FUNDAÇÃO GETULIO VARGAS
ESCOLA BRASILEIRA DE ADMINISTRAÇÃO PÚBLICA E DE EMPRESAS MESTRADO EXECUTIVO EM GESTÃO EMPRESARIAL
An Evaluation of the Food and Drug Administration’s Expedited Pathways
Dissertacao Apresentada A Escola Brasileira De Administracao Publica E De Empresas Para Obtencao Do Grau De Mestre
Brian M. Mayhew Rio De Janeiro 2016
Ficha catalográfica elaborada pela Biblioteca Mario Henrique Simonsen/FGV
Mayhew, Brian Michael
An evaluation of the food and drug administration’s expeditd pathways / Brian Michael Mayhew. - 2016.
78 f.
Dissertação (mestrado) - Escola Brasileira de Administração Pública e de Empresas, Centro de Formação Acadêmica e Pesquisa.
Orientador: Mario Couto Soares Pinto. Inclui bibliografia.
1. Administração de produtos. 2. Produtos novos. 3. Biotecnologia farmacêutica. 4. Alimentos – Adulteração e inspeção. 4. Medicamentos – Adulteração e inspeção. I. Pinto, Mario Couto Soares. II. Escola Brasileira de Administração Pública e de Empresas. Centro de Formação Acadêmica e Pesquisa. III. Título.
CDD – 658.575
Brian M. Mayhew
An Evaluation of the Food and Drug Administration’s Expedited Pathways
Master’s thesis presented to Corporate International Master’s Program, Escola Brasileira de Administracao Publica, Fundacao Getulio Vargas, as a requirement for obtaining the title of Master in Business Management
Dr. Mario Pinto
Rio de Janeiro November 2016
[Approval sheet]
Dedication page
I dedicate my thesis to my wife Holly, my son Chase, my parents, family and friends who have supported me during this program and throughout my life. There is no me without you.
Acknowledgements
I would like to thank Dr. Mario Pinto for his advice and guidance on the development of my thesis topic and the thesis project.
I offer my gratitude to Dr. Christopher Long for his guidance and tutelage regarding thesis design and methodology.
I would like to thank the three academic institutions and the professors involved in this academic program:
Georgetown University McDonough School of Business ESADE University Business and Law School
Fundacao Getulio Vargas
I would like to thank my colleagues at Novartis Regulatory and Development Policy office for their support and guidance: Sharon Olmstead, Gretchen Trout and Chin Koerner.
I would like to thank Novartis Pharmaceutical Corporation for supporting my participation in the program.
TABLE OF CONTENTS
I. INTRODUCTION ...1
II. JUSTIFICATION ...3
III. OBJECTIVES ...10
IV. LITERATURE REVIEW ...11
V. METHODOLOGY ...19
VI. STATISTICAL ANALYSIS …...24
VII. REVIEW OF ANALYSIS ...24
VIII. CONCLUSIONS ...28 IX. RECOMMENDATIONS ...32 X. APPENDIX …...36 XI. GLOSSARY …...57 XII. BIBLIOGRAPHY …...59 XIII. INDEX …...63 XIV. ENDNOTES …...65
LIST OF ILLUSTRATIONS
Figure 1: FDA Expedited Programs ...1
Figure 2: Global Pharmaceutical Launches (2013-2018Est) ...2
Figure 3: Drugs@FDA Example: Lipitor ...20
Figure 4: WUSTL CRIB Example: Lipitor ...20
Figure 5: Darrow and Kesselheim Interactive Graphic ...21
LIST OF TABLES Table 1: FDA Expedited Programs ...4
Table 2: FDA Regulatory Programs – Previous Research Statistics ...18
Table 3: Years of Approval ANOVA ...35
Table 4: Year of Approval: Descriptive Groupings ...……...36
Table 5: Year of Approval: Comparison ...……...36
Table 6: Time Period 1: Tests of Between Subjects Effects ………37
Table 7: Time Period 1: With Select Expedited Programs: Estimates ………37
Table 8: Time Period 2: Tests of Between Subjects Effects ………37
Table 9: Time Period 2: With Select Expedited Programs: Estimates ………37
