departamento de física teórica e experimental
programa de pós-graduação em física
Superparamagnetismo em Jacobsitas
sintéticas
Mateus Bruno Barbosa
Superparamagnetismo em Jacobsitas
sintéticas
Disssertação apresentada ao Programa de Pós-Graduação em Física, do Departamento de Física Teórica e Experimental da Universidade Federal do Rio Grande do Norte, como requisito parcial para a obtenção do título de Mestre em Física.
Orientador: Prof. Dr. José Humberto de Araújo
A meus pais, Joaquim Mateus Barbosa e Mirian da Silva Barbosa.
A Deus.
À minha família, meus pais, meu irmão e minhas irmãs.
Ao meu orientador, Prof. Dr. José Humberto de Araujo, pela orientação e pelo apoio logístico e intelectual durante todo o período do mestrado.
A Armando Araújo, Thatyara Freire e Rodolfo Bezerra, pela ajuda na produção de amostras, técnicas laboratoriais e discussões pertinentes à pesquisa.
Aos colegas das salas César Lattes, Mário Schenberg e Jaime Tionmo, e também aos da sala dos experimentais.
Ao Prof. Dr. Artur da Silva Carriço, pelas recentes e inspiradoras discussões.
Ao Prof. Dr. Joâo Maria Soares pelas medidas de difração de raios-x e espectroscopia Mössbauer relizdas no LAMOP-UERN.
À Capes, pelo apoio financeiro.
rável tudo quanto acontece, porém de modo que nem Deus é o autor do pecado, nem violentada é a vontade da criatura, nem é tirada a liberdade ou a contingência das causas secundárias, antes estabelicidas."
( Capítulo 3 - CONFISSÃO DE FÉ DE
WESTMINSTER)
Neste estudo experimental, amostras sintéticas de Jacobsitas (M nF e2O4)foram
sin-tetizadas pelo método Pechini e calcinadas em atmosfera ambiente e em vácuo de 400 até 700oC. Análises de difração de raio-x (DRX) e microscopia eletrônica de varredura (MEV)
revelaram que a amostra calcinada em400oC é composta por uma fase simples tipo espinélio,
com tamanho médio do cristalito de8,8nmpara amostra calcinada em atmosfera ambiente,
e 20,1 nm para amostra calcinada em vácuo, indicando que o tamanho médio do cristalito pode ser manipulado pelo controle da atmosfera. A curva de magnetização para amostra calcinada a 400oC em atmosfera ambiente revela características de comportamento
superpa-ramagnético, com magnetização de 29.3emu/g num campo máximo de 1.2T. Já a amostra
calcinada em 400oC sob vácuo apresentou magnetização ∼= 67emu/g no campo máximo de
1.5T. A amostra tratada em 500oC, em atmosfera ambiente, acusa além da fase espinélio,
fases secundárias de hematita(F e2O3)e bixbyita(F eM nO3). A curva de magnetização
mos-tra uma queda abrupta na magnetização comparada com as amosmos-tras anteriores. A análise mostra que, para mais altas temperaturas (600oC and 700oC), observou-se apenas a
conti-nuação das fases hematita e bixbyita. A curva de magnetização dessas amostras são linhas retas cortando a origem, consistente com o comportamento antiferromagnético dessas fases. A espectroscopia Mössbauer revelou que para a amostra calcinada em 400oC em atmosfera
ambiente há dois sextetos e um dubleto. Os dois sextetos são atribuídos aos campos hiperfi-nos referentes ao desdobramento magnético no núcleo dos íons F e3+, nos sítios tetraédricos
e octaédricos. O dubleto é atribuído ao comportamento superparamagnético das partículas com diâmetro menor que dc. Já a amostra calcinada em400oC sob vácuo apresenta apenas
dois sextetos.
PALAVRAS-CHAVE: Superparamagnetismo, Jacobsita, Espinélios, M nF e2O4.
In this experimental study sintetic samples of Jacobsites (M nF e2O4) were
synthe-sized by the Pechini method and calcined within ambient atmosphere and afterwards in the vacuum from 400 to 700oC, the range of calcination temperatures. The X-Ray Diffraction
(XRD) and the Scanning Electronic Microscopy (SEM) analysis have shown that the sam-ples treated at 400oC temperature are composed by a simple type of spinel phase, with a
crystallite size of 8.8nm for the sample calcined in ambient atmosphere and 20,1nm for the
sample treated in the vacuum, showing that the cristallite average size can be manipulated by the atmosphere control. The hysteresis loops for the sample calcined at400oC in ambient
atmosphere reveal features of superparamagnetic behavior with magnetization29.3emu/gat
the maximum field of 1.2T. The sample calcined in 400oC under vacuum show
magnetiza-tion∼= 67emu/g at the maximum field of1.5T. The sample treated at500oC, under ambient
atmosphere, has shown besides the spinel phase, secondary phases of hematite (F e2O3) and
bixbyite (F eM nO3). The hysteresis loops demonstrate a sharp drop of the magnetization
compared to the previous sample. The analysis has revealed that for the samples treated in higher temperatures (600oC and700oC) it’s observed the absence of the spinel phase and the
maintenance of the bixbyite and hematite. The hysteresis loops for those samples in accor-dance to the external magnetic field are straight lines crossing the origin, consistent with the antiferromagnetic behavior of the phases.The Mössbauer espectroscopy show to the sample calcined at400oC within ambiente atmosphere two sextet and one doublet. The two sextets
are assigned to the hyperfine fields related to the magnetic deployment in the nuclei ofF e3+
ions, at the tetraedric and octaedric sites. The doublet is assigned to superparamagnetic behavior of the particles with smaller diameter thandc . Now the sample calcined at 400oC
under vacuum only show two sextet.
KEYWORDS: Superparamagnetism, Jacobsite, Spinels, M nF e2O4.
1.1 Movimento de rotação e translação do elétron . . . 3
1.2 Curva de Histerese de um Material Ferromagnético . . . 5
1.3 Dependência da temperatura característica com a susceptibilidade recíproca: (a) para um diamagnético e (b) para um paramagnético [12]. . . 9
1.4 Efeito do campo no momento orbital [11] . . . 10
1.5 Função de Langevin [11] . . . 12
1.6 Espaço de Quantização: (a) Teoria Clássica, (b) e (c) Teoria Quântica [11]. . 15
1.7 Processo de Magnetização . . . 16
1.8 (a) Magnetização espontânea pelo campo molecular; (b) Efeito da temperatura no valor da magnetização espontânea. Curva 1 é a função de Langevin [11] . 18
1.9 Arranjo Antiferromagnético composto de duas sub-redes ferromagneticamente ordenadas [24]. . . 20
1.10 Arranjo Ferrimagnético [9]. . . 22
1.11 Dependência da temperatura da susceptiblidade magnética recíproca para ma-teriais ferro, antiferro e ferrimagnéticos. . . 25
1.12 Magnetização de uma partícula esférica fazendo um ângulo θ com o eixo de
fácil magnetização . . . 27
1.13 Relação do tamanho da nanopartícula e sua histerese magnética, (adaptado de [11]). . . 28
[12]. . . 33
2.2 Definição do parâmetro de deformação u. Meio plano de (110) é mostrado. Atómos de oxigênio (círculos grandes) estão dispostos na direçãoh111idevido a presença dos cátions tetraédricos. No caso ideal, u= 0.375 [12]. . . 34
2.3 Geometria espacial dos orbitaisd (a) dxy, (b) dx2−y2, (c) dz2 [12]. . . 35
