Nota Técnica
*e-mail: gabheerdt@iqm.unicamp.br
VALIDAÇÃO COMPUTACIONAL DE MÉTODOS COMPOSTOS NO ESTUDO DE PROPRIEDADES MOLECULARES
Gabriel Heerdt* e Nelson H. Morgon
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas - SP, Brasil
Recebido em 16/7/10; aceito em 26/11/10; publicado na web em 9/2/11
COMPUTATIONAL VALIDATION OF COMPOSITE METHODS IN THE STUDY OF MOLECULAR PROPERTIES. Composite methods using ONIOM and different basis sets have been used to calculate proton and electron afinities for a set of alcohols at QCISD(T)/6-311++G(2df,p) level of theory. The study was carried out considering HF, MP2 and DFT (25 exchange correlation functional) methods. The calculation performed at ONIOM2(QCISD(T)/6-311++G(2df,p):HF/6-31G(d))//ONIOM2(O3LYP/6-31G(d):HF/6-31G(d)) resulted in the smallest average absolute deviation for AP and AE, 4,75 kJ/mol e 0,43 eV, respectively.
Keywords: ONIOM; proton and electron afinities; composite methods.
INTRODUÇÃO
Nas últimas décadas o aumento no desempenho computacional tem tido um enorme impacto na Química Teórica. Em adição a isso, duas importantes áreas contribuíram para este progresso: o primeiro foi o desenvolvimento e implementação de métodos de correlação eletrônica cada vez mais precisos, tal como o método Coupled Cluster,1,2 e o segundo refere-se à compreensão dos efeitos de grandes conjuntos de base e seu comportamento de convergên-cia no que diz respeito à inclusão de funções de maior momento angular.3 Juntos, esses avanços, levaram a avaliações mais precisas de diversas propriedades moleculares, em especial relacionadas a grandezas termoquímicas.4-10
Mesmo tendo em mãos métodos soisticados e funções de base extensas para descrever qualquer sistema eletrônico, a realização prática de cálculos que envolvam um grande número de partículas ainda é uma tarefa dispendiosa computacionalmente. Algumas es-tratégias têm tido sucesso e tornado possível a abordagem precisa de sistemas químicos relativamente grandes. Dentre elas destacam-se a Teoria do Funcional de Densidade, o método ONIOM e também os métodos compostos.
Métodos compostos
Os métodos compostos surgiram na década de 1980 com o obje-tivo de obter uma energia eletrônica para uma dada geometria mole-cular de equilíbrio de forma aditiva, ou seja, somando termos vindos de cada etapa do cálculo.4 Atualmente, existe uma grande variedade desses métodos sendo mais difundidos os métodos Gaussian-n11-17 e os métodos do conjunto de base completo.18-20
Desde o seu surgimento, devido aos resultados coniáveis para as quantidades termoquímicas e ao tempo computacional reduzido, o número de aplicações desses métodos cresceu rapidamente. Dentre as propriedades que podem ser estudadas por esses métodos estão cálculos envolvendo: ainidades por próton,6,21-24 energias de ioniza-ção,21,25 entalpias de formação,26 energias de dissociação,27-29 energias de atomização,30,31 ainidades por elétron,7,32,33 dentre outras.
Desenvolvidos por Pople e seus colaboradores, a primeira versão das metodologias Gaussian-n (G1)11 surgiu em 1988 descrita como um novo procedimento para obtenção da energia total de moléculas na geometria de equilíbrio. Com o passar dos anos foram sendo adi-cionadas novas correções a essa teoria e também um conjunto maior de moléculas para a validação das mesmas. A teoria encontra-se na quarta geração sendo que cada uma possui suas variações como, por exemplo, a versão G3(MP2).13
Na metodologia G3(MP2) a geometria molecular é obtida pelo método MP2/6-31G(d) considerando todos os elétrons. Uma série de cálculos single point são realizados num nível mais alto de teoria aim de corrigir a energia da geometria de equilíbrio. A energia inal para um sistema qualquer é obtida considerando-se:
E[G3(MP2)] = E[QCISD(T)/6 – 31G(d)] +
∆E(MP2) + ∆E(SO) + E(HLC) + E(ZPE) (1) O termo E[QCISD(T)/6-31G(d)] corresponde a energia resultante do cálculo single point no nível QCISD(T)/6-31G(d). A correção para a perturbação de segunda ordem (∆E(MP2)) é obtida por:
∆E(MP2) = E(MP2/GTLarge) – E(MP2/6 – 31G(d)) (2) Todos os cálculos para correção da energia de equilíbrio são realizados mantendo-se os elétrons do caroço ixos. Com isso um número muito menor de conigurações é calculado e, portanto, o tempo computacional é reduzido signiicativamente.
