER e
ANDERSON, 2001). A inclusão de excipientes na fórmula do medicamento é necessária
para que a formulação possa ser processada ou para que se consiga o desejado efeito
farmacológico ou farmacocinética adequada (MORETON, 1996). Os excipientes mais
Os diluentes são substâncias inertes usadas como “enchimento” para criar volume
desejado (ANSEL et al., 2000). A lactose é um diluente de baixo custo que não reage com a
maioria dos fármacos, quer seja utilizada na forma anidra ou na forma hidratada. A lactose
anidra apresenta a vantagem sobre a lactose hidratada por não levar ao acastanhamento e
descoloração quando em presença de certos fármacos com grupos amina. O amido, que
pode ser obtido de milho, de trigo ou de batata é usado também como diluente em
comprimidos.
A celulose microcristalina é um excipiente para compressão direta com
características de escoamento muito boas. É um material utilizado como diluente em
concentrações elevadas, sendo geralmente combinado com outros excipientes (BANKER e
ANDERSON, 2001).
Os aglutinantes são substâncias usadas para promover aderência das partículas de pó
nas granulações de comprimidos. São citados como exemplo o a carboximetilcelulose
sódica, a etilcelulose, a gelatina, a povidona, o amido pré-gelatinizado, a metilcelulose e
outros (ANSEL et al., 2000).
Os desintegrantes são substâncias adicionadas à maioria das formulações para
facilitar a ruptura ou desintegração dos comprimidos quando estes entram em contato com
líquidos. Os desintegrantes podem atuar promovendo a absorção de água pelo comprimido,
o qual intumesce até à sua desintegração (BANKER e ANDERSON, 2001). O amido é um
desintegrante muito utilizado por ser acessível e apresentar baixo custo (GADALLA et al.,
1989).
Gadalla e colaboradores (1989) estudaram o efeito de diferentes tipos e
comprimidos de aspirina obtidos por dupla compressão. Os autores comprovaram que
nenhum tipo ou concentração dos amidos estudados exerce efeito acentuado na
uniformidade de peso dos comprimidos, mas apresentam desempenhos diferentes quanto ao
tempo de desintegração dos comprimidos; o amido de milho foi o que promoveu a
desintegração dos comprimidos em menores tempos e o amido de arroz em maiores
tempos.
Os lubrificantes são substâncias capazes de transmitir ao sistema particulado,
propriedades deslizantes (facilitar o deslizamento, ou seja, o escoamento do sistema
particulado do alimentador para a câmara de compressão) e/ou antiaderentes (minimizar ou
anular a tendência de aderência do sistema particulado aos punções e matrizes) facilitando,
portanto, a compressão e a ejeção dos comprimidos da máquina (PRISTA et al., 2002).
Entre os mais utilizados, citamos o talco, os carbowaxes, os estearatos e gorduras.
Comprimidos podem ser definidos como formas farmacêuticas sólidas unitárias
obtidas por compactação em equipamentos adequados. O processo físico de compactação
pode ser definido como a compressão e consolidação de um sistema bifásico (pó e gás)
quando é aplicada uma força. A compressão traduz-se por uma redução do volume do pó,
como resultado do deslocamento da fase gasosa, enquanto que, a consolidação tem como
resultado o aumento da força mecânica do material devido a interações entre as partículas
(MARSHALL, 2001).
Quando se aplicam forças mecânicas sobre uma mistura de pós, verifica-se
normalmente uma alteração do empacotamento das partículas do pó e na maioria dos casos,
este é o mecanismo principal que explica a redução do volume no início do processo
provoca uma deformação dessas partículas, em maior ou menor grau. Se, por remoção da
força aplicada a deformação é reversível, isto é, se o material se comporta como borracha,
diz-se que ocorreu uma deformação elástica (MARSHALL, 2001).
Quando as forças aplicadas ultrapassam o limite elástico, ou ponto de cedência
observa-se uma deformação que não é reversível após a remoção da força. A redução do
volume desses materiais resulta numa deformação plástica (MARSHALL, 2001).
Este mecanismo predomina em materiais para os quais a tensão de corte é inferior à
força tênsil ou de ruptura (Figura 8). Quando a tensão de cisalhamento é superior ao seu
limite de cedência, as partículas fraturam-se em vez de se deformarem e os fragmentos
resultantes ajudam a preencher os vazios existentes. A predisposição de um material para se
deformar de uma maneira determinada depende da organização dos seus átomos ou
moléculas e da presença de planos de fratura fracos nessa estrutura (MARSHALL, 2001).
1.2.1. Características físicas dos comprimidos
O peso do comprimido é regulado em função do enchimento volumétrico da câmara
de compressão, considerando no ajuste, um peso teórico previamente determinado
(RUDNIC e SCHWARTZ, 1995).
Trata-se de um parâmetro de consideração primordial na produção de comprimidos,
pois está diretamente relacionado à uniformidade de dose (considerando-se uniformidade
de mistura) e também indiretamente implica na uniformidade dos demais parâmetros físicos
de qualidade como friabilidade, dureza e desintegração (MARSHALL e RUDNIC, 1990).
Por mais moderno e automatizado que seja o equipamento de compressão, é
impossível manter um mesmo peso em todas as unidades produzidas. Por esta razão,
existem normas farmacopéicas que estabelecem limites de variação de peso. A Tabela 1
mostra os limites de variação de peso segundo a Farmacopéia Brasileira, (1988).
Tabela 1: Limites de variação de peso
Peso médio dos comprimidos (mg) % diferença permitida
Até 80 ± 10
80-250 ± 7,5
Maior que 250 ± 5
A espessura desejada para um comprimido deve ser combinada com o volume do
material colocado na matriz, com o diâmetro dessa matriz e com a pressão aplicada ao
enchimento pelos punções. Para produzir comprimidos de espessura uniforme durante a
produção e entre produções da mesma formulação, é preciso ter o cuidado de empregar o
aplicada afeta não só a espessura do comprimido, mas também sua dureza e, este último
fator é, provavelmente, o mais importante (ANSEL et al., 2000).