2.4 Ordenamento de longo alcance em espninélios inversos, resultado de uma ocu-pação alternada de sítios octaédricos por dois tipos de cátions. Apenas cátions octaédricos são apresentados.[12][37] . . . 37
2.5 Ordamento de longo alcance em espinélios com razão 1:3 de cátions em sítios octaédricos.[12][37] . . . 38
3.1 Esquema de um Magnetômetro de Amostra Vibrante,[23] . . . 44
3.2 Absorção ressonante por um núcleo atômico. (Adaptado de [19]) . . . 45
3.3 Gráfico da emissão γ de um núcleo.(Adaptado de [19]) . . . 46
3.4 Recuo de um núcleo após emissão γ.(Adaptado de [19]) . . . 46
3.5 Esquema simplificado da Espectroscopia Mössbauer (Adaptado de [19]). . . . 47
3.6 Representação esquemática do MEV [41] . . . 49
3.7 Esquema da varredura do feixe de elétrons [41] . . . 50
4.1 Difratograma de Raio-X da amostra calcinada em400oC sob atmosfera ambiente 53 4.2 Difratograma de Raio-X da amostra calcinada em500oC sob atmosfera ambiente 54 4.3 Difratograma de Raio-X da amostra calcinada em600oC sob atmosfera ambiente 55 4.4 Difratograma de Raio-X da amostra calcinada em700oC sob atmosfera ambiente 55 4.5 Difratograma de Raio-X da amostra calcinada em400oC sob vácuo . . . . . 56
4.6 Difratogramas de Raio-X das amostras calcinadas em 500oC sob vácuo . . . 57
4.7 Imagens obtida pelo MEV das amostras calcinadas em400e500oC em atmos-fera ambiente . . . 58
4.9 Curva Magnetizaçãovs Campo aplicado das amostras calcinadas a 400 e 500 em atmosfera ambiente . . . 59
4.10 Curva Magnetizaçãovs Campo aplicado das amostras calcinadas a600e700oC
em atmosfera ambiente . . . 60
4.11 Curva MvsH das amostras calcinadas a 400 e500oC em vácuo . . . . 61
4.12 Fit da Curva MvsH da amostra calcinada a400oC em vácuo . . . . 62
4.13 Espectro Mössbauer da amostra calcinada em 400oC sob atmosfera ambiente. 63
4.14 Espectro Mössbauer da amostra calcinada a 400oC em vácuo. . . . 64
4.1 Informações das amostras calcinadas sob atmosfera ambiente . . . 54
4.2 Informações das amostras calcinadas sob vácuo . . . 56
4.3 Informações dos parâmetros do fit da função de Langevin adaptada. . . 62
4.4 Informações dos parâmetros da espectroscopia Mössbauer da amostra calci-nada a 400oC em atmosfera ambiente. . . . 63
4.5 Informações dos parâmetros da espectroscopia Mössbauer da amostra calci-nada a 400oC em vácuo. . . . 64
Agradecimento ii
Resumo iv
Abstract v
Lista de Figuras viii
Lista de Tabelas ix
1 Conceitos de Magnetismo 2
1.1 Fenômenos Magnéticos . . . 6
1.1.1 Diamagnetismo . . . 7
1.1.2 Paramagnetismo . . . 8
1.1.3 Ferromagnetismo e Antiferromagnetismo . . . 16
1.1.4 Ferrimagnetismo . . . 22
1.1.5 Superparamagnetismo . . . 26
2 Ferritas 31 2.1 Óxidos Magnéticos . . . 31
2.2 Estrutura Espinélio . . . 32
3.2 Tratamento Térmico . . . 41
3.3 Difratometria de Raios-X . . . 41
3.3.1 Refinamento Rietveld . . . 42
3.4 Magnetometria de Amostra Vibrante . . . 43
3.5 Espectroscopia Mössbauer . . . 45
3.6 Microscopia Eletrônica de Varredura (MEV) . . . 49
4 Resultados 52 4.1 Difratometria de Raio-X . . . 53
4.2 Microscopia Eletrônica de Varredura (MEV) . . . 58
4.3 Magnetometria de Amostra Vibrante (MAV) . . . 59
4.4 Espectroscopia Mössbauer . . . 63
5 Conclusão 65
Referências bibliográficas 67
"Porque em esperança fomos salvos.
Ora a esperança que se vê não é
espe-rança; porque o que alguém vê como o
esperará? Mas, se esperamos o que não
vemos, com paciência o esperamos."
Romanos 8: 24 - 25
Este trabalho tem como objetivo sintetizar, caracterizar e estudar as propriedades magnéticas da ferrita de manganês(M nF e2O4), com dimensões nanométricas. Esse composto
surge naturalmente na crosta terrestre e recebe o nome de Jacobsita. Tem sido utilizado em vários setores da pesquisa, desde a indústria até medicina, [1], [2], [3], [4], [5], [6]. Por meio do método Pechini, preparam-se dois lotes de amostras. No primeiro lote fez-se tratamento térmico em atmosfera ambiente, calcinando as amostras em 400, 500, 600 e 700oC. No
segundo lote, o tratamento térmico foi feito a vácuo, calcinando as amostras em400e500oC.
Utilizou-se a técnica de difração de raio-x, para determinar as fases presentes nas amostras calcinadas nas diferentes temperaturas e atmosferas, e a técnica de espectroscopia Mössbauer para identificar a localização dos sítios de F e3+, bem como o estado em que
ele se encontra. Também fez-se uso da técnica de microscopia eletrônica de varredura para vizualizar a formação cristalina da substância e, por fim, pela técnica de magnetometria de amostra vibrante, obteve-se a curva de magnetização vs campo aplicado, com o intuito de analisar as propriedades magnéticas: remanência, campo coercitivo e magnetização de saturação.
Cap´ıtulo
Conceitos de Magnetismo
"O que você sabe não tem valor. O
valor está no que você faz com o que
sabe."
Bruce Lee
Quando dividimos um ímã ao meio, observamos a formação de outro ímã; não im-porta quantas divisões sejam feitas, sempre haverá um novo ímã, ou seja, não conseguiremos separar os pólos norte e sul, ou seja, obter o que é chamado de monopolo magnético. A unidade fundamental do magnetismo é chamada de dipolo magnético. Um átomo pode agir como um dipolo magnético. Para produzir um campo magnético, fazemos uma corrente elé-trica circular em uma espira condutora. Por analogia, um átomo funciona assim: os elétrons orbitam ao redor do núcleo.
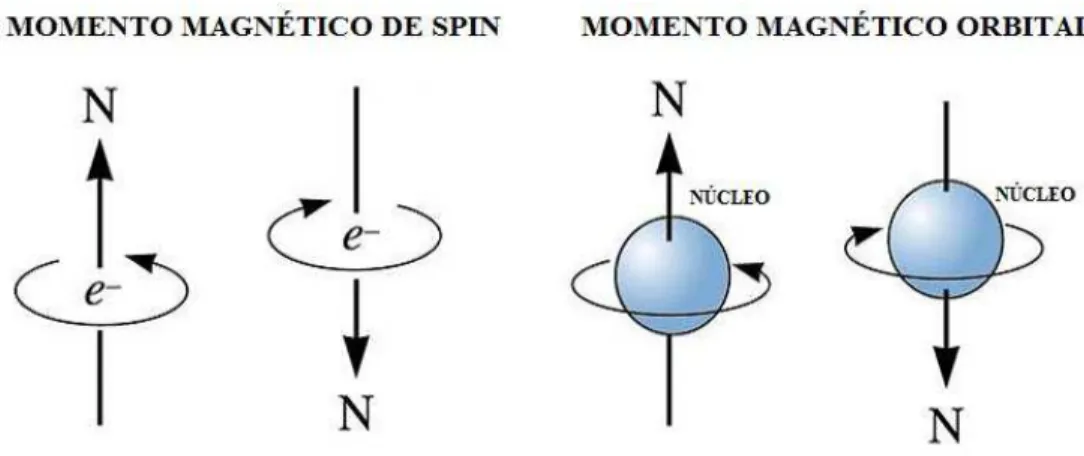
Pensando ainda em um elétron circulando em volta do núcleo, imagina-se que ele gire em torno do seu próprio eixo, como ocorre com os planetas do sistema solar, que possuem um movimento de translação e de rotação. Esses movimentos provocam dois momentos de dipolo magnético: o orbital ou de translação e o intrínseco ou de rotação (o spin), como mostra a figura 1.1. Esses dois movimentos são as fontes do magnetismo nos átomos; portanto, notamos que o comportamento magnético dos átomos provém do movimento dos elétrons. Embora o núcleo possua magnetismo, esse é desprezível se comparado ao dos elétrons [7].
Figura 1.1: Movimento de rotação e translação do elétron
O momento magnético total do átomo é a soma dos momentos orbital e de spin. O momento orbital é normal ao plano da órbita e o de spin é paralelo ao eixo de rotação. O momento associado a esses movimentos é uma grandeza vetorial e pode ser representado da seguinte maneira:
µtotal =µorbital+µspin =−
e
2m(L+ 2S) (1.1)
Devido ao caráter vetorial da soma dos momentos magnéticos, há duas possibilidades de comportamento:
1: Os elétrons estão alinhados de tal maneira que ocorre um cancelamento mútuo e o átomo como um todo não apresenta um momento magnético. Esse caso se aplica aos materiais diamagnéticos.
2: O cancelamento dos momentos magnéticos dos elétrons é apenas parcial. Logo o átomo apresenta um momento magnético líquido diferente de zero. As substâncias que se comportam dessa maneira podem ser principalmente paramagnéticas, ferrimagnéticas, ferromagnéticas e antiferromagnéticas.