A correção de spin-órbita [∆E(SO)] é adicionada para átomos a partir do terceiro período da tabela periódica, sendo seu valor obtido experimentalmente através de técnicas espectroscópicas ou então a partir de cálculos mais precisos.34,35
A energia de ponto zero [E(ZPE)] é corrigida pelo escalona-mento (0,8929) das frequências provenientes do cálculo no nível HF/6-31G(d).
E por im, o termo E(HLC) (higher level correction) é adicionado com o objetivo de ajustar deiciências remanescentes na energia inal. Ele é calculado considerando-se as expressões:
E(HLC) = – Cnβ – D(nα – nβ) (4)
Os termos A, B, C e D são escolhidos de tal modo que os valores inais de algumas propriedades resultem no menor erro possível em relação aos valores experimentais, e valem 9,279; 4,471; 9,345 e 2,021 mhartree, respectivamente. A Equação 3 é utilizada para moléculas e a Equação 4 para átomos e seus íons. Os termos nα e nβ representam os números de elétrons α e β, sendo nα ≥ nβ.
A partir da energia G3(MP2), dada pela Equação 1, é possível obter os valores para entalpia e energia livre e com isso explorar uma variedade de propriedades termodinâmicas.13
O método ONIOM
A partir de meados da década de 1970, surgiram métodos conhe-cidos como híbridos.36-38 Esses métodos consistem na combinação de diferentes tipos de aproximações, ab initio, semiempírico e mecânica molecular, tentando assim aproveitar as vantagens de cada uma e contornando algumas de suas limitações.
A primeira implementação desta nova geração de métodos icou conhecida como IMOMM, Integrated Molecular Orbital and Mo-lecular Mechanics.39 Após o sucesso desse método, novos estudos foram feitos para se obter maior versatilidade nos cálculos de estrutura eletrônica. Surgiu então o método ONIOM, Our own n-layered Inte-grated Molecular Orbital and Molecular Mechanics,40 que pode ser descrito como uma superposição de cálculos como numa “cebola”. Segundo o esquema teórico desse método, qualquer sistema molecular pode ser dividido em diferentes níveis ligados seguindo uma ordem mais conveniente ao problema em questão. Cada parte pode ser tratada sobre qualquer nível de cálculo e ao integrar-se os resultados obtidos nestas partes, obtém-se uma extrapolação atingindo valores de energia mais precisos sobre todo o sistema.41
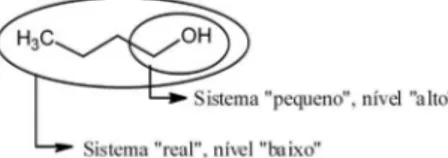
Quando dois níveis diferentes de aproximação são integrados o método ONIOM segue o modelo apresentado na Figura 1.