Após a sua obtenção, os comprimidos podem ser submetidos a processos de
revestimento, acondicionamento, transporte e, por estas razões, é importante que
apresentem uma adequada resistência às solicitações mecânicas tais como, pressão,
rolamento, choque e atritos (RUDNIC e SCHWARTZ, 1995).
A resistência mecânica é, geralmente, avaliada pela medida da dureza e da
friabilidade. A dureza avalia a resistência do comprimido ao esmagamento sob pressão
radial ou axial, ou seja, a força requerida para fraturar o comprimido. A friabilidade indica
a porcentagem de perda de peso dos comprimidos em função da liberação de partículas,
quando eles são submetidos a choques, rolamentos e atritos produzidos por processos de
agitação ou rolamento e queda (BANKER e ANDERSON, 2001).
Por outro lado, aceita-se como princípio geral que um fármaco para ser absorvido
pelo organismo tem que estar em solução. Para a maioria dos comprimidos, antes do
fármaco se dissolver, terá que ocorrer a quebra do comprimido em pequenas partículas ou
grânulos, um processo conhecido como desintegração (BANKER e ANDERSON, 2001).
É importante salientar que a desintegração não implica na completa dissolução da
forma farmacêutica, nem mesmo do princípio ativo, porém, é fato que a dissolução de um
fármaco a partir de um comprimido fragmentado é mais rápida e completa (MARSHALL e
RUDNIC, 1990).
O tempo de desintegração adequado para cada comprimido dependerá do tipo de
ação desejada, havendo uma variação desde tempos extremamente curtos como 5 minutos
1.3. Técnicas termoanalíticas
Um conjunto de técnicas muito utilizado para verificar a interação entre os
componentes da formulação é a análise térmica. A análise térmica compreende um
conjunto de técnicas nas quais propriedades físicas de uma substância é medida como
função da temperatura, enquanto a substância é submetida a um programa controlado de
temperatura (GIOLITO e IONASHIRO, 1988).
A DSC é a técnica que pode fornecer informações qualitativas e quantitativas sobre
o estado físico-químico do fármaco. A ausência do pico de fusão do fármaco em
termogramas, assim como não alteração da temperatura de transição vítrea, pode indicar se
o fármaco encontra-se em estado amorfo ou em estado cristalino menos ordenado (MU e
FENG e 2001).
De acordo com a definição de análise térmica citada anteriormente, algumas
técnicas como a difração de raios-X ou espectroscopia no infravermelho podem, quando
utilizadas de maneira específica, fornecer informação termoanalítica, mas esse caso não
será considerado na utilização da técnica de difração de raios-X a seguir (GIOLITO e
IONASHIRO, 1988).
1.4. Difração de raios-X
A difração de raios-X é fundamentada na condição de interferência construtiva
2dhkl senθθθθ = nλλλλ Equação [1]
onde : n = número de ondas
d= distância entre os planos de índice de Miller hkl
λ= comprimento de onda no feixe monocromático
θ= ângulo de difração
Esta é a lei fundamental da cristalografia de raios-X e estabelece que para um dado
comprimento de onda (λ), o raio refletido emergirá apenas nos ângulos (θ) para os quais a
relação acima é satisfeita. Cada pico de difração é produzido por um certo conjunto de
planos que satisfaça esta condição. Como conjuntos semelhantes de planos estão dispostos
no cristal de acordo com a sua simetria, os arranjos dos pontos no padrão de Laue reflete a
simetria do cristal, permitindo a identificação do composto cristalino (KLUG e
ALEXANDER, 1974).
Através da Equação de Scherrer ( Equação 2) que permite calcular o tamanho dos
cristais isolados, uma relação pode ser estabelecida entre Le a largura à meia altura de um
determinado pico.
θ β
λ
cos
K
L= Equação [2]
onde :
L = dimensão dos cristais
ȕ= largura a meia altura
ș = ângulo de incidência
Supondo que os átomos da estrutura cristalina definem uma série de conjuntos de
planos p paralelos igualmente espaçados por uma distância d, seja uma onda plana, de
comprimento de onda Ȝ, incidente sobre um conjunto de planos paralelos p separados de
uma distância d, uma relação entre Lhkl = pde a largura à meia altura pode ser estabelecida.
Como o tamanho do cristalito está relacionado com a extensão do grau de cristalinidade,
quanto maior o tamanho do cristalito, maior o número de planos cristalográficos nas
direções hkl e portanto maior o grau de cristalinidade.
1.5. Processos de obtenção de comprimidos
Os processos de obtenção de comprimidos podem ser divididos em dois grupos:
compressão direta e compressão após processo de granulação (ARNAUD et al., 1998,
MARSHALL e RUDNIC, 1990)
Se, por um lado, a compressão direta apresenta vantagens importantes como número
reduzido de operadores, menor número de etapas durante o processamento, menor número
de equipamentos, menor área de produção e processo totalmente executado a seco, por
outro lado, apresenta certas limitações importantes que restringem sua escolha como
método para a produção de comprimidos. São poucos os fármacos que se apresentam como
substâncias cristalinas que reúnem o conjunto de características que possibilitam a
compressão direta. Muitos materiais apresentam atração intermolecular relativamente fraca
o que dificulta a compressão. Mesmo que estas substâncias apresentem aptidão para coesão,
dificuldade para desintegrar e liberar o fármaco. Se a desintegração não é problemática,
outros componentes a serem incluídos na formulação podem interferir na compressibilidade
do fármaco e impossibilitar a aplicação do método. Aplicar a técnica de compressão direta
para produzir comprimidos que contenham fármacos não compressíveis é uma tarefa em
que nem sempre se logra êxito. Se o fármaco é de dose elevada, isto pode implicar na
adição de quantidades tão elevadas de diluentes de compressão direta que levaria à
obtenção de comprimidos de grandes dimensões que dificultam a deglutição, além de
encarecer o produto. Por sua vez, como os fármacos de dose baixa necessitam do uso de
diluentes que são substâncias inertes usadas como “enchimento” para criar volume
desejado e, se misturas uniformes de fármacos com diluentes não podem ser obtidas, o
processo torna-se pouco viável. Já a mistura uniforme de excipientes para a compressão
direta com fármacos de doses intermediárias torna o processo bastante atrativo para
produzir comprimidos (BANKER e ANDERSON, 2001).