Quando se estuda magnetismo, nota-se a existência de três grandezas importantes para a descrição das propriedades magnéticas que encontramos na matéria: o campo mag-nético H~, a indução magnética B~ e a magnetização M~. A magnetização é uma grandeza
vetorial macroscópica [7]. Ao pegarmos um elemento de volume ∆V, isto é, um pedaço
pe-queno do ponto de vista macroscópico, porém de tamanho suficiente para conter uma grande quantidade de átomos, e fazermos a soma de todos os momentos magnéticos (P
por unidade de elemento de volume:
~
M = lim
∆V→∞
1 ∆V
n
X
i=1
~µi (1.2)
Pode haver, nos materiais, tanto momentos de dipolo magnético intrínseco como momentos de dipolo magnético induzido. O segundo caso ocorre devido à presença de um campo magnético externo, que provoca alterações nos dipolos magnéticos elementares, tanto permanentes quanto induzidos, gerando assim um campo de indução que mudará o campo inicial. Os campos magnéticos são formados por cargas elétricas em movimento. Quando consideramos o vácuo como o meio onde acontece o fenômeno , observamos que a indução magnética é diretamente proporcional ao campo magnético:
~
B =µ0H~ (1.3)
em que, µ0 = 4πx10−7Hm−1 é a permeabilidade magnética no vácuo. No vácuo, essas duas
grandezas estão relacionadas somente à densidade de corrente elétrica da fonte. Quando um material se submete à presença de um meio magnético, tanto o campoHquanto a induçãoB
serão influenciados pela magnetizaçãoM do meio. A magnetização vista microscopicamente resulta da soma dos momentos magnéticos e essa soma tem de ser diferente de zero. Para que haja magnetização, é necessário que a média dos momentos magnéticos aponte para a mesma direção. Os momentos de dipolo magnético que consideramos como correntes microscópicas são fontes da indução magnética B. O vetor campo magnético está associado somente às correntes macroscópicas [8]. Vale ressaltar que M e B têm a mesma dimensão. Podemos relacionar essas três grandezas da seguinte maneira:
~
B =µ0(H~ +M~) (1.4)
Experimentalmente, observa-se uma relação entre a magnetização e o campo, para materiais isotrópicos e lineares [7]:
~
B =χmH~ (1.5)
A grandeza que surge dessa relação χm é adimensional e foi denominada
que será chamado paramagnético; caso a susceptibilidade seja negativa, a indução se enfra-quecerá pela presença do material, que se chamará diamagnético. A susceptibilidade é função da temperatura, com a qual, às vezes, varia drasticamente. Apesar disso, para materiais pa-ramagnéticos e diamagnéticos, temos que|χm|<1; já para materiais ferromagnéticos, temos
χm ≫1.
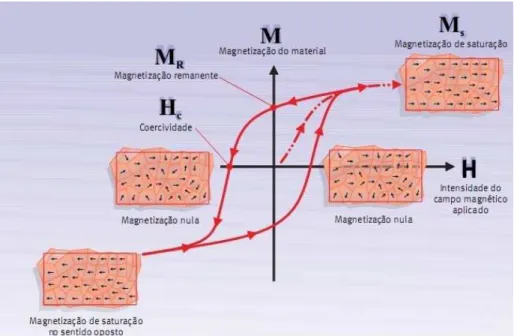
Quando magnetizamos uma substância ferromagnética, que em princípio está des-magnetizada, colocando-a na presença de um campo magnético, os momentos de dipolo magnéticos dessa substância se alinham de acordo com o campo aplicado numa relação li-near, que ocorre até haver a saturação provocada pelo alinhamento magnético total, gerando assim um ímã permanente, sem a presença de corrente (observe a figura 1.2). A primeira curva (pontilhada), chamada de curva virgem, é a resposta a um campo externo a partir de um estado desmagnetizado.
Figura 1.2: Curva de Histerese de um Material Ferromagnético
Para reduzir a remanência a zero temos de aplicar ao material um campo contrário também chamado de desmagnetizante (-H). O campo coercitivo ou desmagnetizante, neces-sário para reduzir a remanência até zero, é chamado coercividade. Ao aumentarmos o campo desmagnetizante, criamos uma magnetização com polaridade contrária. Dessa forma, ao se magnetizar novamente o material com um campo H, temos a repetição do processo origi-nando uma curva fechada que chamamos de Histerese Magnética (a palavra histerese vem do grego e significa atraso). A histerese magnética consiste na capacidade que cada material possui de manter suas propriedades magnéticas [7], [10].
Os materiais que possuem uma alta coercividade são denominados magneticamente duros e dão origem aos ímãs permanentes. O nome advém do fato de que os primeiros ímãs conhecidos foram os aços, que também são mecanicamente duros. Sendo assim, para ímãs permanentes, uma melhoria em suas propriedades significa aumento da remanência e, tam-bém, da coercividade; os materiais moles como o ferro possuem características opostas. Essas duas classes de materiais possuem grande aplicação na indústria. Os transformadores, gera-dores e motores de corrente alternada necessitam da menor coercividade possível, enquanto os de corrente contínua necessitam de ímãs com altos valores de remanência e coercividade.
1.1 Fenômenos Magnéticos
1.1.1
Diamagnetismo
O diamagnetismo é resultado do movimento orbital dos elétrons, que produz um campo magnético. Não se gera campo magnético externo porque, para cada órbita, há dois elétrons circulando em direções opostas. A teoria clássica desse fenômeno foi elaborada pelo físico francês Paul Lagenvin [1872-1946] e publicada em 1905, em um trabalho notável [11]. O diamagnetismo está presente em todos os tipos de materiais, não obstante, esse fenômeno é em geral mascarado por um comportamento paramagnético ou ferromagnético mais intenso, que costuma ocorrer simultaneamente ao diamagnetismo.
Na presença de um campo magnético, podemos ver o diamagnetismo como con-sequência da Lei de Lenz atuando em escala atômica; haverá uma alteração na velocidade orbital dos elétrons no sentido de evitar qualquer mudança no campo magnético produzido pelo átomo. Dessa forma, as correntes eletrônicas em cada átomo são modificadas de tal maneira que tendem a enfraquecer o campo; para isso, opõem-se a ele. As substâncias dia-magnéticas não apresentam um momento magnético externo, ou seja, polos magnéticos como um ímã permanente. Todavia, quando submetidas a um campo externo, tendem a se afastar da região em que esse campo é mais forte, diz-se, então, que possui magnetismo negativo.
O resultado coletivo de todos os átomos do material é uma reação contrária ao campo; logo, a tendência observada macroscopicamente é o afastamento com relação ao campo aplicado. Materiais que possuem esse tipo de comportamento magnético, apresentam a susceptibilidade negativa e crescente com o número de elétrons por átomo. Nada nesse modelo sugere uma forte dependência térmica da susceptibilidade, e isso também está de acordo com os experimentos realizados [11]. Em geral, compostos orgânicos são diamagnéti-cos. Valores típicos da susceptibilidade de materiais diamagnéticos estão entre−1×10−7 e
−2×10−6. Os supercondutores são diamagnéticos perfeitos com χ=−1, [12].
Em uma abordagem quântica, tem-se de forma simplificada o pressuposto de que as camadas eletrônicas estão preenchidas. Sendo assim, não há momento angular orbital e de spin:
Consequentemente, têm-se:
µB(L+gS)·H= 0 (1.7)
Assumindo que o campo H é paralelo ao eixo z,
H= (0,0, H) (1.8)
devido a
H×ri =H·
−yi
xi 0 (1.9) obtemos:
(H×ri2)2 =H2(x2i +yi2) (1.10)
Consequentemente, uma mudança de energia do estado fundamental ocorre ao termo diamagnético que equivale a
∆E0 =
e2H2
8m
X
i
h0|x2i +y2i|0i (1.11)
em que |0i é a função de onda no estado fundamental
1.1.2
Paramagnetismo
As substâncias paramagnéticas possuem susceptibilidade magnética de intensidade compa-rável à dos materiais diamagnéticos, todavia é positiva e depende do inverso da temperatura absoluta. Conforme a Lei de Curie, temos:
χm =
C
T (1.12)
constatações de Pierre Curie foram todas experimentais.
A teoria clássica do paramagnetismo foi postulada anos mais tarde por Langevin e qualitativamente é simples. Supõe-se que, em um paramagneto, os átomos possuem um momento magnético ~µ devido ao não cancelamento dos momentos orbitais e de spin, uma
vez que as subcamadas eletrônicas desses átomos estão parcialmente preenchidas. Na ausên-cia de um campo magnético, a magnetização do material é zero. Ao colocar um material paramagnético perante um campo magnético externo aplicado H~, os momentos de dipolo
magnético orientaram-se paralelamente ao campo externo. Se o campoH~ for não uniforme,
o material paramagnético é atraído da região onde o campo magnético é menos intenso para a região onde o campo magnético é mais intenso.
Figura 1.3: Dependência da temperatura característica com a susceptibilidade recíproca: (a) para um diamagnético e (b) para um paramagnético [12].
da susceptibilidade.