A equação para a energia inal de um sistema contendo 2 camadas é escrita como:
E = EH,Smodel + EL,Real – EL,Smodel (5)
em que H (high) e L (low) referem-se aos níveis de teoria “alto” e “baixo” de aproximação, respectivamente, enquanto SModel (small) e Real (real) referem-se aos sistemas de tamanho “pequeno” e “real”, respectivamente. Por exemplo, pode ser realizado um cálculo no nível alto QCISD(T)/6-31G(d) para a parte pequena, e no nível baixo um cálculo semi-empírico PM3 para as partes pequeno e real, extrapolan-do os resultaextrapolan-dos obtém-se uma aproximação extrapolan-do cálculo no nível alto para a parte real, ou seja, QCISD(T)/6-31G(d) para toda a molécula. A teoria do funcional de densidade
Em 1927 Llewellen H. Thomas e Enrico Fermi propuseram uma alternativa relativamente simples para a resolução da equação de Schrödinger,42 conhecida como a aproximação de Thomas-Fermi. Nesta aproximação, ao invés da função de onda, considera-se a
den-sidade eletrônica ρ(r). Assim, a energia total do sistema (E) passa a ser escrita como um funcional de ρ(r), ou seja, E[ρ(r)]. Essa teoria passou a ganhar mais espaço em 1964-65 quando Kohn, Sham e Hohenberg formalizaram a Teoria do Funcional de Densidade.43,44
O sucesso da formalização de Kohn, Sham e Hohenberg depende da qualidade do funcional de troca-correlação de cada método. Com ilosoias diferentes em suas aproximações, diversos funcionais têm sido desenvolvidos (Tabela 1). Na primeira geração de funcionais a energia de troca-correlação é obtida a partir da descrição da densidade eletrônica como um sistema de gás de elétrons homogêneo, ela é conhecida como LDA (Local Density Approximation). Os funcionais do tipo GGA (Generalized Gradient Approximation) expressam o funcional de troca-correlação em termos do gradiente de carga total. Ao adicionarem-se no gradiente de densidade eletrônica termos que dependam da densidade de energia cinética têm-se os funcionais conhecidos como meta-GGA. Uma última maneira de representar a densidade eletrônica é adicionando uma fração de energia de troca Hartree-Fock, esses métodos são conhecidos como híbridos.45,46
Propriedades termodinâmicas
O estudo de processos em que ocorre a transferência de elétrons e prótons é muito difundido devido à importância dessas reações nas mais diversas áreas, como em processos biológicos,47-50 nanoestru-turas,51,52 entre outros.
Ainidades por próton e eletrônica
A ainidade por próton (AP) é deinida como sendo a variação de entalpia ocorrida numa reação de desprotonação de um sistema neutro (AP = –∆H6):
HX→X– + H+ (6)
A ainidade por elétron (AE) é deinida como sendo a variação de entalpia ocorrida numa reação em que um ânion (X-) perde um elétron icando neutro (X) (AE = ∆H7):
X–→X + e– (7)
METODOLOGIA COMPUTACIONAL
Na validação de um método composto capaz de descrever as ainidades por próton e eletrônica foi utilizado um grupo de 10 monoalcoóis saturados, de cadeia alifática, linear ou ramiicada e com números de carbonos variando entre 2 até 9. Os alcoóis: etanol; 1,1-dimetil-etanol; 1-pentanol; 3-pentanol; 3-metil-2-butanol; 2-etil-1-butanol; 2-hexanol; 2-metil-2-pentanol; 2-heptanol; 1-nonanol;
Figura 1. Diagrama esquemático do método ONIOM
Tabela 1. Métodos utilizados para o estudo das propriedades moleculares
B1LYP PBE1PBE MPW3PBE
B3LYP* tHCTH HCTH
B3P86 tHCTHhyb HCTH147*
B3PW91 X3LYP HCTH93
B972 BMK M06
B98 B1B95 M06-2X
BhandH MPW1LYP VSXC
BhandHLYP MPW1PBE HF
O3LYP mPW1PW91 MP2
foram escolhidos aim de validar um método para ser aplicado em cálculos de propriedades moleculares do colesterol, um álcool policíclico.
Os modelos testados foram: variações para o método ONIOM: ONIOM 1, ONIOM 2, sem ONIOM; funções de base dupla-ζ (dupla-zeta-valência) para as camadas alta (dupla-ζ-a) e baixa (dupla-ζ-b) do ONIOM; funções de base tripla-ζ (tripla-zeta-valência) para a camada alta do ONIOM; métodos DFT, MP2 e HF para a camada alta. A camada alta do método ONIOM refere-se ao melhor nível de teoria aplicado, enquanto, na camada baixa o nível de teoria aplicado foi o método Hartree-Fock(HF) em todo procedimento. A escolha do método HF deve-se ao fato dele ser um método ab initio pouco custoso computacionalmente.
