Alguns aspectos da eletroxidação de monóxido de carbono em superfícies monocristalinas...
Texto
Imagem

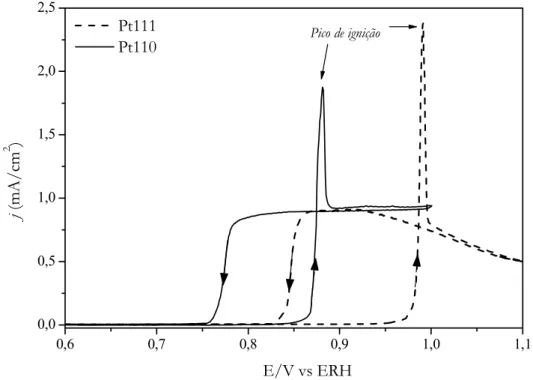


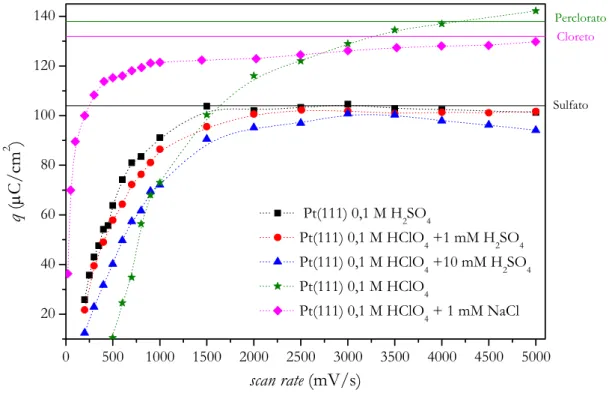
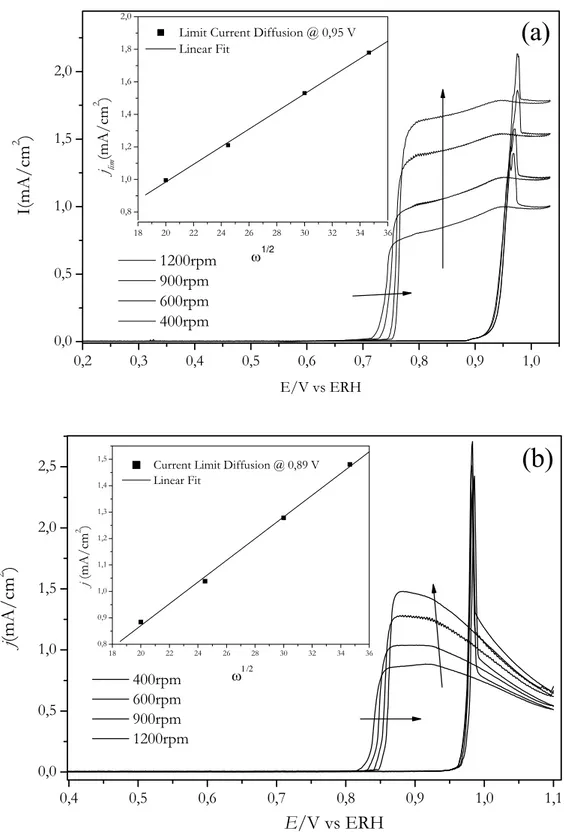

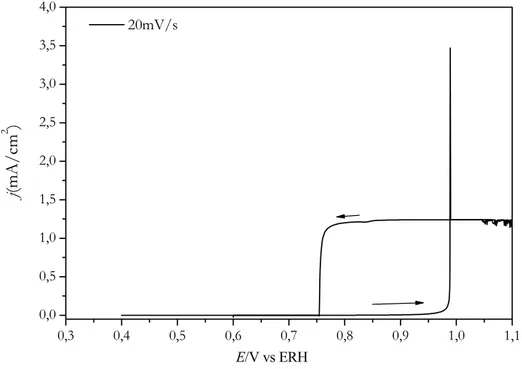


Documentos relacionados
O Ministério do Reino, aler- tado para a situação, ordena ao administrador-geral do distrito de Évora a inventa- riação e a guarda da referida livraria 58 e, em resposta,
Porém, a partir dos meados do século XIV, e até antes para algumas regiões como o Baixo-Alentejo ou o concelho de Santarém, medidas defensivas, procurando conservar as matas para
This highly sensitive approach permits identification and quantification of a wide variety of amino acid misincorporations, but does not produce a comprehensive view of
[r]
Objetivou-se com este estudo avaliar a qualidade de leite pasteurizado com inspeção estadual pela pesquisa de estafilococos coagulase positiva, sua
A Vector Network Analyser is used to generate the transmitted signal and to collect the received signals, being both in the frequency range of 100 MHz to 3 GHz.. Figure 3.4:
The focus of this thesis was to determine the best standard conditions to perform a laboratory-scale dynamic test able to achieve satisfactory results of the
— Publicado o ajuste de casamento, e não appare- cendo opposição no correr dos doze dias, dará o official do registro aos nubentes certificado de habilitação para a
