The Diels-Alder reaction at the beginning of the twenty-first century.
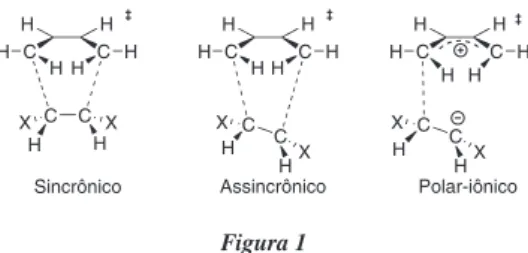
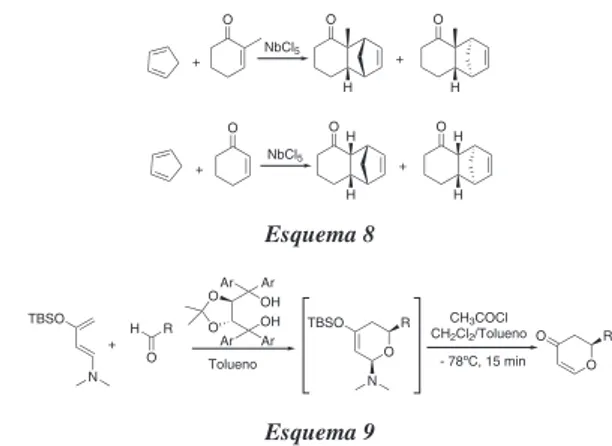
Texto
Imagem
Documentos relacionados
acyliminium ion cyclization cascade. The key step in this process involves the generation of an amino-substituted furan by a Pummerer induced cyclization reaction. After the
Key steps are Pd-catalyzed Stille coupling reaction between a vinyl iodide and a vinylstannane followed by an intramolecular Diels-Alder cycloaddition reaction to afford the
Diels-Alder reactions using 7c and methyl angelate (reaction 1) or methyl tiglate (reaction 2) as dienophiles..
We report a synthesis of the 1,6-disubstituted eudesmane sesquiterpene carbon skeleton, based upon a multicomponent Diels-Alder reaction.. Keywords: eudesmane,
The viscosity increase is likely due to the Diels-Alder reaction between the olefin and the diene on the alkenyl moiety of cardanol- aza-dicarboxylate ester, leading
A reação de cicloadição entre a mono-oxima tosilada de para -benzoquinona ( 38 ) com o ciclopentadieno ( 74 ) também pode formar quatro produtos, uma vez que a cicloadição pode
Paul Veyne has suggested in 1971 that Sociology lacked a study object. Three quarters of a century after Durkheim’s Rules , it had yet to discover social types and orders