As etapas de cálculos realizadas para obtenção da energia para o estado fundamental foram:
- otimização e freqüência - método/dupla-ζ-a:HF/dupla-ζ-b; - energia -QCISD(T)/dupla-ζ-a:HF/dupla-ζ-b;
- energia:método/tripla-ζ:HF/dupla-ζ-b.
A energia para o estado fundamental a partir desses cálculos pode ser escrita como:
E0 = E2 + E3 – E1 + ZPE (8) sendo E2, E3 e E1 as energias inais obtidas nas etapas de cálculos apresentadas. Ao subtrair-se a energia obtida na etapa 1 (E1) das energias das etapas 2 e 3 (E2 e E3) obtém-se uma extrapolação para a energia no nível QCISD(T)/ tripla-ζ. Foram omitidas as correções de escalonamento de ZPE e HLC, pois o objetivo foi veriicar a qualidade do que está sendo testado sem “mascarar” os resultados.
As variáveis, variações ONIOM, conjuntos de funções de base e métodos, foram testadas isoladamente, ou seja, enquanto uma era testada as outras eram mantidas ixas, dessa forma foi excluída a correlação entre elas, tornando-se o procedimento mais objetivo. Na Tabela 1 são apresentados os métodos testados, as funções de base escolhidas são apresentadas a seguir:
- dupla-ζ(a/b): 6-31G(d), cc-pVDZ;
- tripla-ζ:6-311++G(2df,p), aug-cc-pVTZ, GTLarge. As variações escolhidas para o método ONIOM foram: - sem ONIOM - sempre se utilizou o método e a base da camada alta para toda a estrutura;
- ONIOM 1 - as estruturas foram dividas em duas camadas, sendo que a hidroxila e o carbono diretamente ligado a ela e os átomos de hidrogênio eventualmente ligados a esses átomos foram deinidos como pertencendo à camada alta, o restante da molécula foi deinido como camada baixa;
- ONIOM 2 - também foram deinidas duas camadas, a hidroxila, o carbono diretamente ligado a ela e os carbonos ligados a ele bem como os hidrogênios utilizados para completar valência desses átomos foram deinidos como sendo da camada alta e o restante da molécula da camada baixa.
Todos os cálculos foram feitos utilizando-se os programas Gaussian0353e NWCHEM-5.1,54 sendo o último utilizado apenas para cálculos envolvendo funcionais de densidade não presentes no programa Gaussian03.
RESULTADOS E DISCUSSÃO
Em todos os resultados foram aplicados métodos estatísticos a im de analisar diferenças signiicativas ou não entre os dados obtidos, podendo assim conirmar qual fornece os melhores valores. O método lsd (least square difference) foi utilizado para comparar médias, e ele é deinido como:
(9) onde tα é o valor tabelado de t de student para 95% de coniança, MSerro
é o quadrado médio dos resíduos da ANOVA e n o número de repetições em cada tratamento.
Além disso, o tempo computacional foi levado em conta, já que o objetivo também foi o de se estabelecer uma relação ótima entre custo computacional e qualidade dos resultados.
Funções de base tripla-zeta-valência(tripla-ζ)
Quando testados os conjuntos de funções de base tripla-ζ obtiveram-se os resultados apreobtiveram-sentados na Tabela 2 para ainidades por próton e elétron.
Aplicando-se o teste de comparação de médias lsd para AP tem-se:
onde MStotal é a somatória de todas as variâncias, MStrat a somatória das
variâncias dos tratamentos, a o número de tratamentos e n o número de repetições em cada tratamento, no caso são três funções de base (a=3) e dez moléculas (n=10). Somando o valor obtido de 4,34 kJ/mol com o menor desvio absoluto entre os tratamentos, tem-se um intervalo (4,76-9,10 kJ/mol) no qual os desvios são considerados equivalentes a 95% de coniança.