Figura 1.4: Efeito do campo no momento orbital [11]
Considere uma unidade de volume de um determinado material com n átomos, em que cada um possui momento magnéticoµ. Esse momento é representado por um vetor que
atravessa o centro de uma esfera de raio unitário. Calculemos o número dn de momentos
inclinados em um ângulo entreθ eθ+dθ em relação ao campoH. Na ausência de um campo H, o número de vetores µ atravessando uma unidade de área na superfície da esfera será o
mesmo em qualquer ponto dessa superfície, e dn é simplesmente proporcional a dA, que é
2πsinθdθ para uma esfera de raio unitário [11], como podemos observar na figura 1.4. Mas,
quando o campo é aplicado, os momentos se alinham na sua direção e cada momento passa a ter uma energia potencial
Ep =−µHcosθ (1.13)
Num estado de equilíbrio térmico com temperatura T, a probabilidade de um átomo
ter a energiaEp será eEp/kT, em que k é a constante de Boltzmann. O número de momentos
entreθ e θ+dθ é proporcional a dA multiplicado pelo fator de Boltzmann, isto é,
Em que K é o fator de proporcionalidade e é encontrado fazendo
Z n
0
dn=n
Por comodidade, vamos chamar α =µH/kT
2πK
Z π
0
eαcosθsinθdθ=n (1.15)
O momento magnético total na direção do campo aplicado por unidade de volume é conhecido como magnetização M, que é dada por:
M =
Z n
0
µcosθdn
em queµcosθé a contribuição de cada momento magnético. Substituindo a equação
1.14 na expressão acima, temos:
M = 2πKµ
Z π
0
eαcosθsinθcosθdθ (1.16)
= nµ
Rπ
0 e
αcosθsinθcosθdθ
Rπ
0 eαcosθsinθdθ
Fazendo x= cosθ e dx=−sinθdθ, temos:
M = nµ
R−1 1 xe
αx
R−1
1 eαx
=nµ e
α+e−α
eα−e−α −
1
α
!
M =nµ cothα− 1 α
!
(1.17)
nµ é o momento máximo possível a um material. Quando ocorre o perfeito
alinha-mento de todos os moalinha-mentos magnéticos paralelamente com o campo aplicado, tem-se a saturação do material. Denotamos nµ=M0. Assim
M M0
= cothα− 1
que é mais conhecida como função de Langevin e pode ser expressa por meio de uma série
L(α) = α 3 −
α3
45+ 2α5
945 −... (1.19)
que é válida somente para α ≤1. Se α for grande, L(α) tende a 1; se α <0,5, é uma linha
reta com inclinação 1/3, como podemos observar na figura 1.5.
Figura 1.5: Função de Langevin [11]
Da Função de Langevin, temos dois resultados:
1. A saturação ocorrerá se α(= µH/KBT) for suficientemente grande. Isso faz sentido
2. Para α pequeno, M varia linearmente com H; é o que observamos sob condições
nor-mais.
A teoria de Langevin leva à Lei de Curie. Para α pequeno, L(α) =α/3, na equação
1.19, temos:
M = nµα
3 =
nµ2H
3kT (1.20)
Portanto,
χν =
M
H =
nµ2
3kT,
χm =
χν
ρ =
nµ2
3ρkT
em que ρ é a densidade. Todavia n =N ρ/A, em que N é o número de Avogrado; e A, a massa atômica. Assim
χν =
N µ2
3AkT = C T
Am2
m3Am−1[adimensional](SI)
e
χm =
N µ2
3ρAkT = C ρT
Am2
KgAm−1 =
m3
Kg(SI) (1.21)
que é a Lei de Curie, com a constante de Curie dada por
C = N µ
2
3Ak (1.22)
A teoria de Langevin para paramagnetismo, que leva à lei Curie, baseia-se na su-posição de que os portadores individuais de momento magnético (átomos ou moléculas) não interagem uns com os outros, mas são ativados apenas pelo campo aplicado e pela agitação térmica.
Na teoria quântica do paramagnetismo, as principais conclusões da teoria clássica são modificadas, mas não drasticamente. Em substâncias paragméticas, a energia de cada momento magnético µ em um campo H é −µHcosθ, em que θ pode assumir apenas
deter-minados valoresθ1, θ2, ... e valores intermediários não são permitidos, o que não acontece na
1.6. As regras do espaço de quantização em geral são expressas em termos do momento angular em vez do momento magnético; devemos, pois, considerar a relação entre momento angular orbital e momento angular de spin.
µorbital =
eh
4πm (1.23)
Sabendo que p=h/2π, temos, então:
µorbital =
e
2mporbital (1.24)
O momento angular decorrente do spin é sh/2π, em que s= 1/2. Temos, assim:
µspin =
eh
4πm = e
mpspin (1.25)
Combinando as duas últimas equações, obteremos uma forma geral para o momento angular, que é
µ=g(e/2m)(p) (1.26)
em que o fator g é 1 para momento orbital e 2 para momento de spin.
O momento angular total de um átomo é a combinação das contribuições do momento orbital e do momento de spin, que é representado por J. Com isso, o momento magnético
efetivo de um átomoµef será:
µef =g
eh
4πm
!
p
J(J + 1) Am2(SI) (1.27)
Sabendo que o magneton de Bohr é µB=eh/4πm, temos
µef =g
p
J(J+ 1)µB (1.28)
de magnetons de Bohr:
nef =g
p
J(J+ 1) (1.29)
Devido à quantização espacial, o momento efetivo pode ter valores discretosθ1, θ2, ...
para o campo. Em vez de especificar esses ângulos, especificaremos os valores de µH, que é
o componente deµef na direção de H:
µH =gMJµB (1.30)
Em que MJ é um número quântico associado aJ [11]. Há 2J+ 1 valores deMJ e o
maior valor deµH égJµB.
Figura 1.6: Espaço de Quantização: (a) Teoria Clássica, (b) e (c) Teoria Quântica [11].
Os valores de J podem ser um número inteiro ou semi-inteiro e vão de J = 1/2 até
J =∞. Isso significa:
• J = 1/2corresponde à contribuição somente do spin, ou seja,L= 0→J =S = 1/2, de
modo que o fator de Landé g = 2. Assim, os valores de MJ diminuem de +J para −J
em valores unitários. Nesse caso, temos+1/2e−1/2, em que os momentos magnéticos µH são µB e −µB, paralelo e antiparalelo ao campo aplicado, como mostrado na figura
• J = ∞ corresponde a um número infinito de orientações. Isso é equivalente à distri-buição clássica, como mostra a figura 1.6a.
1.1.3
Ferromagnetismo e Antiferromagnetismo
Pierre Weiss formulou a hipótese de que haveria um campo molecular que agiria nas subs-tâncias ferromagnéticas, tanto abaixo quanto acima datemperatura de Curie, e, complemen-tando, um ferromagneto desmagnetizado estaria divido em regiões microscópicas chamadas dedomínios. Cada domínio está magnetizado espontaneamente até o valor de saturaçãoMS
direcionado aleatoriamente, culminando numa magnetização total nula. Sendo assim, o pro-cesso de magnetização consiste em tornar esses multidomínios um único monodomínio com magnetização resultante direcionada no sentido do campo aplicado, conforme percebemos na figura 1.7.
Figura 1.7: Processo de Magnetização
No ferromagnetismo, os átomos do material possuem uma forte interação que tende a manter seus momentos magnéticos alinhados em uma única e exclusiva direção, mesmo na ausência de campo externo aplicado. Esse efeito é geralmente causado pela interação de troca. Para um ferromagneto submetido a um campo externo H, o hamiltoniano que soluciona esse problema é
H=−X
ij
JijSi·Sj+gJµB
X
j
Para vizinhos próximos, Jij >0.
Para a hipótese de Weiss de campo molecular, usa-se um hamiltoniano dessa forma:
H =gJµB
X
i
Si·(H+Hm) (1.32)
Assumindo que o único campo a agir em um material é o campo molecular Hm
(proporcional à magnetização), temos
Hm =γM (1.33)
em que γ é a constante de campo molecular. Para ferromagnetos, γ >> 1, devido à
influência da interação na interação de troca. Definimos a magnetização relativa como1:
M Ms
=BJ(y) (1.34)
com BJ(y)sendo a função de Brillouin e
y= gJµBJ(H+γM)
kT . (1.35)
A magnetização espontânea em materiais ferromagnéticos varia conforme a tempe-ratura, cujo limite é atemperatura de Curie (Tc). Acima de Tc, os materiais ferromagnéticos
tornam-se paramagnéticos.
Na figura 1.8 (a), percebe-se que a magnetização produzida pelo campo molecular é a intersecção entre as curvas. Na origem, temos um ponto de instabilidade, e a magnetização pode ir tomando os valores0, A, B, E, ...até chegar no ponto estávelP. Esse ponto é estável,
pois a partir dele, mesmo na ausência de campo, a magnetização não mudará.
Agora, veremos o comportamento desses materiais mediante a variação da tempera-tura, isto é, como a magnetização de saturaçãoMS varia com a temperatura, e observaremos
em qual temperatura esses materiais tornam-se paramagnéticos. Do paramagnetismo, temos
1
Figura 1.8: (a) Magnetização espontânea pelo campo molecular; (b) Efeito da temperatura no valor da magnetização espontânea. Curva 1 é a função de Langevin [11]
α=µH/kT e consideramos que a magnetização relativa é dada pela função de Langevin
M M0
=L(α) = coth(α)− 1
α. (1.36)
Caso não haja campo aplicado, temos:
α= µHm
kT =
µγM
kT =
µγM kT
M M0
, (1.37)
M M0
= kT
µγM0
!