Além destas dificuldades e por causa das características inadequadas de escoamento
e compressão, os fármacos freqüentemente necessitam ser granulados antes de serem
comprimidos. Assim, é comum o emprego do processo de granulação como uma das fases
da produção de comprimidos (MURAKAMI et al., 2001).
A granulação é a operação unitária farmacêutica de aglomeração de partículas que
implica em aumento de tamanho das partículas de pós ou da mistura de pós. O processo de
granulação objetiva obter partículas coesas compostas pelos componentes da mistura dos
pós e como conseqüência, melhorar as características de compressão incluindo fluidez e
compressibilidade (BADAWY et al., 2000). Assim, o emprego da granulação melhora o
compressão mais eficiente às custas de menor esforço do equipamento de comprimir e
resulta em produtos de maior resistência mecânica (MURAKAMI et al., 2001; SYMECKO
e RHODES, 1995).
A técnica utilizada para preparar granulados compostos por fármacos sensíveis ao
calor, à umidade, ou a ambos, é a granulação por via seca. Este processo envolve a
compactação dos componentes da fórmula do comprimido em máquinas de comprimir ou
em compactadores, seguida de processo de moagem, uniformização do tamanho dos
grânulos e adição da fase lubrificante antes da compressão final. Quando a mistura inicial
dos pós é submetida à compressão em máquinas de comprimir, são aplicadas altas pressões
através dos punções e obtêm-se compactos conhecidos como slugs. São os slugs que serão
submetidos à moagem para a obtenção dos grânulos. Eventualmente, uma única
pré-compressão pode não ser suficiente para conferir ao granulado as características desejáveis;
neste caso, outras compressões seguidas de moagem, calibração e adição de lubrificante
devem ser executadas (BANKER e ANDERSON, 2001).
A preparação de granulados por via úmida principia-se pela obtenção de mistura
homogênea das substâncias pulverulentas da fórmula. Em seguida, umedece-se a mistura
com um líquido e obtém-se a massa que irá atravessar as malhas do equipamento de
granulação para a obtenção do granulado. Os grânulos assim formados devem ser secos até
adquirirem teor de umidade adequado (BANKER e ANDERSON, 2001).
Os grânulos podem ser secos em estufas de circulação forçada de ar (secadores de
leito estático). Nas estufas, não existe movimento das partículas do sólido submetido à
secagem (RANKELL et al., 2001). Assim, o sistema de circulação forçada de ar consegue
estufa, pois a circulação forçada provoca a sua renovação constante por novas camadas de
ar mais seco. Por outro lado, a circulação forçada aumenta a velocidade da corrente de ar
que passa sobre o material a secar, o que permite que a secagem se processe mais
facilmente quando comparado com uma estufa sem a circulação forçada de ar (PRISTA et
al., 2002).
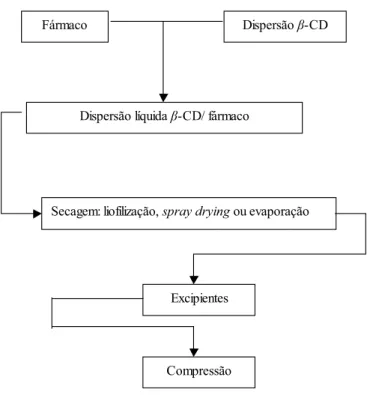
No processo usual de obtenção de comprimidos contendo fármacos e β-CD, as
dispersões líquidas de fármaco/β-CD são submetidas a processos de secagem por
liofilização, spray-drying ou evaporação para eliminação do solvente e o material seco é
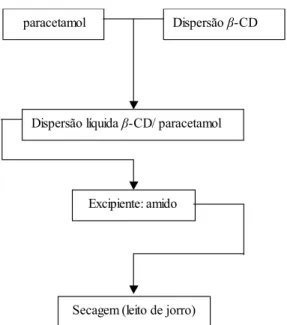
incorporado a excipientes de comprimido. A Figura 9 representa o esquema do processo
usual de obtenção de comprimidos contendo CDs.
Figura 9: Esquema do processo usual de obtenção de comprimidos contendo CDs
Fármaco Dispersão ȕ-CD
Dispersão líquida ȕ-CD/ fármaco
Secagem: liofilização, spray drying ou evaporação
Excipientes
Tasic e colaboradores (1992), após secagem da dispersão líquida de paracetamol/β
-CD em spray-dryer, incorporaram vários excipientes à dispersão sólida obtida. Os autores
incorporaram Avicel® PH101 como diluente para compressão direta, o Aerosil 200 como
deslizante. O Ac-Di-Sol, que é um derivado da carboximetilcelulose reticulada, foi
empregado como desintegrante e o estearato de magnésio como lubrificante na dispersão
sólida. Com a incorporação destes excipientes, os autores obtiveram comprimidos com
características físicas adequadas.
A secagem pelo método de spray drying envolve a formação de um material sólido
particulado através da atomização de um líquido, que pode apresentar características físicas
diversas (solução, suspensão), no interior de uma câmara de secagem, na qual também é
inserida uma corrente gasosa (MASTERS, 1985).