Para AE o valor obtido para o teste de lsd foi de 0,07 eV. Tanto para os desvios absolutos de AP quanto AE pode-se concluir que não existem diferenças entre as funções de base tripla-ζ, isso porque nenhum desvio se encontra fora dos intervalos de coniança obtidos. Assim, o tempo computacional entra como critério para escolha da melhor função de base. Observa-se uma grande vantagem da função 6-311++G(2df,p) com relação as demais funções tripla-ζ.
Tabela 2. Desvios absolutos de AP (kJ/mol) e AE (eV) para as funções de base tripla-ζ
Método 6-311++G(2df,p) aug-cc-pvTZ GTLarge
AP AE AP AE AP AE
B3LYP 5,11 ± 4 0,41 ± 0,06 4,74 ± 4 0,40 ± 0,06 5,31 ± 3 0,42 ± 0,06
HCTH147 5,15 ± 4 0,41 ± 0,06 4,79 ± 3 0,34 ± 0,04 4,64 ± 3 0,42 ± 0,06
Média 5,13 ± 4 0,41 ± 0,06 4,76 ± 3 0,40 ± 0,05 4,98 ± 3 0,42 ± 0,06
Tempo relativo* 0,39 1,00 0,70
Funções de base dupla-zeta-valência(dupla-ζ)
As diferentes funções de base dupla-ζ testadas nesse trabalho fo-ram utilizadas nas camadas alta e baixo do método ONIOM, portanto, foram deinidas as melhores com relação aos desvios médios absolu-tos e tempos computacionais para cada camada. Na Tabela 3 têm-se os resultados para as funções de base dupla-ζ para a camada alta.
Os valores de lsd obtidos para AP e AE, respectivamente, são iguais a 5,58 kJ/mol e 0,08 eV. Sendo assim, conclui-se que a base 6-31G(d) possui resultados signiicativamente melhores para AP, além de um tempo computacional menor.
Na Tabela 4 são apresentados os resultados para as funções de base dupla-ζ para a camada baixa. Os valores de lsd obtidos, 5,40 kJ/mol para AP e 0,08 eV para AE, deixam claro que os desvios médios absolutos não podem ser considerados diferentes. Analisando o tempo de processamento observa-se que a função de base 6-31G(d) leva uma expressiva vantagem e por isso mostra ser a mais adequada.
Métodos para a camada alta
Na Tabela 5 são apresentados os desvios médios absolutos para
AP em cada método testado. O valor de lsd calculado a partir desses dados é igual a 4,43 kJ/mol. Já o menor erro encontrado, para o método O3LYP, é de 4,75 kJ/mol. Assim, conclui-se que as médias aceitas são aquelas cujos valores estão entre 4,75-9,18 kJ/mol.
Os resultados obtidos para a AE são mostrados na Tabela 6. Para a comparação dos desvios absolutos pelo método lsd foram utilizados apenas os métodos aceitos na etapa anterior, ou seja, os resultados de AP. O valor obtido para esses métodos foi de 0,07 eV. Desta forma, são aceitos como sendo equivalentes desvios médios absolutos com valor máximo de 0,44 eV.
Como pode ser observado dos 27 métodos testados apenas 8 (Tabela 6) foram aceitos em ambos os testes de comparação aplicados. ONIOM
Os resultados obtidos para AP e AE, considerando-se como teste os níveis da variável ONIOM, são apresentados na Tabela 7. Com os valores de lsd, 4,29 kJ/mol para AP e 0,08 eV para AE, pode-se con-cluir que não há diferença entre as variações para o método ONIOM. O tempo computacional signiicativamente mais baixo quando utili-zadas as variações ONIOM1 e ONIOM2 indicam a versatilidade da utilização do método ONIOM nesse estudo.