α (1.38)
Nota-se que a magnetização relativa é uma função linear de α e sua inclinação é
proporcional à temperatura absoluta. Observa-se, no gráfico da figura 1.8 (b), uma plotagem da equação 1.36 para T2, que, com o aumento da temperatura, rotaciona no sentido
anti-horário até a linha 2 e, com mais um aumento de temperatura, rotaciona para a linha 3. Não há magnetização espontânea quando a temperatura é T3. A curva 3 tangencia a função
de Langevin na origem; portanto, T3 = Tc, que é a temperatura de Curie. Em qualquer
temperatura mais alta que T3, não haverá magnetização espontânea, pois o material será
T3, que é a mesma da função de Langevin, 1/3. Temos, assim:
kTc
µγM0
= 1 3
Tc =
µγM0
3k (1.39)
A inclinação da reta, que representa o campo molecular, é em qualquer temperatura:
kT µγM0
= T
3T c (1.40)
Entretanto, a inclinação da curva determina a intersecçãoP com a curva de Langevin
e, portanto, o valor deM/M0. Isso significa que todo material ferromagnético que apresenta
diferentes valores deM0 eTc tem os mesmos valores deM s/M0para qualquer valor particular
deT /Tc. Isso é geralmente chamado delei de estados correspondentes.
Ao aplicar um pequeno campoHemT ≥Tc, acontecerá uma pequena magnetização,
de modo que a aproximação para a função de Brillouiny <<1 pode ser empregada. Assim,
M Ms ≈
gJµB(J+ 1)
3k
H+γM T
!
(1.41)
Isso nos leva a
M Ms ≈
Tc
λMs
H+γM T
!
(1.42)
Rearranjando os termos, temos:
M Ms
1− Tc
T
!
≈ µMTcH
s
(1.43)
Portanto,
χ= lim
H→0
µ0M
H ∝
1
T −Tc
(1.44)
Suscintamente, pode-se explicitar a teoria quântica do ferromagnetismo desta ma-neira: a magnetização ocorre devido a spins paralelos e não a momentos de dipolo magnético orbitais. Portanto, os elétrons das subcamadas mais externas de átomos de ferromagnetos se orientam de maneira que seus spins sejam paralelos, reduzindo assim a energia do átomo. Isso implica dizer que dois elétrons 3d se encontram mais afastados, em média de seus spins se forem paralelos em vez de antiparalelos (devido ao princípio de exclusão de Pauli), e, uma vez mais afastados a energia de repulsão coulombiana mútua é menor. Logo, o momento dipolar magnético de spin permanente ocorre devido à interação entre as coordenadas de spin e de espaço impostas pela exigência quântica referente à troca de coordenadas de partí-culas indistinguíveis. Por isso, o acoplamento de spin é às vezes considerado consequente de uma forte interação de troca que tem lugar no átomo. Há interação de troca entre átomos adjacentes em uma rede cristalina de átomos de ferro; essa interação pode levar o sistema a um estado de menor energia; o mesmo pode acontecer, também, quando os spins de pares adjacentes de átomos forem paralelos.
De forma análoga, pode-se explicar o comportamento antiferromagnético: momentos magnéticos vizinhos estão alinhados antiparalelamente. Com isso, há duas possibilidades de descrição a partir do modelo de Weiss.
1. Uma interação de troca negativa entre vizinhos próximos é considerada;
2. A rede é dividida em duas sub-redes (ver figura 1.9):
- cada sub-rede exibe um arranjo ferromagnético e
- há uma orientação antiparalela da magnetização entre as sub-redes.
Mediante o modelo de Weiss, pressupondo-se que não haja campo magnético externo e que o campo molecular da sub-rede ↑ Hm(+) é proporcional à magnetização da sub-rede ↓
M− e vice-versa, temos:
Hm(+) =−|λ|M−
Hm(−) =−|λ|M+
A magnetização das sub-redes podem ser escritas, portanto, como
M± =MsHJ −
gµBJ|λ|M∓
kT
!
(1.45)
As duas sub-redes têm orientação antiparalela em relação uma a outra, mas a mag-nitude das suas magnetizações são iguais:
|M+|=|M−|=M
e, portanto,
M =MsHJ −
gµBJ|λ|M
kT
!
. (1.46)
O campo molecular segue esse modelo em cada sub-rede e desaparece em tempe-raturas acima da temperatura de Néel (TN), que é a temperatura de transição, definida 2
por:
TN =
gµB(J+ 1)|λ|Ms
3k =
n|λ|µ2
ef
3k (1.47)
A magnetização das sub-redes estarão em direções opostas; com isso, a magnetização líquida M++M− será nula. Será, contuto, não nula para temperaturas abaixo de TN.
Para T > TN, o efeito de um pequeno campo aplicado será semelhante ao de um
2
ferromagneto, e a susceptibilidade será dada por:
χ= lim
H→0
µ0M
H ∝
1
T +TN
, (1.48)
que é a lei de Curie com uma modificação: troca-se o termo −Tc por +TN.
Pode-se ter uma forma geral para expressar a susceptibilidade
χ∝ 1
T −θ (1.49)
em que θ é a temperatura de Curie Weiss. Seθ = 0, o material é paramagnético; se θ >0, o material é ferromagnético e espera-se θ=Tc; e se θ <0, o material é
antiferromag-nético e espera-se θ=−TN [25].
1.1.4
Ferrimagnetismo
Os materiais ferrimagnéticos possuem dois tipos diferentes de íons magnéticos que também se orientam antiparalelamente, mas, como existem dois tipos de íons com momentos magnéticos diferentes, a magnetização resultante não é nula. Há, então, uma magnetização espontânea à temperatura ambiente. A estrutura do material contém componentes do tipo "spin para cima"e "spin para baixo", o que promove um momento magnético resultante não nulo em um dos sentidos, como mostra a Figura 1.10.
Os efeitos magnéticos externos das ferrites são intermediários entre o ferromagne-tismo e o antiferromagneferromagne-tismo. Os ferrimagnéticos apresentam condutividade elétrica muito baixa, o que é vantajoso para determinadas aplicações, como na detecção de frequências ele-tromagnéticas altas, devido à ausência de correntes de Foucault apreciáveis, e sem as perdas de energia resultantes. Há uma temperatura a partir da qual materiais ferrimagnéticos se tornam paramagnéticos; tal temperatura é conhecida com Temperatura de Curie.
O campo molecular é descrito de forma análoga ao caso antiferromagnético3. Em
decorrência de questões de simetria, temos:
v2 =w1 <0 (1.50)
devido ao acoplamento antiferromagnético. Mas v1 6=w2. Vamos definir
v2 =w1 =−v, com v >0 (1.51)
e
v1 =κv
w2 =ǫw
Sendo assim, pode-se estudar um sistema ferromagnético como uma função da razão das constantes de campo molecular, em vez dos seus valores absolutos. Portanto, tem-se
Hm(+) =κvM+−vM− (1.52)
H(−)
m =−vM−+ǫvM+ (1.53)
Dessa forma, a magnetização das sub-redes será
3
M+=ngJµBJ·HJ
gJµBJµ0
kT ·(H+κvM+−vM−)
!
(1.54)
M−=ngJµBJ·HJ
gJµBJµ0
kT ·(H−vM++ǫvM−)
!
(1.55)
Acima da temperatura de transição, recaímos na função de Brillouin
BJ(y) =
J+ 1
3J y (1.56)
que resulta em:
M+ =
ng2
Jµ2BJ(J+ 1)µ0
3kT ·(H+κvM+−vM−)
M+ =
c+
T (H+κvM+−vM−) (1.57)
com c+ sendo
c+ =
ng2
Jµ2BJ(J+ 1)µ0
3k (1.58)
e, para outra sub-rede, temos, de forma análoga
M−=
c−
T (H−vM++ǫvM−) (1.59)
A solução dessas equações é dada por
M+ =
c+T −c+c−ǫv−c+c−v
T2−v(κc
++ǫc−)T +c+c−v2(κǫ−1)·
H (1.60)
M− =
c−T −c+c−σv−c+c−v
T2−v(κc
++ǫc−)T +c+c−v2(κǫ−1)·
Portanto, a magnetização total M =M++M− será:
M = (c++c−)T −c+c−v(2 +κ+ǫ)
T2−v(κc
++ǫc−)T +c+c−v2(κǫ−1)
(1.62)
Agora, calculamos a susceptibilidade inversa
µ0
1
χ = H
M =H·
T2−v(κc
++ǫc−)T +c+c−v2(κǫ−1)
(c++c−)T −c+c−v(2 +κ+ǫ)
(1.63)
Introduzindo os parâmetros adequados, θ, χ0 e σ, chegamos a:
µ0
1
χ = T c++c−
+ 1
χ0
+ σ
T −θ (1.64)
Pode-se observar na figura 1.11 que o comportamento de sistemas ferromagnéticos e antiferromagnéticos para a susceptibilidade inversa é linear; todavia, para sistemas ferri-magnéticos, tem-se um caráter hiperbólico. Tomando limiteT → ∞para a equação anterior
temos
1
χ ∝ T c++c−
+ 1
χ0
(1.65)
bem como a intersecção com o eixo T, que determina a temperatura críticaTc:
1
χ(Tc) = 0 (1.66)
Devido a diferentes magnitudes das magnetizações das sub-redes, frequentemente a magnetização total tem um comportamento consideravelmente complexo4, mas não
entrare-mos em detalhes neste trabalho.