Table 10: Time Period 2: Pairwise Comparison ………...……….38
Table 11 Time Period 3: Tests of Between Subjects Effects ...……….38
Table 12 Time Period 3: With Select Expedited Programs: Estimates ………39
Table 13 Time Period 3: Pairwise Comparison ………...……….39
Table 14 Time Period 4: With Select Expedited Programs: Estimates ………40
Table 15 Time Period 4: Pairwise Comparison ………...……….40
Table 16: Expedited Program: ANOVA ………...42
Table 17: Number of Expedited Programs: Descriptives ...42
Table 18: Expedited Programs: Comparative Results ...42
Table 19: Accelerated Approval: Descriptives ...43
Table 20: Accelerated Approval: T-Test ...43
Table 21: Breakthrough Therapy: Descriptives ...43
Table 22: Breakthrough Therapy: T-Test ...43
Table 23: Fast Track: Descriptives ...43
Table 24: Fast Track: T-Test ...44
Table 25: Priority Review: Descriptives ………...44
Table 26: Priority Review: T-Test ...………...44
Table 27: Expedited Pathways ANOVA ...44
Table 28: Expedited Pathway Combinations ...44
Table 29a: Post Hoc Analyses: Expedited Combination ...45
Table 29b: Post Hoc Analyses: Expedited Combination ...46
Table 29c: Post Hoc Analyses: Expedited Combination ...47
Table 30: Therapeutic Areas Descriptives ...47
Table 31: Post Hoc Analyses: Therapeutics Comparative Analyses ...49
Table 32 Bone Therapy Area: Tests of Between Subject Effects ...….……….….54
Table 33 Bone Therapy Area With Expedited Program: Estimates .……….54
List of Symbols, Abbreviations and Acronyms
Accelerated Approval (Accelerated, A, AA) Brazillian Health Regulatory Agency (ANVISA) Breakthrough Therapy Designation (BTD, B) End of Phase II (EOPII)
European Medicines Agency (EMA) Fast Track Designation (FT, F)
Food and Drug Administration (FDA or “the Agency”) Investigational New Drug (IND)
Mechanisms of Action (MOA) New Drug Application (NDA) Office of New Drugs (OND) Orphan Drug Designation (Orphan)
Pharmaceutical and Medical Device Agency (PMDA) Pre-Investigation New Drug Application (pre-IND) Pre-New Drug Application (pre-NDA)
Prescription Drug User Fee Act (PDUFA) Priority Review (PR, P)
Qualified Infectious Disease Product (QIDP) Standard Development and Review (SDR) Therapeutics Goods Agency (TGA) United States (US)
ABSTRACT
Biopharmaceutical development is characterized by challenging regulations, intense competition and significant costs that result in the need for biopharmaceutical companies to consistently produce innovation biopharmaceutical products. The United States Congress has sought to provide a balanced environment that combines significant regulatory oversight by the US Food and Drug Administration (FDA) with market-based incentives (patent protection, exclusivity) and expedited pathways (accelerated approval, breakthrough designation, fast track designation, and priority review) that seek to quickly identify and move innovative new medicines through
development that will address unmet medical need and treat serious or life-threatening diseases or conditions. While FDA’s expedited programs are believed to accelerate the development of innovative drug products, the programs have not been formally measured against their intended purpose: more efficient development and regulatory reviews. This thesis research project attempts to effectively measure FDA’s expedited programs by cataloguing FDA approvals from 1987-2015, measuring development and regulatory review time, and drawing conclusions and making recommendations based on the statistical analyses generated from the project.