CONCLUSÃO
Os resultados das avaliações das diferentes funções de base deixam claro que as funções de base de Pople, 6-31G(d) e 6-311++G(2df,p) mostram-se mais adequadas, principalmente pelo tempo computacional. Pode-se concluir que o método ONIOM1 consegue descrever tão satisfatoriamente as propriedades aqui estu-Tabela 3. Desvios médios absolutos para AP (kJ/mol) e AE (eV) para as
funções de base dupla-ζ da camada alta
Método 6-31G* cc-pvDZ
AP AE AP AE
B3LYP 5,11 ± 4 0,41 ± 0,06 11,74 ± 5 0,32 ± 0,06
HCTH147 5,15 ± 4 0,41 ± 0,06 14,23 ± 5 0,35 ± 0,07
Média 5,13 ± 4 0,41 ± 0,06 12,98 ± 5 0,34 ± 0,07
Tempo relativo 0,47 1,00
Tabela 4. Desvios médios absolutos para AP (kJ/mol) e AE (eV) para as funções de bases base dupla-ζ da camada baixa
Método 6-31G* cc-pvDZ
AP AE AP AE
B3LYP 5,11 ± 4 0,41 ± 0,06 6,79 ± 4 0,39 ± 0,06
HCTH147 5,15 ± 4 0,41 ± 0,06 7,23 ± 4 0,39 ± 0,06
Média 5,13 ± 4 0,41 ± 0,06 7,01 ± 4 0,39 ± 0,06
Tempo relativo 0,26 1,00
Tabela 5. Resultados de AP (kJ/mol) para os métodos testados na camada alta em ordem de qualidade
Método Desvios Método Desvios Método Desvios
O3LYP 4,75 ± 4 M06 6,18 ± 3 B3PW91 7,81 ± 4
B1LYP 4,85 ± 4 mPW3PBE 6,42 ± 3 HCTH 7,96 ± 4
B3LYP 5,11 ± 4 B98 6,50 ± 3 BHandH 8,28 ± 3
HCTH147 5,15 ± 4 mPW1LYP 7,16 ± 4 B972 8,63 ± 4
HCTH93 5,40 ± 3 PBE1PBE 7,34 ± 3 B3P86 9,32 ± 3
tHCTH 5,64 ± 3 B1B95 7,42 ± 4 BHandHLYP 10,19 ± 4
VSXC 5,64 ± 4 mPW1PW91 7,50 ± 4 MP2 10,28 ± 4
X3LYP 5,74 ± 4 tHCTHhyb 7,63 ± 3 BMK 13,31 ± 4
M06-2X 5,96 ± 4 mPW1PBE 7,73 ± 4 HF 23,31 ± 4
Tabela 6. Resultados de AE (eV) para os métodos testados na camada alta em ordem de qualidade de AP
Método Desvios Método Desvios Método Desvios
O3LYP* 0,43 ± 0,08 M06 0,48 ± 0,05 B3PW91 0,51 ± 0,06
B1LYP* 0,42 ± 0,06 mPW3PBE 0,48 ± 0,06 HCTH* 0,37 ± 0,06
B3LYP* 0,41 ± 0,06 B98 0,48 ± 0,06 BhandH 0,53 ± 0,06
HCTH147* 0,41 ± 0,06 mPW1LYP* 0,38 ± 0,06 B972 0,53 ± 0,06
HCTH93 0,45 ± 0,06 PBE1PBE 0,51 ± 0,06 B3P86** 0,53 ± 0,06
tHCTH 0,47 ± 0,06 B1B95 0,52 ± 0,06 BHandHLYP** 0,55 ± 0,06
VSXC* 0,39 ± 0,08 mPW1PW91 0,51 ± 0,06 MP2** 0,28 ± 0,07
X3LYP* 0,40 ± 0,06 tHCTHhyb 0,50 ± 0,06 BMK** 0,54 ± 0,09
M06-2X 0,49 ± 0,06 mPW1PBE 0,51 ± 0,06 HF** 0,70 ± 0,06
Tabela 7. Desvios absolutos de AP (kJ/mol) e AE (eV) para o método ONIOM
Método ONIOM1 ONIOM2 Sem ONIOM
AP AE AP AE AP AE
B3LYP 5,69 ± 3 0,45 ± 0,06 5,11 ± 4 0,41 ± 0,06 6,66 ± 3 0,49 ± 0,07
HCTH147 7,15 ± 3 0,49 ± 0,06 5,15 ± 4 0,41 ± 0,06 6,76 ± 3 0,47 ± 0,07
Média 6,42 ± 3 0,47 ± 0,06 5,13 ± 4 0,41 ± 0,06 6,71 ± 3 0,48 ± 0,07
Tempo relativo 0,32 0,57 1,00
dadas quanto os métodos ONIOM2 ou sem utilizar ONIOM, com uma redução de tempo computacional muito signiicativa.