1.1.5
Superparamagnetismo
O termo superparamagnetismo foi introduzido porBean e Livingston em 1959 [13], com vistas a descrever o comportamento magnético de partículas magnéticas de dimensões de escala nanométrica. Nessa escala, existem efeitos de tamanho, de confinamento e de superfície que influenciam as propriedades magnéticas das nanopartículas. Na suposição inicial, considerou-se que os momentos magnéticos no interior de uma partícula considerou-se movimentam coerentemente, apontando na mesma direção mediante a aplicação de um campo magnético externo, ou seja, o momento magnético total pode ser representado por meio de um único vetor clássico de magnitude µ [14]. A redução de tamanho da nanopartícula considerada monodomínio é
suficiente para que o seu momento magnético não fique estável no eixo magnético preferencial durante um tempo típico de medida.
De acordo com o artigo de Bean e Livingston, no superparamagnetismo, há dois fenômenos: alinhamento tipo Langevin de macrospins em um campo externo e bloqueio superparamagnético ou congelamento da magnetização reversa. Na ausência de um campo externo, a característica deLangevin em ter magnetização em temperatura ambiente torna-se importante em partículas por volta de 2nm. Em contrapartida, o bloqueio
superparamag-nético ocorre entre3nm (materiais magnéticos duros) e30nm(materiais magnéticos moles),
[15].
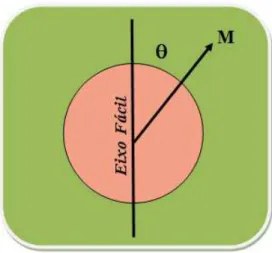
A direção do momento magnético de cada partícula está determinada pela minimi-zação da energia de anisotropia do sistema (amostra) que, no caso de ser uniaxial, pode ser expressa da seguinte maneira:
E =KaV sin2θ (1.67)
em que Ka é a constante de anisotropia, V é o volume da partícula e θ é o ângulo
entre o momento magnético da partícula e o eixo de fácil magnetização, como indica a figura 1.12. Sendo assim, percebemos que o momento magnético da partícula possui dois estados de mínima energia, um paraθ = 0 e outro para θ =π. Os estados de mínima energia estão
4
Figura 1.12: Magnetização de uma partícula esférica fazendo um ânguloθ com o eixo de fácil
magnetização
separados por uma barreira de energia igual aKaV.
Se a temperatura do sistema aumenta, aumenta também a energia térmicakT. Para
uma determinada temperatura, o valor da energia térmica pode vir a ser comparável ou mesmo maior que a barreira de energia KaV. Nesse caso, a magnetização da partícula (em
equilíbrio térmico) não estará mais bloqueada em uma direção, mas apresentará flutuações entre os dois estados de mínima energia de forma muito rápida. Nesse caso, dizemos que o sistema de nanopartículas está em um estado superparamagnético, pois poderá ser descrito por um modelo paramagnético efetivo, em que os momentos magnéticos são os de cada partícula como um todo, o momento magnético resultante.
Em um sistema no estado superparamagnético, as partículas possuem um tempo de relaxação que caracteriza asflutuações do momento magnético, que é essencialmente o tempo médio para reverter o momento magnético de um estado de equilíbrio para outro. Esse tempo, primeiramente introduzido por Néel, pode ser descrito pela lei de Arrhenius [16]:
τ =τ0e
KaV kB T
(1.68)
O tempo de relaxação τ é determinado por uma frequência de tentativas de saltos τ0 da ordem de 1010Hz, em que kB é a constante de Boltzmann, T a temperatura, V o
crítico para o superparamagnetismo é diretamente proporcional à temperatura; portanto, ao obtermos uma distribuição de partículas com diferentes tamanhos e elevando sua temperatura elas vão se tornando cada vez mais superparamagnéticas. Por exemplo, Um aglomerado de partículas esféricas de Cobalto, com68Å de diâmetro e tempo de relaxaçãoτ = 0.1s, chegaria
rapidamente ao equilíbrio térmico. Todavia, se aumentarmos o diâmetro da partícula para 90Å, o valor de τ chega a3,2×109s , aproximadamente 100 anos. Podemos concluir, então,
que o momento magnético torna-se tão instável que acaba demorando muito mais para sofrer uma reversão [17], mesmo com uma alteração tão pequena no diâmetro da partícula.
Com base nisso, pode-se afirmar que o efeito superparamagnético é observado em uma determinada partícula quando esta, em dada temperatura, tem seu tempo de relaxação inferior o tempo necessário para se realizar a medida. Caso contrário, diz-se comumente que a partícula encontra-se no estado bloqueado. Podemos chegar a esse estado bloqueado de duas diferentes maneiras: primeiro reduzindo a temperatura da nanopartícula gradualmente até atingirmos o alinhamento do momento magnético da partícula, conforme a nossa necessidade; a outra forma possível é aumentando o tamanho da nanopartícula de tal forma que a barreira potencial de energia magnética será da ordem de tamanho da partícula, não criando assim um obstáculo. Observamos a relação do alinhamento magnético da nanopartícula e sua histerese na figura 1.13.
Figura 1.13: Relação do tamanho da nanopartícula e sua histerese magnética, (adaptado de [11]).
com o seu momento magnético instável, observamos que sua histerese é fechada, as duas curvas estão superpostas (curva vermelha na figura 1.13). Com isso, podemos observar que a coercividade ou Campo Coercitivo é zero, isto é, não é necessário aplicar campo magnético externo para desalinhar o momento magnético da partícula.
Já para as partículas com pseudomonodomínio ou multidomínio (curva azul na figura 1.13) observamos que o Campo Coercitivo é muito alto, ou seja, exige-se um campo magnético alto para se desmagnetizar a amostra. Materiais desse tipo são muito necessários na produção, por exemplo, de discos rígidos de computadores, uma vez que as informações gravadas nesses materiais, não devem se perder em caso de contato com campo magnético externo.
Outro fator importante para a confirmação de superparamagnetismo nessas amostras é a comparação do tamanho médio do cristalito com o diâmetro críticodcsuperparamagnético
para tais amostras. O tempo de relaxação é essencialmente o tempo necessário para reverter o momento magnético de um estado de equilíbrio para outro. Esse tempo característico depende da energia da barreira KV e da temperatura. Da equação 1.67 temos:
τ =τ0
KaV
kBT
(1.69)
Em nanopartículas, comportamento magnético depende também do tempo de me-didaτm, que varia desde valores altos (tipicamente102s), até baixos valores (10−8s), no caso
da espectrocopia Mössbauer.
Para altas temperaturas ou pequenos volumes,kBT ≫KaV, tem-seτm ≫τ →Regime
Superparamagnético. kBT ≪KaV, tem-se τm ≪τ →Regime Bloqueado.
Definindo o volume crítico a uma temperatura constante To ao requerer τm =τ
lnτ = lnτ0+
KaVcrit
kBT0
=
ln 102
...
ln 10−8 .
(1.70)
Assumindo que τm = 100s [42], tem-se:
Vcrit ≈
25kT Ka
(1.71)
Sabe-se queKa para ferrita de manganês é 0,056J/cm3 [43], tendo-se, assim,
Vcrit ≈
25·1,38·10−23m2Kgs−2K−1˙300K
0,056Kgm2s−2cm−3
Vcrit ≈
10350·10−23
0,056 ˙10−21nm−3
Vcrit ≈1850nm3
Considerando que a nanopartícula possui forma esférica, pode-se calcular o raio e consequentemente o diâmetro crítico dcrit para M nF e2O4.
dcrit = 2rcrit = 2·
3
r
3Vcrit
4π
Cap´ıtulo
Ferritas
"The grass was greener. The light was
brighter. The taste was sweeter. The
nights of wonder. With friends
sur-rounded. The dawn mist glowing. The
water flowing. The endless river.