Os secadores de leito fluidizado são equipamentos em que as partículas sólidas são
parcialmente suspensas numa corrente de gás ascendente. As partículas são elevadas caindo
então de maneira aleatória de modo que a mistura resultante do sólido e do gás atua como
um líquido em ebulição (RANKELL, 2001).
O secador do tipo leito de jorro foi desenvolvido, no ano de 1954, por Gisher e
Mathur. Esse equipamento foi inicialmente projetado para a secagem de grãos de trigo em
um processo que permite a aplicação de maiores quantidades de calor sem a ocorrência de
perda da qualidade do material. Em seguida, os pesquisadores em questão, prevendo o
grande potencial de aplicação da nova técnica, iniciaram um estudo mais profundo sobre o
regime fluidodinâmico de jorro e concluíram que: “O mecanismo de fluxo dos sólidos
entretanto, o leito de jorro possui as mesmas aplicações do leito fluidizado, só que para
partículas de dimensões mais elevadas” (MATHUR e EPSTEIN, 1974).
O leito de jorro é constituído por uma câmara de secagem cilíndrica conectada a
uma base cônica, a qual possui em sua extremidade inferior um orifício de reduzida
dimensão, através do qual o fluido de jorro é alimentado ao sistema (PALLAI et al., 1995).
O regime de jorro é estabelecido pela entrada de um jato de fluido em um leito de
sólidos particulados. Após a entrada do fluido, normalmente constituído por ar, observa-se
aceleração das partículas sólidas com a formação de um canal central diluído, onde as
mesmas apresentam elevada velocidade. Essa região é denominada de região de jorro. Ao
redor do canal central, verifica-se a presença de um leito denso de partículas, que se
deslocam contra o fluxo ascendente de ar, traçando uma trajetória parabólica em relação à
região central do equipamento. A região que compreende esse leito deslizante recebe a
denominação de ânulo ou região anular. A desaceleração das partículas provenientes da
região de jorro ocorre após as mesmas atingirem a superfície do leito (região de fonte), e
resulta em sua queda sobre a região anular. Por fim, essas partículas descreverão uma
trajetória anular de volta para a região de jorro, seja após atingirem a base do leito (próximo
ao orifício de entrada do ar) seja através de fluxo cruzado (na interface jorro-ângulo)
(MATHUR, 1974).
A Figura 10, ilustra esquematicamente a movimentação cíclica e ordenada das
Figura 10: Diagrama esquemático do regime de jorro
Lin e Kao (1989) obtiveram partículas sólidas de ȕ-CD e paracetamol utilizando a
técnica do spray-drying. As partículas apresentaram características de escoamento e
compressibilidade ruins devido ao tamanho de partículas formado pelo processo de
spray-drying. Tendo em vista os problemas de escoamento, compressibilidade e a necessidade da
incorporação de vários excipientes para obtenção de comprimidos de fármaco/ ȕ-CD no
processo usual, este trabalho objetiva avaliar novos processos de obtenção de materiais com
características de escoamento e compressibilidade adequadas através da incorporação da
2. OBJETIVOS
O objetivo principal deste trabalho foi avaliar processos de obtenção de
comprimidos com a incorporação da dispersão de paracetamol e β-CD diretamente em
excipiente.
Para tanto, este trabalho foi planejado nas seguintes etapas:
• Avaliação da influência da β-CD na solubilidade do paracetamol;
• Incorporação da dispersão líquida de paracetamol e β-CD em excipientes,
granulação e secagem em leito estático (estufa), denominado processo I;
• Avaliação dos granulados e comprimidos obtidos pelo processo I;
• Incorporação da dispersão líquida de paracetamol e β-CD em excipiente e secagem
em leito fluidizado (leito de jorro), denominado processo II;
3. MATERIAIS 3.1. Matérias-primas
• paracetamol – Henrifarma- Lote NR 0206003
• β -ciclodextrina-Kleptose® -Roquette
• amido de milho- Henrifarma- Lote FEO 43
• celulose microcristalina- Henrifarma- Lote NR 71066
• lactose monohidratada- Synth- Lote 86770
3.2. Equipamentos
• Agitador magnético Phoemix®AP-56
• Balança analítica Ohaus® - AS200
• Balança de infra-vermelho Mettler® (PL200/LP11)
• Centrífuga Du Pont®– Sorvall TC 6
• Desintegrador Erweka® (modelo ZT-502)
• Difratômetro de raio-X -Siemens® D-5000
• DSC 50-SHIMADZU®
• Durômetro Schleuniger Pharmatron® 6D
• Espectrofotômetro HP® 8453
• Estufa com circulação de ar Fabbe® (modelo 170)
• Friabilômetro tipo Roche, Erweka® (modelo TA 20)
• Granulate Flow tester Erweka® (GWF)
• Leito de jorro –Faculdade Ciências Farmacêuticas Ribeirão Preto-USP
• Micrômetro digital Mitutoyo®
• Microscópio eletrônico de varredura JSM T33OA- JEOL®
• Microscópio Estereoscópio Leica® ( modelo MZAPO)
• Microscópio óptico DMRXA (Leica®)
• Purificador de água Milli-Q Plus- Millipore®
• Tamises números: 18, 20, 30, 50, 60, 80, 120 e coletor
• Tapped Volumeter Erweka® ( SVM 12)
• Ultrasom Branson®-modelo 1210
4. MÉTODOS
4.1. Metodologia analítica
A partir de uma solução aquosa de paracetamol na concentração de 50µg/ml, foram
preparadas diluições para obter soluções de concentrações de 2,5; 5,0; 7,5; 10,0; 12,5 e
15,0µg/ml. Colocaram-se 2,0 ml das soluções em uma cubeta de quartzo e as leituras das
absorvâncias foram realizadas em espectrofotômetro em comprimento de onda 243 nm.