Com relação aos tipos de funcionais de troca-correlação testados foi possível observar vantagens de alguns funcionais em relação a outros, mas principalmente uma enorme vantagem do método DFT comparando aos métodos HF e MP2 (no caso da AP).
Conclui-se que a conformação ONIOM2, os conjuntos de funções de base de Pople, 6-311++G(2df,p) e 6-31G(d), e o método O3LYP resultam em desvios médios absolutos menores para AP e AE, 4,75 kJ/mol e 0,43 eV, respectivamente, e vantagem em tempo computa-cional sendo mais adequados para descrever os sistemas em questão. AGRADECIMENTOS
G. Heerdt agradece à CAPES pela bolsa de estudo. Ao CNPq e FAPESP pelo apoio inanceiro e ao Instituto de Química da UNI-CAMP pela infraestrutura.
REFERÊNCIAS
1. Raghavachari, K.; Trucks, G. W.; Pople, J. A.; Head-Gordon, M.; Chem. Phys. Lett. 1989, 157, 479.
2. Pople, J. A.; Head-Gordon, M.; Raghavachari, K.; J. Chem. Phys.1987,
87, 5968.
3. Dunning, T. H. Jr.; Peterson, K. A.; Woon, D. E.; P. v. R Schleyer, eds.;
Encyclopedia of computational chemistry, Wiley: New York, 1998. 4. Lima, J. C. B.; Morgon, N. H.; Quim. Nova2010, 33, 195.
5. Morgon, N. H.; Souza, A. R.; Sambrano, J. R.; J. Mol. Struct.2006, 759, 189.
6. Morgon, N. H.; Int. J. Quantum Chem.2006, 106, 2658.
7. Morgon, N. H.; Riveros, J. M.; Int. J. Mass Spectrom.2001, 210, 173. 8. Feller, D.; Peterson, K. A.; J. Chem. Phys.1998, 108, 154.
9. Feller, D.; Dixon, D. A.; J. Chem. Phys.2001, 115, 3484.
10. Ruden, T. A.; Helgaker, T.; Jorgensen, P.; Olsen, J.; Chem. Phys. Lett.
2003, 371, 62.
11. Curtiss, L. A.; Raghavachari, K.; Fox, D. J.; Head-Gordon, M.; Pople, J. A.; J. Chem. Phys.1989, 90, 5622.
12. Curtiss, L. A.; Raghavachari, K.; Redfern, P. C.; Rassolov, V.; Pople, J. A.; J. Chem. Phys.1998, 109, 7764.
13. Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.; Rassolov, V.; Pople, J. A.; J. Chem. Phys.1999, 110, 4703.
14. Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.; Pople, J. A.; Chem. Phys. Lett.1999, 313, 600.
15. Curtiss, L. A.; Redfern, P. C.; Rassolov, V.; Kedziora, G.; Pople, J. A.;
J. Chem. Phys.2001, 114, 9287.
16. Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.; J Chem Phys. 2007,
126, 084108.
17. Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.; J Chem Phys.2007,
127, 124105.
18. Montgomery, J. A. Jr.; Frisch, M. J.; Ochterski, J. W.; Petersson, G. A.;
J. Chem Phys. 1999, 110, 2822.
19. Montgomery, J. A. Jr.; Frisch, M. J.; Ochterski, J. W.; Petersson, G. A.;
J. Chem Phys.2000, 112, 6532.
20. Ochterski, J. W.; Petersson, G. A.; Montgomery Jr., J. A.; J. Chem. Phys.
1996, 104, 2598.
21. Namazian, M.; Coote, M. L.; J. Chem. Thermodyn.2008, 40, 1116. 22. Gronert, S.; Simpson, D. C.; Conner, K. M.; J. Am. Soc. Mass Spectrom.