INTRODUCTION
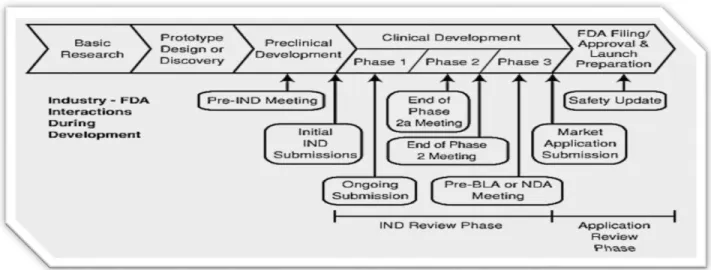
The global biopharmaceutical market is valued at more than $1 trillion and expected to grow to $1.3 trillion by 2018, with the United States (US) estimated to account for approximately one third of the global market.1 In the US, the industry is characterized by significant barriers to entry, stringent regulatory requirements, and significant competition.2 Additionally, the drug development process is long and onerous with several stages of research and development, followed by review and an approval decision by a regulatory health authority (for example: the Food and Drug Administration (FDA) in the US; See Figure 1). The drug development process includes important steps to correctly identify a candidate drug product, assess its safety and efficacy in patients, and ensure that the drug sponsor can manufacture the product under specified quality measures. These requirements represent significant costs: current estimates indicate that the average cost to bring a product to market is $2.870 billion and takes approximately 11 years.3 Figure 1. FDA’s Drug Development and Regulatory Process
Source: US FDA White Paper Prescription Drug User Fee Act (PDUFA)4
Given this challenging dynamic, biopharmaceutical companies seek to continually identify innovative products, so called “first in class” or “best in class products” to better meet the needs of patients.5,6 Cohen noted that the “pharmaceutical industry is motivated to innovate, not just
incrementally or sequentially but also to produce pioneering innovations that drive profits and provide competitive advantages.”7 According to IMS Health, innovative products will continue
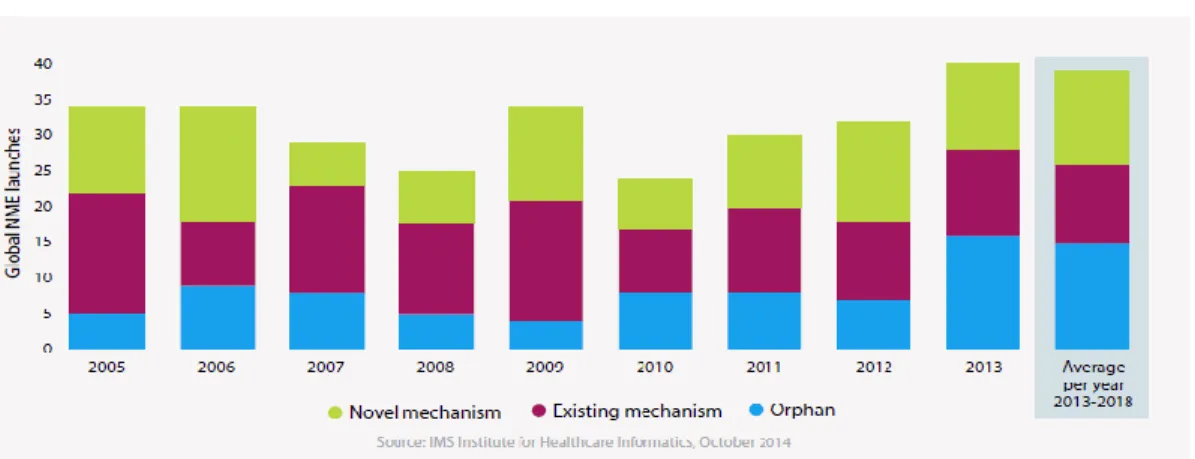
to drive global biopharmaceutical launches through the introduction of novel mechanisms of action (MOAs) or via products for orphan indications (See Figure 2).8
Figure 2. Global Pharmaceutical Launches (2013-2018 Est)
Pharmaceutical innovation is largely predicated on the ability of companies to identify, develop, manufacture, and commercialize new products that meet the needs of patients. Indeed, the US FDA (or “the Agency”) has commented that “innovation in the pharmaceutical industry plays a vital role in improving public health.”9 Further, FDA monitors and reports on the number of
innovative drug approvals each year as Dr. John Jenkins, Director, Office of New Drugs (OND), Center for Drug Evaluation and Research (CDER), FDA, commented in his annual update that 41% (17 drugs) of medicines approved in 2014 were first-in-class treatments, “one indicator of the degree of a drug’s innovation.”10 FDA has also studied pharmaceutical innovation to identify
better measurements for innovation.11 Clearly, the ability to introduce innovative products to the
market is vitally important.