Elaborou-se um gráfico relacionando as absorvâncias com a concentração de paracetamol.
Com os valores experimentais, por regressão linear, calculou-se a equação da reta e o
coeficiente de correlação.
4.2. Determinação da solubilidade do paracetamol em solução aquosa de ȕ-CD
O método para avaliação da solubilidade de fases descrito por Higuchi e Connors
(1965), é fundamentado nas alterações de solubilidade da molécula-hóspede por adição de
CD. Excesso de paracetamol foi adicionado a concentrações variadas de CD de 0 até
16mM, que corresponde a solubilidade aquosa da mesma. Os frascos foram lacrados,
protegidos da luz e submetidos à agitação constante sob temperatura controlada de 25ºC,
por 48 h. Finalizada a agitação, as amostras foram centrifugadas a 3.000 rpm por 15
minutos em Centrífuga Du Pont ® Sorvall TC 6, filtradas em membrana de acetato de
celulose (0,45µm) e a concentração do paracetamol determinada por espectrometria na
região do UV. Com os valores obtidos, construiu-se um diagrama de solubilidade.
4.3. Obtenção do granulado em estufa (Processo I)
150 ml de uma dispersão líquida de ȕ-CD de concentração 18,5 mg/ml foi preparada
em água destilada a 70ºC e após total solubilização da ȕ-CD foi adicionado paracetamol na
concentração de 18,5 mg/ml. A dispersão ȕ-CD/ paracetamol ficou sob agitação magnética
até atingir a temperatura ambiente.
A Figura 11 representa o primeiro processo (Processo I), com a incorporação da
dispersão líquida de ȕ-CD/paracetamol em excipientes, obtenção do granulado e secagem
em leito estático (estufa).
Figura 11: Esquema do processo (I) para obtenção de grânulos secos em leito estático (estufa).
paracetamol dispersãoβ-CD
dispersão líquida β-CD/ paracetamol
excipiente: amido, celulose ou lactose
Granulação/ tamis 18
Três excipientes, amido de milho, lactose monohidratada ou celulose microcristalina
doravante denominados de amido, lactose e celulose, foram gradualmente umedecidos com
a dispersão até obtenção de massa úmida, apropriada para granular. Os lotes foram
preparados com as respectivas massas de excipientes e quantidades de dispersão aquosa de
ȕ-CD/ paracetamol utilizadas conforme a Tabela 2.
Tabela 2: Composição das amostras
Excipientes Massa (g) Dispersão ȕ-CD/
paracetamol (ml)
Água (ml)
AMIDO 150 50 40
CELULOSE 150 50 30
LACTOSE 250 50 --
A massa úmida obtida foi granulada em um tamis 18 (abertura de malha = 1,00
mm). A secagem dos granulados foi realizada em estufa provida de circulação forçada de ar
(Fabbe ®-modelo 170), mantida a 30º C, por 24 horas, até que o teor de umidade residual
fosse menor que 5%. Os grânulos secos foram calibrados em tamis 18 (abertura de malha
=1,00 mm), tamis 120 (abertura de malha = 0,125 mm) e montados sobre o coletor. O
material que ficou retido no coletor foi considerado como “pó fino” e não foi utilizado.
Finalizado o processo, os granulados foram denominados GLE- amido, GLE- celulose,
GLE- lactose.
A compressão do GLE- amido, GLE- celulose e GLE- lactose foi conduzida em
máquina de comprimir de excêntrico Erweka® Korsh EKO, montada com jogo de matriz e
punções planos de 12 mm de diâmetro e ajustada para produzir comprimidos de peso
compressão do GLE- amido, GLE- celulose, GLE- lactose o equipamento foi ajustado no
nível 10 da escala do equipamento.
4.4. Obtenção do granulado em leito jorro (Processo II)
250ml de uma dispersão líquida de ȕ-CD de concentração 18,5 mg/ml foi preparada
em água destilada a 70ºC e após total solubilização da ȕ-CD foi adicionado paracetamol na
concentração de 18,5 mg/ml. A dispersão ȕ-CD/ paracetamol ficou sob agitação magnética
até atingir a temperatura ambiente e incorporada a 200g de amido. O amido de milho foi
escolhido como excipiente devido aos resultados apresentados pelos granulados obtidos
pelo processo I, por apresentar um baixo custo e levando-se em conta também que a CD é
obtida a partir do amido. A Figura 12 representa o segundo processo (Processo II) com a
incorporação da dispersão líquida de ȕ-CD/paracetamol em excipiente e secagem em leito
fluidizado (leito de jorro).
Figura 12: Esquema do processo II para obtenção de grânulos secos em leito de jorro paracetamol Dispersão ȕ-CD
Dispersão líquida ȕ-CD/ paracetamol
Excipiente: amido
O secador tipo leito de jorro utilizado neste trabalho é composto por uma coluna
cilíndrica de 140 mm de diâmetro e 830 mm de altura, acoplada a uma base cônica com
ângulo interno de 60º e altura de 95 mm. O diâmetro do orifício de entrada do ar é de
22mm e a dispersão foi introduzida no sistema através de gotejamento controlado por uma
bomba peristáltica digital (Masterflex®) com fluxo de 10 ml/min.
O sistema operacional, esquematizado na Figura 13, é constituído de um
compressor, responsável pela alimentação do ar que vai promover o movimento de jorro no
leito. O ar passa por um aquecedor e tem sua temperatura fixada e controlada (HW 2000,
COEL LTDA).
Esferas de vidro de 2,6 mm de diâmetro foram empregadas como material inerte. A
altura máxima de jorro estável e a velocidade mínima de jorro foram estabelecidas através
do emprego dos métodos clássicos (MATHUR e EPSTEIN,1974).
A vazão do ar é controlada por uma válvula gaveta e medida por uma placa de
orifício calibrada. Estabelecido o jorro, iniciou-se o aquecimento do ar. Atingida a
temperatura desejada, iniciou-se o processo de granulação. As condições operacionais
empregadas são encontradas na Tabela 3.