Fore-ver and eFore-ver. "
High Hopes - PINK FLOYD
2.1 Óxidos Magnéticos
Os óxidos magnéticos geralmente são compostos por elementos dos grupos de transição, nos quais estão os elementos que possuem as camadas eletrônicas mais internas não preenchidas. Os elétrons das camadas mais externas contribuem na ligação química, e os elétrons das camadas internas não preenchidas são responsáveis pela variedade de propriedades magnéti-cas, em decorrência do desemparelhamento dos spins. Tais estados eletrônicos que produzem magnetismo são pouco afetados pelo ambiente, uma vez que sua valência é bem definida.
A força do acoplamento spin-órbita é determinante para estabelecer até que ponto o momento orbital contribui para as propriedades magnéticas. Os spins também interagem com a rede; nessa interação, o elétron sofre influência de dois campos separadamente: o campo elétrico cristalino captura ou extingue o momento orbital, mediante o Efeito Stark, e a interação de troca ordena os spins [22].
Os elementos que produzem efeitos magnéticos mais significantes, quando
dos com oxigênio, são os que possuem camadas internasdn efnincompletas. Neste trabalho,
nosso foco de interesse são os elementos pertencentes ao grupo 3dn, mais conhecido como
série do ferro, que se inicia no Escândio Sc21 e termina no Cobre Cu29.
2.2 Estrutura Espinélio
Ferritas Espinélio constitui um grande grupo de óxidos que possuem estrutura semelhante ao espinélio natural M gAl2O4. Existem muitos espinélios sintéticos comercializados, mas sem
dúvida o espinélio mais conhecido e também um dos mais utilizados é a magnetita F e3O4,
que é um óxido natural.
Espinélios possuem estrutura estável e predominantemente iônica; todavia, sítios ocupados por cátions são influenciados por vários fatores, dentre eles: efeitos de ligações covalentes e energias de estabilização de campo do cristal de cátions de metais de transição. A primeira estrutura espinélio foi determinada a princípio por Bragg (1915) e Nishikawa (1915). A estrutura ideal é cúbica de face centrada (cfc), composta por um conjunto de átomos de oxigênio; nela, 1/8dos sítios tetraédricos e 1/2 dos sítios octaédricos intersticiais são ocupados por cátions. Adotaremos A, para representar os sítios tetraédricos, e B, para
os sítios octaédricos.
A célula unitária contém 8 unidades de fórmulaAB2O4, é formada por8sítiosA,16
sítiosBe32oxigênios. Pode-se descrever a célula unitária tomando os sítiosAcomo origem da célula unitária. Por conveniência, divide-se a célula unitária em oito cubos de arestaa/2 para mostrar a disposição dos sítios A eB, como podemos observar na figura 2.1.
Os átomos de oxigênio geralmente não estão localizados em posições exatas das sub-redescfc. As posições detalhadas são determinadas pelo parâmetro u, que reflete o ajuste da estrutura a fim de acomodar diferenças nas razões de raios dos cátions nos sítios tetraédricos e octaédricos.
O parâmetro u é definido na figura 2.2 e tem valor de0.375para um arranjo ideal de face centrada de átomos de O, tomando como célula unitária a da figura 2.1. Pode-se usar,
outrossim, o centro de simetria para definir o parâmetro, localizado em (0.125,0.125,0.125),
como origem da célula unitária. Nesse caso, o valor ideal deu é0.25.
A situação ideal é dificilmente realizada, e os reais valores de u, para maioria dos
Figura 2.1: Estrutura Espinélio. Célula unitária divida em octantes. Cátions nos sítios tetraédricosA, cátions nos sítios octaédricos B e átomos O em dois octantes [12].
tetraédricos são forçados a moverem-se na direção[111]para ceder espaço aos cátionsA, que em geral são maiores que o espaço ideal permitido pelo arranjo de face centrada do oxigênio, mas sem alterar a simetria total 43m. O octaedro torna-se menor e assume a simetria 3m.
A média radial dos cátions afeta primeiramente o parâmetro a, enquanto a razão radial de
cátions tetraédricos e octaédricos determina principalmente o valor de u. Se o parâmetro
da rede é tomado como a média ponderada das projeções dos comprimentos das ligações tetraédricas e octaédricas na célula unitária, o parâmetro pode ser descrito por [12]:
a = 8(lig. tet.)
3√3 +
8(lig. oct.)
3 (2.1)
Essa expressão é responsável por aproximadamente96.7%das variações do parâmetro de rede de 149 óxidos espinélio [26].
No espinélio M gAl2O4, os cátionsAl eM g ocupam respectivamente os sítios
octaé-drico e tetraéoctaé-drico. A distribuição de cátion usual é:
Figura 2.2: Definição do parâmetro de deformação u. Meio plano de (110) é mostrado. Atómos de oxigênio (círculos grandes) estão dispostos na direçãoh111idevido a presença dos cátions tetraédricos. No caso ideal,u= 0.375 [12].
em que os colchetes indicam a ocupação dos sítios octaédricos e os parênteses a dos sítios tetraédricos. Essa distribuição é chamadanormal. SeDrepresenta um cátion divalente,
eT um trivalente, tem-se outra distribuição
(T)[DT]O4 (2.3)
que é chamada espinélio inverso. Em vários casos, encontra-se uma distribuição intermediária
(D1−δTδ)[DδT2−δ]O4 (2.4)
na qual δ é o grau de inversão, com valor zero para distribuição normal e 1 para distribuição inversa. Em muitos casos, o grau de inversão depende da técnica de preparo, em especial a taxa de resfriamento após a sinterização.
mais interessantes e persistentes problemas da química de cristais.
Os fatores que contribuem para a energia total da rede em espinélios são:
1. Energia elástica;
2. Energia eletrostática (Madelung);
3. Energia de estabilização do campo cristal;
4. Efeitos de polarização.
As duas primeiras energias, em geral, são suficientes para determinar a energia total da rede em óxidos de metais que não são de transição. A energia elástica está relacionada ao grau de distorção da estrutura do cristal, que, por sua vez, está relacionado às diferenças de raio iônico, assumindo que os íons adotam uma forma esférica. Cátions menores, com raio entre 0.225 e 0.4, ocupam sítios tetraédricos, já cátions de raio entre 0.4 e 0.73 ocupam sítios octraédricos. Essa distribuição leva a um mínimo de tensão na rede, uma vez que cátions trivalentes são geralmente menores que os divalentes. As contribuições eletrostática e elástica estão embasadas no pressuposto da simetria esférica dos íons com apenas interações coulombianas, o que está longe de ser o caso para cátions de metais de transição em espinélios ferrimagnéticos.
Figura 2.3: Geometria espacial dos orbitaisd (a) dxy, (b) dx2−y2, (c) dz2 [12].
carga dos orbitais d, figura 2.3, interage com a distribuição de carga do ambiente no qual o íon de transição é inserido. Os cinco orbitaisd (dxy, dyz, dzx, dz2, d
x2−
y2)já não têm a mesma
energia, mas estão dividos conforme a simetria do campo eletrostático produzido pelos ânions do sítio particular da rede. As bases físicas para essa divisão são simplesmente a repulsão eletrostática entre os elétrons d e os elétrons dos orbitais em torno dos ânions.
A polarização pode ser considerada o grau de distorção da densidade de carga ele-trônica ao redor do íon e pode surgir de várias maneiras. Dois casos extremos seriam: um efeito insignificante que dá origem a um vínculo puramente covalente e a remoção eficaz de um elétron de um íon em direção ao seu vizinho dará origem a um vínculo puramente iônico. Em relação aos íons dos metais de transição em espinélios, espera-se que apenas íons esferica-mente simétricos(d5 e d10)possam indicar uma tendência para covalência. Nesse caso, sítios
tetraédricos são, pois, preferidos. Cátions que revelam afinidade covalente para ambientes tetraédricos são F e3+, Ga3+, In3+ e, mais fortemente, Zn2+ e Cd2+. Espinélios formados
pelos primeiros cátions citados são inversos, enquanto os formados pelos últimos tendem a ser espinélios normais [12].
O grau de inversão, δ, não é exatamente zero ou um em muitas ferritas e pode ser
modificado por meio da técnica de preparação. Em altas temperaturas (próximas ao ponto de fusão, por exemplo), seria esperada uma simples distribuição estatística de todos os cátions nos sítios do cristal (δ = 2/3). Um resfriamento muito rápido pode extinguir estados em alta
temperatura. Se a redistribuição dos cátions é lenta, estados de alta temperatura permane-cem em temperatura ambiente. Em muitos casos, uma simples distribuição de Boltzmann tem evidenciado uma boa concordância com os resultados experimentais. A distribuição de Boltzmann pode ser descrita da seguinte maneira:
(2−δ)(1−δ)
δ2 =e
−E
kT (2.5)
em que E é a energia de ativação para a troca de sítio entre um cátion trivalente
e um divalente, k é a constante de Boltzmann e T é a temperatura. A termodinâmica da
redistribuição de cátion é bem conhecida pela variação da taxa de resfriamento entre0.01oC/s
e1000oC/s; uma variação contínua na configuração de cátion é obtida emCoF e
2O4 [27]. Um
valor típico deE é0.8±0.05eV para uma ferrita inversa, N iF e2O4 [28], e0.14eV para uma
ferrita mista, M gF e2O4 [29]. A distribuição das seguintes ferritas tem sido investigada por
Esses mecanismos de redistribuição de cátions (os cinéticos), entretanto, são comple-xos e podem ser afetados significativamente pela presença de F e2+ [33]. Quando uma
con-centração substancial deF edivalente estava presente num espinélio inverso, a redistribuição
cinética independia da vacância da concentração de cátions, tamanho de grão e estequiome-tria deO. Ferritas que contêm F eapenas no estado trivalente, a exemplo deM gF e2O4, que
tem mostrado, em vez disso, uma clara dependência no tamanho do grão e de outros fatores associados com nucleação e mecanismos de crescimento [34].