2009, 20, 2116.
23. Range, K.; Riccardi, D.; Cui, Q.; Elstner, M.; Work, D. M.; J.Phys. Chem.2005, 7, 3070.
24. Richard, R. M.; Ball, D.W.; J. Molec. Model, 2008, 14, 21.
25. Jalbout, A.F.; Darwish, A. M.; Alkahby, H. Y.; J. Mol. Struct.2002, 585, 205.
26. Wilcox, C. F.; Attygalle, A. B.; J. Mol. Struct.1998, 434, 207. 27. Zheng-Xin, T.; Xiao-Hong, L.; Xian-Zhou, Z.; J. Mol. Struct.2009, 907,
126.
28. Jursic, B. S.; J. Mol. Struct.1998,422, 253.
29. Zhao, J.; Cheng, X.; Yang, X.; J. Mol. Struct.2006,766, 87. 30. Jalbout, A. F.; Fernandez, S.; Chen, H.; J. Mol Struct.2002, 584, 143. 31. Miller, T. M.; Arnold, S. T.; Viggiano, A. A.; Int. J. Mass Spectrom.
2003,227, 413.
32. Namazian, M.; Siahrostami, S.; Coote, M. L.; J.Fluorine Chem.2008,
129, 222.
33. Wang, L.; Int. J. Mass Spectrom.2007, 264, 84.
34. Curtiss, L. A.; McGrath, M. P.; Blaudeau, J-P.; Davis, N. E.; Binning, R. C. Jr.; Radom, L.; J. Chem Phys. 1995, 103, 6104.
35. Xie, J.; Zare, R. N.; Chem. Phys. Lett.1989, 159, 399. 36. Warshel, A.; Karplus, M.; J. Am. Chem. Soc.1972, 94, 5612. 37. Warshel, A.; Levitt, M.; J. Mol. Biol.1976, 103, 227.
38. Field, M. J.; Bash, P. A.; Karplus, M.; J. Comput. Chem.1990, 11, 1970. 39. Maseras, F.; Morokuma, K.; J. Comput. Chem.1995, 16, 1170. 40. Svensson, M.; Humbel, S.; Froese, R. D. J.; Matsubara, T.; Sieber, S.;
Morokuma, K.; J. Phys. Chem. 1996, 100, 19357.
41. Dapprich, S.; Komaromi, I.; Byun, K. S.; Morokuma, K.; Frisch, M. J.;
J. Mol. Struct.1999, 1, 461.
42. Thomas, L. H.; Proc. Comb. Phil. Soc.1927, 23, 542; Fermi, E.; Rend. Accad. Lincei1927, 6, 602.
43. Hohenberg, P.; Kohn,W.; Phys. Rev. B1964, 136, 864. 44. Kohn, W.; Sham, L. J.; Phys. Rev.1965, 140, 1133.
45. Dykstra, C. E.; Frenking, G.; Kim, K. S.; Scuseria, G. E.; Theory and Applications of Computational Chemistry: The First Forty Years, Else-vier: Amsterdan, 2005.
46. Zhang, I. Y.; Luo, Y.; Xu, X.; J Chem Phys.2010, 133, 104105. 47. Bensasson, R. V.; Zoete, V.; Dinkova-Kostova, A. T.; Talalay, P.; Chem.
Res. Toxicol.2008, 21, 805.
48. Librando, V.; Alparone, A.; Tomaselli, G.; J. Molec. Model.2008, 12, 489.
49. Higashi, N.; Tanimoto, K.; Nishioka, M.; Ishikawa, K.; Taya, M.; J. Biochem.2008, 144, 77.
50. Morgan, J. E.; Gennis, R. B.; Maeda, A.; Photochem. Photobiol., B
2008, 84, 1038.
51. Bhattacharya, S. K.; Kshirsagar, A.; Eur. Phys. J. D.2008, 48, 355. 52. Reinard, M. S.; Johnston, M. V.; J. Am. Soc. Mass Spectrom.2008, 19,
389.
K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03, Revision E.02; Gaussian, Inc., Wallingford CT, 2004.