Tabela 3: Condições operacionais empregadas na granulação do material
Hmax = altura máxima de jorro; H/ Hmax = relação entre a altura utilizada (14 cm) e a altura
máxima de jorro estável determinada (28 cm); Vmj = velocidade mínima de jorro; V/Vm=
relação entre a velocidade empregada e a velocidade mínima de jorro; Ti = temperatura de
entrada; Ts = temperatura de saída.
A vazão do ar foi calculada através do emprego de uma placa de orifício conectada
a um tubo em U contendo mercúrio como fluido manométrico, segundo a equação 3:
onde:
V0= vazão volumétrica do ar (m3/s)
A = área do orifício (0,00005102m2)
LEITO DE JORRO
Hmax (m) H/Hmax Vmj (m3/min ) V/Vmj Ti (ºC) Ts (ºC)
0,28 0,33 0,0252 1,2 80 61
(
)
(
4)
0
1 2
β ρ
ρ −
⋅ ⋅ ⋅
= A Ca g h
Ca = coeficiente de calibração (0,61)
ȡ = densidade do ar (Kg/ m3)
ȡm = densidade do fluido manométrico (Kg/ m3)
g = aceleração da gravidade (m/s2)
h = altura da coluna de mercúrio (0,144m)
ȕ = relação entre o diâmetro do orifício e do tubo (0,40625)
Finalizado o processo, o granulado foi recolhido do coletor indicado na Figura 13 e
denominado GLJ- amido.
A compressão do GLJ- amido foi conduzida em máquina de comprimir de
excêntrico Erweka® Korsh EKO, montada com jogo de matriz e punções planos de 12 mm
de diâmetro e ajustada para produzir comprimidos de peso teórico de 0,500g. O
equipamento possui uma escala arbitrária de força (0 a 10). Para a compressão do GLJ-
amido o equipamento foi ajustado no nível 8 da escala do equipamento.
4.5. Avaliação dos granulados obtidos pelos processos I e II 4.5.1. Umidade
A perda por dessecação do GLE- amido, GLE- celulose, GLE- lactose e do GLJ-
amido foi determinada por método gravimétrico, empregando-se balança analítica com
sistema de secagem por infravermelho. Amostras de aproximadamente 1g foram pesadas
em bandejas de alumínio taradas, dessecadas por temperatura entre 75 e 100ºC, sendo a
4.5.2. Análise morfológica
A análise morfológica do GLE- amido, GLE- celulose e GLE- lactose foi realizada
por captação de imagens , através de Lupa DMRXA (Leica®) utilizando o programa Leica®
Qwin Image Systems e aumento de 80 vezes.
A análise morfológica da CD, paracetamol e GLJ- amido foi realizada por
fotomicrografias obtidas por microscopia eletrônica (Microscópio eletrônico de varredura
JSM T33OA- JEOL®) de varredura. As amostras foram distribuídas sobre uma fita
dupla-face aderida a um suporte de metal, revestidas sob atmosfera de argônio, com ouro coloidal
e analisadas. As imagens fotomicrografadas obtidas foram avaliadas.
4.5.3. Distribuição Granulométrica
A determinação da granulometria (tamanho e distribuição), dependendo do tamanho
da partícula, pode ser realizada por tamisação, microscopia óptica e eletrônica, por
espalhamento de luz, por difração de laser entre outros métodos (BARBER, 1993).
A determinação da granulometria do GLE- amido, GLE- celulose e GLE-lactose foi
realizada seguindo a metodologia prevista na Farmacopéia Brasileira (1988). O
procedimento foi desenvolvido mecanicamente empregando-se um vibrador de tamises
(Produtest®), acionado no ponto 8 da escala arbitrária (0 a 10) do equipamento e tamises
padronizados superpostos, partindo-se das aberturas de malha maior diâmetro ao menor.
Neste ensaio, cerca de 70g do GLE- amido, GLE- celulose e GLE-lactose foram colocados
no tamis de maior malha e submetido à vibração, repetindo-se o procedimento em
triplicata, durante 15 minutos. Os tamises utilizados, com suas respectivas aberturas de
Tabela 4: Número dos tamises e abertura da malha empregados na determinação
granulométrica dos materiais secos em leito estático.
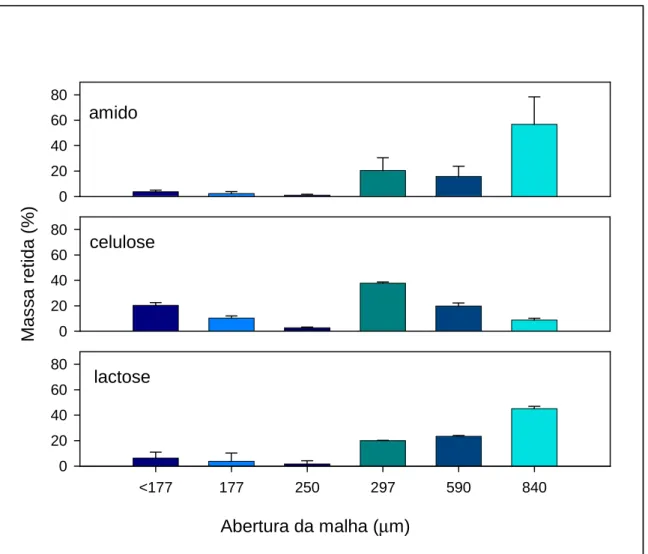
Número do tamis Abertura da malha (mm)
20 0,840 30 0,590 50 0,297 60 0,250 80 0,177 Padrão <U.S.> proposto pelo National Bureau of Standars
A determinação da distribuição do tamanho das partículas do GLJ- amido foi
realizada por captação de imagens, através de microscópio óptico DMRXA (Leica®),
utilizando o programa Leica® Qwin Image Systems para quantificação. Foram medidos os
diâmetros de uma amostragem mínima de 200 partículas, usando o método de Feret
(PRISTA et. al., 2002).