Some, like Munos, have concluded that innovation is fostered by a “demanding regulatory apparatus, such as the United States and the UK.”12 Thus, the legislative and regulatory
environment within the United States has been designed to encourage innovation by offering incentives for novel product development (e.g., patent protection, market exclusivity, research tax credits) but also fostering competition and maintaining significant barriers to entry via high regulatory requirements to conduct drug discovery, clinical research, and product
commercialization.13 Moreover, Congress has enacted laws over the past 35 years that have granted FDA the authority to speed the development, review and approval decisions of novel and innovative medicines. So-called “expedited programs” these programs were enacted to help FDA identify and facilitate the development of drugs to address unmet medical needs or treat serious or life threatening diseases. FDA’s expedited programs (accelerated approval, breakthrough
product and to shorten FDA’s regulatory review of marketing authorization applications. Said another way, the programs are meant to decrease the time it takes to research and develop products so that patients in need of novel drug products can access them more quickly. In all, support for these programs is shared across stakeholders, including Congress, FDA, patient advocates and industry. In fact, Friends of Cancer Research (FOCR), a prominent patient advocacy organization, played a key role in the enactment of the breakthrough designation, the newest expedited program.14
However, while product innovation in the biopharmaceutical industry is critical, it is unclear whether the enactment of FDA’s expedited pathways has led to a discernable impact on the number of innovative products in the marketplace. While it is assumed that FDA’s expedited programs speed development and review of novel therapeutics, research has not yet been conducted to specifically determine whether FDA’s four expedited programs have indeed led to shorter development and regulatory review times. The research question at hand is whether FDA’s expedited programs have facilitated the development and review of innovative products as measured by clinical development and regulatory review. This research proposal will analyze the time biopharmaceutical products are reviewed and approved under FDA’s regulatory authority to determine whether the expedited pathways are associated with shorter development and review times. It is expected that the expedited programs should decrease the amount of time required for clinical development and regulatory review given the increased FDA-sponsor engagement and communication and the potential to reduce the amount of clinical required as a result of the expedited programs.
I. JUSTIFICATION
Modern drug development is beset by patent expirations, increased cost of research and
development, high failure rate, pricing constraints, and increased regulations.15 As part of FDA’s regulatory requirements, drug sponsors must submit and receive clearance for Investigational New Drug Applications (INDs) before clinical trials can begin. When clinical trials are
completed, sponsors then submit a New Drug Application (NDA) – i.e., marketing authorization application - which should include all of the necessary data required for FDA to make a decision about whether the drug should be allowed to enter the US market. Taken together, the time from the IND submission to the NDA approval would be the clinical development and regulatory
review time. According to FDA, standard development times range from 4-8.5 (or more) years and its standard regulatory review of marketing authorization applications is 10 months.16 Published literature has demonstrated that the clinical development time averages around 7.5 years.17
To help balance these challenges and support product innovation in the pharmaceutical industry, U.S. Congress has passed several laws to encourage FDA to enable development and approval of innovative products more efficiently. So called expedited programs, they attempt to reduce the clinical development time for each product and the time FDA takes to review the marketing authorization application. Each of FDA’s expedited programs has their own characteristics, i.e., evidentiary requirements to achieve each designation, processes for requesting and obtaining the designations and timing of when a designation can be requested (see Table 1 for more
information).