No que concerne a ferritas inversas (ou em vários casos em que há mais de um tipo de cátions ocupando o mesmo conjunto de sítios cristalográficos), fenômenos ordenados podem ser esperados [35]. Nessas ferritas, a ocupação dos sítios octaédricos por dois tipos de cátions (divalente e trivalente) pode levar à ordem de longo alcance, na qual sucessivas camadas (001) de sítios octaédricos são ocupadas alternadamente por cátions divalentes e trivalentes. Nesse caso, existem duas sub-redes iônicas nos sítios octaédricos [36] (figura 2.4).
Há, outrossim, evidências de que quando a razão de dois cátions nos sítios B é 1 : 3, a ordem de longo alcance pode ser estabelecida. Cada linha de cátions octaédricos nas direções h110i contém cátions B em forma ordenada (figura 2.5). Um exemplo disso é (F e)[Li0.5F e2.5]O4 [38], cuja estrutura é essencialmente cúbica. A transição para a
distribui-ção aleatória deLi+ eF e3+ nos sítios octaédricosB ocorre entre1000 e1028K. Um terceiro
tipo de ordenamento de cátion pode ocorrer quando existe a taxa de cátion 1 : 1 em sítios tetraédricos, nos quais os cátions alternam-se. Essa superestrutura tem sido observada em
Li0.5F e0.5[Cr2]O4 [39].
Cap´ıtulo
Procedimento Experimental
"I have finally found a place to live
In the presence of the Lord."
Presence of the Lord - Eric Clapton
A obtenção das amostras de ferrita de manganês foi realizada por meio do método
Pechini, também conhecido como Método dos Precursores Poliméricos, que consiste em um processo de polimerização através da mistura de nitratos, facilitando a manipulação das propriedades físicas e estruturais desejadas para o material resultante. Tal processo produz óxidos mistos cujas estruturas apresentam uma organização tridimensional dos constituintes, resultando em materiais na forma de pó, o que o torna bastante eficaz devido à possibilidade do controle do tamanho das partículas, da cristalinidade e da porosidade. Por esse motivo, os materiais fabricados pelo método possuem alta pureza, homogeneidade e temperaturas de processamento baixas, quando comparados a outros métodos equivalentes. Tais fatores são de incontestável relevância, pois influenciam nas propriedades ópticas, magnéticas, catalíticas e mecânicas do produto resultante.
Inicialmente, o sistema consiste em uma suspensão de partículas coloidais, cujo ta-manho varia entre1e100nm, dispersas em um solvente. Essas partículas se ligam formando
cadeias ramificadas e tridimensionais chamadas de microgel. Essas regiões crescem até ocu-parem metade do volume total; nessa situação, a viscosidade aumenta e, assim, o sistema atinge o ponto de gel e passa a se comportar como um fluido estático. Nesse estágio do processo, o sistema é formado por uma rede de partículas coloidais ou cadeias poliméricas e, a partir desse momento, as cadeias estruturais crescem conjuntamente, criando uma rede contínua por todo o sistema. O gel continua sendo aquecido até chegar à textura de flocos,
formando o precursor que será utilizado para calcinação [18], [19].
3.1 Material Utilizado
• 1 becker
• 1 balança de precisão
• 1 agitador magnético com variação de temperatura • 1 pipeta
• Nitrato de Ferro III: F e(N O3)3+ 9H2O
• Nitrato de Manganês: M n(N O3)2+ 4H2O
• Ácido Nítrico: H3C6H5O7H2O
• Etileno Glicol: C2H6O2
• Água destilada
O processo de produção da Ferrita de Manganês (M nF e2O4) ocorreu no Laboratório
de Magnetismo e Materiais Magnéticos do DFTE-UFRN, seguindo as etapas discriminadas:
1. Pesagem dos nitratos;
2. Diluição dos nitratos no becker com 300ml de água destilada, já no agitador magnético;
3. Adição de ácido cítrico com a espátula, após a diluição completa dos nitratos;
4. Adição de etileno glicol com a pipeta;
5. Manutenção do medidor de temperatura em 100oC, até obter uma solução uniforme;
6. Aumento da temperatura em100oCa cada 30 minutos, até obter a temperatura máxima
de 300o do agitador para acelerar o processo de polimerização e exaustão dos nitratos;
7. Espera até que a solução chegue à textura de flocos;
3.2 Tratamento Térmico
O tratamento térmico é efetuado com o intuito de eliminar toda a matéria orgânica restante na amostra, como também de sabermos em qual temperatura a fase (jacobsita) desejada se encontra e fazermos o controle do tamanho dos cristalitos.
Antes da calcinação, as amostras são maceradas com vistas a uniformizar o tamanho dos grãos. Tal maceramento é efetuado em um almofariz de ágata; logo depois, as amostras são levadas a uma mufla modelo LT0212 e, então, calcinadas à temperatura controlada. As amostras de Ferrita de Manganês (M nF e2O4) foram calcinadas em 400oC, 500oC, 600oC e
700oC, tanto em atmosfera ambiente quanto em vácuo.
3.3 Difratometria de Raios-X
Com o objetivo de investigar as características estruturais das amostras produzidas, bem como verificar se a produção das amostras se o tratamento térmico geraram ferritas de man-ganês, utilizamos uma ferramenta poderosa chamada difração de Raios X (a difratometria de raios-x, que é a melhor opção para a determinação estrutural de um sólido).
Essa técnica é de fundamental importância para o estudo da matéria condensada, pois permite a investigação do arranjo ordenado dos sólidos, parâmetros de rede, defeitos estruturais, simetria cristalina, distância interplanar, etc. Baseia-se no princípio de que os cristais são formados por átomos ordenados de forma periódica em três dimensões [19], [20].
Quando o comprimento de onda (λ) de uma onda eletromagnética é mensurável com
as dimensões atômicas da rede, pode-se verificar que as relações de fase entre os espalhamentos tornam-se periódicas e que efeitos de difração dos raios-x podem ser observados em vários ângulos. Considerando-se dois ou mais planos de uma estrutura cristalina, se a diferença entre os caminhos ópticos for um número inteiro (n) de comprimento de onda (λ), haverá
uma superposição construtiva (um feixe de raios-x será observado); caso contrário, haverá superposição destrutiva. Isso pode ser descrito matematicamente segundo a lei de Bragg: [19], [21]
2dsinθ =λn (3.1)
![Figura 1.3: Dependência da temperatura característica com a susceptibilidade recíproca: (a) para um diamagnético e (b) para um paramagnético [12].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15558572.100216/22.918.281.682.456.801/figura-dependência-temperatura-característica-susceptibilidade-recíproca-diamagnético-paramagnético.webp)
![Figura 1.5: Função de Langevin [11] Da Função de Langevin, temos dois resultados:](https://thumb-eu.123doks.com/thumbv2/123dok_br/15558572.100216/25.918.155.803.359.863/figura-função-langevin-função-langevin-temos-dois-resultados.webp)
![Figura 1.6: Espaço de Quantização: (a) Teoria Clássica, (b) e (c) Teoria Quântica [11].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15558572.100216/28.918.270.695.511.761/figura-espaço-de-quantização-teoria-clássica-teoria-quântica.webp)
![Figura 1.9: Arranjo Antiferromagnético composto de duas sub-redes ferromagneticamente ordenadas [24].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15558572.100216/33.918.203.757.837.1008/figura-arranjo-antiferromagnético-composto-duas-redes-ferromagneticamente-ordenadas.webp)

![Figura 1.13: Relação do tamanho da nanopartícula e sua histerese magnética, (adaptado de [11]).](https://thumb-eu.123doks.com/thumbv2/123dok_br/15558572.100216/41.918.167.789.684.971/figura-relação-tamanho-nanopartícula-sua-histerese-magnética-adaptado.webp)
![Figura 2.3: Geometria espacial dos orbitais d (a) d xy , (b) d x 2 −y 2 , (c) d z 2 [12].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15558572.100216/48.918.199.733.708.949/figura-geometria-espacial-dos-orbitais-a-d-xy.webp)