Para a construção do gráfico de distribuição do tamanho de partículas o número de
classes foi calculado, utilizando-se a equação 4 (VIEIRA, 1980):
k= 1 + 3,22log n Equação [4]
onde:
k: número de classes
n: é o número de dados
4.5.4. Escoamento
Para estudar as características de escoamento do GLE- amido, GLE- celulose e
o ângulo de escoamento e o tempo necessário para o material escoar. Foram utilizados os
diâmetros de abertura de 12 mm e 9 mm. Para estudar as características de escoamento do
GLJ- amido utilizou-se somente o diâmetro de abertura do funil de 12 mm.
4.5.5. Densidades aparentes bruta e compactada, índice de compressibilidade percentual e fator de Hausner
As densidades aparentes do GLE- amido, GLE- celulose, GLE- lactose e GLJ-
amido foram determinadas, indiretamente, através das medidas de seus volumes aparentes.
Para o ensaio da densidade aparente bruta, o material foi colocado em uma proveta
de 250 mL do equipamento Tapped Volumeter Erweka ® (SVM 12), determinando-se a
massa em balança semi-analítica Ohaus®, modelo PL 400 (HANCOCK et al., 2001). A
densidade aparente bruta foi calculada através da equação 5:
Vb m
db= Equação [5]
onde:
db = densidade bruta m = massa (g)
Vb = volume bruto (mL)
Para a determinação da densidade aparente compactada, procedeu-se da seguinte
forma: o material foi colocado em uma proveta de 250mL do equipamento Tapped
Volumeter (Erweka® SVM 12); após a colocação do material na proveta, esta foi inserida
material. Este procedimento foi repetido até que o volume aparente sofresse uma redução
inferior a 2%, considerando-se volume compactado aquele lido na penúltima determinação
e determinando-se a massa em balança semi-analítica Ohaus®, modelo PL 400
(HANCOCK et al., 2001). A densidade aparente compactada foi calculada através da
seguinte equação 6:
Vc m
dc= Equação [6]
onde:
dc = densidade compactada (g/ml) m = massa (g)
Vc = volume compactado (ml)
A partir dos valores de densidades aparentes brutas e compactadas, foi possível
calcular o índice de compressibilidade percentual, IC%, através da equação 7 (VACHON e
CHULIA, 1998).
100
IC%= ⋅
dc dc - db
Equação [7]
onde: IC% = Índice de Compressibilidade Percentual (%)
Segundo a metodologia referendada por Guo (1985), o fator de Hausner foi
determinado através do quociente entre as densidades de compactação e bruta, conforme
equação 8:
db dc
FH = Equação [8]
onde: FH = fator de Hausner
dc = densidade aparente compactada (g/mL) db = densidade aparente bruta (g/mL)
4.5.6. Difração de raios X do GLJ- amido
A identificação da estrutura cristalina do paracetamol, da ȕ-CD, da mistura física e
do GLJ- amido foi realizada por difração de raios-X. Os difratogramas foram obtidos em
um difratômetro de raios-X para policristais Siemens®D-5000, radiação CuKα
monocromatizada por cristal grafite. Os dados foram tratados usando-se o programa
Winmetric
e o refinamento dos parâmetros cristalográficos foi feito a partir dos dados de
difração (2θ e intensidade). A velocidade de varredura usada foi de 0,3 segundos a cada
0,02º em um intervalo de 20º até 120º.
4.5.7 Obtenção das curvas de DSC do GLJ- amido
Aproximadamente 2,6mg de paracetamol, ȕ-CD, amido, mistura física e GLJ-
amido foram colocados em cadinho de alumínio selado (DSC 50-SHIMADZU®). As curvas
4.6. Avaliação dos comprimidos obtidos a partir dos processos I e II 4.6.1. Aspecto
Foram observados os caracteres visuais da forma farmacêutica obtida em relação à
forma geométrica, superfície, coloração e presença de partículas ou material estranho à
formulação.
4.6.2. Variação do peso
Foram pesados 20 comprimidos em balança analítica Ohaus®(modelo AS 200) e
calculados a média e o desvio padrão.
4.6.3. Espessura
A espessura dos comprimidos foi medida em Micrômetro digital Mitutoyo®
(precisão de 0,001 mm) em 20 unidades, em triplicata.
4.6.4. Resistência mecânica
Para avaliar a resistência mecânica dos comprimidos à pressão radial, mediu-se a
dureza de 20 comprimidos através do Durômetro Schleuniger Pharmatron® (modelo 6D).
Para o teste de friabilidade empregou-se friabilômetro tipo Roche, Erweka® (modelo TA
20). O teste foi realizado através da determinação do peso de 20 comprimidos isentos de pó
e estes são submetidos a 100 quedas livres de uma altura de cerca de 13 cm dentro do
tambor rotativo do equipamento. Depois do rolamento, determinou-se o peso dos
comprimidos isentos de pó, exprimindo-se a friabilidade em função da percentagem de pó
4.6.5. Tempo de desintegração
Determinou-se o tempo necessário para os comprimidos desintegrarem. Foram
analisadas 18 unidades em aparelho para medir tempo de desintegração Erweka® (modelo
ZT-502), de acordo com os critérios estabelecidos pela Farmacopéia Brasileira, 1988. O
equipamento é constituído por 6 tubos de vidro com 7,5 cm de comprimento, abertos e
fixos em redes de 10 “mesh” no fundo dos mesmos. Para determinar o tempo de
desintegração, colocou-se um comprimido em cada tubo e o suporte dos cestos foi
mergulhado num recipiente de 1 litro com água, a 37ºC ± 1º C de modo que os
comprimidos permanecessem 2,5 cm abaixo da superfície do líquido durante o seu
movimento ascendente não se aproximando mais do que 2,5 cm do fundo do recipiente.