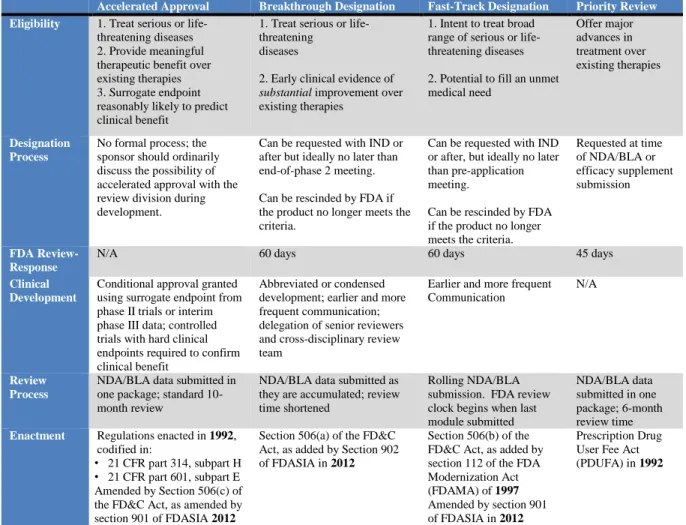
Table 1. FDA Expedited Programs
Accelerated Approval Breakthrough Designation Fast-Track Designation Priority Review Eligibility 1. Treat serious or life-
threatening diseases 2. Provide meaningful therapeutic benefit over existing therapies 3. Surrogate endpoint reasonably likely to predict clinical benefit
1. Treat serious or life-threatening
diseases
2. Early clinical evidence of
substantial improvement over
existing therapies
1. Intent to treat broad range of serious or life-threatening diseases
2. Potential to fill an unmet medical need Offer major advances in treatment over existing therapies Designation Process
No formal process; the sponsor should ordinarily discuss the possibility of accelerated approval with the review division during development.
Can be requested with IND or after but ideally no later than end-of-phase 2 meeting.
Can be rescinded by FDA if the product no longer meets the criteria.
Can be requested with IND or after, but ideally no later than pre-application meeting.
Can be rescinded by FDA if the product no longer meets the criteria.
Requested at time of NDA/BLA or efficacy supplement submission FDA Review-Response
N/A 60 days 60 days 45 days
Clinical Development
Conditional approval granted using surrogate endpoint from phase II trials or interim phase III data; controlled trials with hard clinical endpoints required to confirm clinical benefit
Abbreviated or condensed development; earlier and more frequent communication; delegation of senior reviewers and cross-disciplinary review team
Earlier and more frequent Communication
N/A
Review Process
NDA/BLA data submitted in one package; standard 10-month review
NDA/BLA data submitted as they are accumulated; review time shortened
Rolling NDA/BLA submission. FDA review clock begins when last module submitted
NDA/BLA data submitted in one package; 6-month review time Enactment Regulations enacted in 1992,
codified in:
• 21 CFR part 314, subpart H • 21 CFR part 601, subpart E Amended by Section 506(c) of the FD&C Act, as amended by section 901 of FDASIA 2012
Section 506(a) of the FD&C Act, as added by Section 902 of FDASIA in 2012
Section 506(b) of the FD&C Act, as added by section 112 of the FDA Modernization Act (FDAMA) of 1997 Amended by section 901 of FDASIA in 2012
Prescription Drug User Fee Act (PDUFA) in 1992
Notably, the programs are not exclusive, so a single product can benefit from one, two, three or all of the programs.
A. FDA Expedited Programs – Background
In 1992, Congress enacted legislation that authorized FDA to grant priority review designations with an eye toward shortening the time required for FDA’s regulatory review of products that treat serious conditions and unmet medical needs (i.e., novel or innovative products). That same year, FDA implemented the regulations for the accelerated approval pathway regulations for drugs and biologics. These expedited pathways were followed by the fast track designation in 1997, which enabled sponsors developing products to treat serious conditions or unmet medical needs the ability to receive additional oversight and communication with FDA regulatory staff, among other benefits.19 More recently, Congress passed legislation (Food and Drug
Administration Safety and Innovation Act (FDASIA)) granting FDA the authority to implement the breakthrough therapy designation program. FDA’s breakthrough designation follows closely to the fast track designation, but is based on early clinical data that demonstrates significant
improvement over currently marketed therapies (i.e., innovative products). According to Dr.