4.7. Análise estatística
Os resultados foram expressos estatisticamente por meio da análise de variância,
Teste F e Teste Tukey utilizando o programa Sisvar, desenvolvido pelo Departamento de
5. RESULTADOS E DISCUSSÃO
5.1. Metodologia analítica
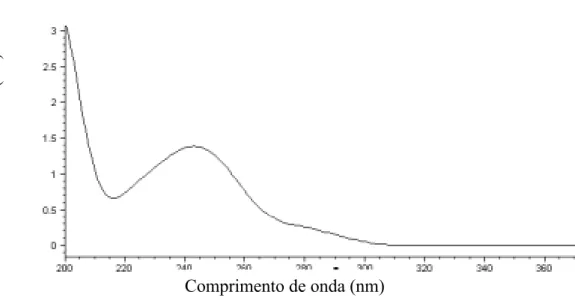
A metodologia analítica empregada foi a espectrofotometria sendo que a Figura 14
mostra que o comprimento de onda de máxima absorção do paracetamol foi de 243nm,
resultado obtido a partir de uma varredura de uma solução aquosa de paracetamol de
concentração 20 µg/ml em espectrofotômetro na faixa de comprimento de onda de 200 a
400nm.
Figura 14: Espectro de absorção do paracetamol em solução aquosa na região do UV.
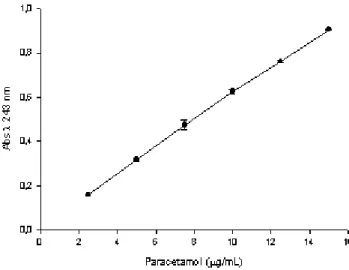
Através da análise dos valores de absorvâncias correspondentes às concentrações
conhecidas de paracetamol em água destilada, expressos na Tabela 5, foi construída a curva
analítica do paracetamol como mostra a Figura15, em 243 nm, que corresponde ao comprimento
de onda de máxima absorção do paracetamol na região do UV.
Intensidade (u.a)
Tabela 5. Valores das absorvâncias das soluções aquosa do paracetamol obtidas por
espectrofotometria no Ȝ = 243 nm .
Concentração
(µg/ml)
Abs 1 Abs 2 Abs 3 x σ CV
2,5 0,1552 0,1605 0,1624 0,159 0,0037 2,327
5,0 0,3240 0,3173 0,3108 0,317 0,0066 2,082
7,5 0,4668 0,4578 0,4995 0,475 0,0219 4,611
10,0 0,6295 0,6347 0,6153 0,627 0,0100 1,595
12,5 0,7540 0,7682 0,7634 0,762 0,0072 0,944
15,0 0,9075 0,9017 0,9072 0,906 0,0033 0,364
Figura 15: Curva analítica do paracetamol em água destilada (equação da
reta: y = 0,06469x + 0,00536, r2= 0,9997)
5.2. Determinação da solubilidade do paracetamol em solução aquosa deȕ-CD.
A determinação da solubilidade do paracetamol em soluções aquosas de
concentrações crescentes foi útil para avaliar a influência da ȕ-CD na quantidade de
Na Figura 16, está representada uma isoterma de solubilidade, sendo evidente o
aumento da concentração de paracetamol em água com o concomitante aumento da
concentração de ȕ-CD, indicando que as moléculas estão interagindo. Baseado nos dados
da literatura, o aumento da solubilidade do paracetamol foi verificado até valores próximos
ao limite de solubilidade da ȕ-CD em água, que é de cerca de 16mM (FROMMING e
SZEJTLI, 1994). A análise deste experimento evidencia uma isoterma do tipo AP, com um
discreto desvio positivo de linearidade que ocorre próximo ao limite de solubilidade da ȕ
-CD em água, sugerindo que mais de uma molécula de -CD é complexada com uma
molécula-hóspede.
Figura 16: Isoterma de solubilidade
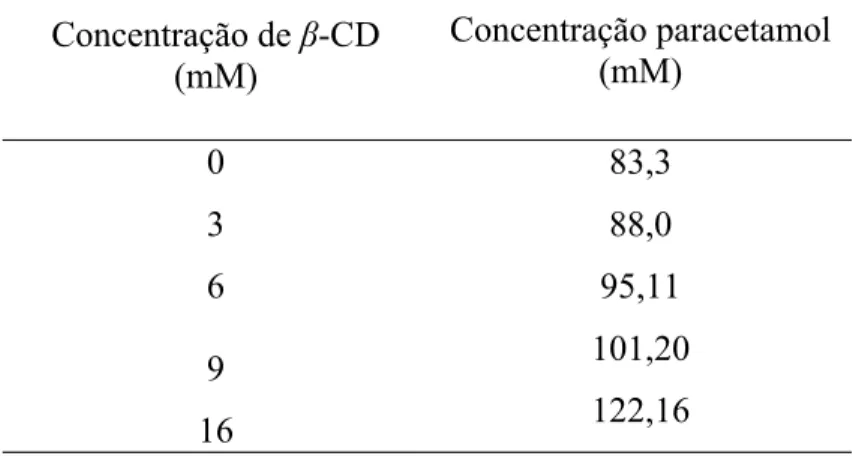
A Tabela 6 apresenta os resultados das concentrações do paracetamol em soluções
aquosas de concentrações crescentes de ȕ-CD. Verificou-se um aumento aproximadamente
de 46% no valor da solubilidade do paracetamol em solução aquosa de ȕ-CD 16 mM em
relação à solubilidade do paracetamol em água.
-2 0 2 4 6 8 10 12 14 16 18
80 90 100 110 120 130 Co ncent ração par acet am ol ( m M )