Janet Woodcock, CDER Center Director at FDA, the purpose of FDASIA was, in part, to introduce legislative changes that granted the US Food and Drug Administration (FDA) the authority to establish new and enhance existing innovative regulatory pathways.20
A recent study by Kesselheim et al demonstrated that, of the 774 drugs approved from 1987 to 2014, 9% received Accelerated Approval, 19% received Fast Track designation, and 43% received Priority Review. 21 Additionally, FDA designated 111 products as breakthrough
therapies from over 330 requests received (including both CDER and CBER).22 Recent statistics provided by FDA indicate that 52 breakthrough therapies have been approved as of June 30, 2016, in only its fourth year of existence.23
More detail about each expedited program follows to provide an understanding of the nuances of each expedited program.
1. Accelerated Approval of New Drugs (Subpart E) or Biologics (Subpart H) for Serious or Life-Threatening Illnesses
Accelerated Approval was enacted in 1992 to allow FDA to approve a new drug or biologic that
treats a serious or life-threatening illness and provides meaningful therapeutic benefit to patients over existing treatments based on whether the drug has an effect on a surrogate or an intermediate clinical endpoint.24 Many observers point to the pressure from physicians and the patient
community during the AIDS crisis of the late 1980s and early 1990s as the inflection point that compelled the creation of the accelerated approval program.25 The program was intended for
drugs that seek to treat diseases or conditions with endpoints that would take a longer period of time to study, such as HIV/AIDS or oncology. The use of a surrogate endpoint instead of
traditional clinical endpoints would decrease the duration of clinical development and thus enable patients to access the treatments sooner than would otherwise be possible under standard
development and review. If a product is approved under accelerated approval, the sponsor must then conduct post-approval studies to confirm the drug’s clinical benefit. If the confirmatory trials do not verify the clinical benefit or if a sponsor does not complete the post-approval trials, then FDA can withdraw marketing approval for the drug. Additionally, FDA can restrict the accelerated approval if, for example, the Agency determines that the drug can only be used safely when prescribed by specially trained physicians.
In terms of usage, a recent study demonstrated that accelerated approval is one of the least used expedited pathways where 9% of the 774 drugs approved from 1987 to 2014 received
Accelerated Approval26 . The point was emphasized in FDA’s guidance on Expedited Pathways where the Agency attempted to increase its use by broadening the inclusion criteria to focus on a drug’s ability to offer clinically important improvements from a patient or public health
perspective rather than a drug’s ability to offer a direct advantage over other existing therapies.
2. Breakthrough Therapy Designation
Breakthrough Therapy designation (BTD) was enacted in 2012 as part of FDASIA (legislative
vehicle for PDUFA V). BTD is intended to expedite the development and review of drugs for serious or life-threatening conditions where preliminary clinical evidence demonstrates the drug may have substantial improvement on at least one clinically significant endpoint over available
therapy.27 A breakthrough designation provides all of the features provided by fast track program (See subsection 3. Fast Track Designation) features as well as more intensive FDA engagement (especially senior management) throughout the development process. A breakthrough designation can be rescinded by the FDA if the Agency determines that the product no longer meets the qualifying criteria (Note: four designations have been rescinded to date).28 The breakthrough
designation offers additional values for stakeholders, such as:
Reputational value for sponsor’s portfolio
Potential to bring innovative products to patients more quickly
Strongly supported by patient groups, e.g., FOCR and National Organization for Rare Diseases (NORD)
The breakthrough pathway also requires a commitment from FDA/CDER to involve senior management during drug development.29 Such engagement includes: 1) earlier and potentially more frequent meetings with the sponsor and the CDER review team throughout the development of the drug, 2) providing timely advice and interactive communication with the sponsor to ensure program design feasibility; and 3) the involvement of senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review.30
Breakthrough has proven to be more popular with sponsors than initially envisioned by FDA. The Agency has publicly stated that breakthrough has become a resource drain on CDER staff, especially senior CDER officials, due to the increased involvement of these officials earlier in drug development.31 CMC and manufacturing scale up have, at times, had difficulty keeping pace with
the truncated development and review times due to rapidly moving through the nonclinical and clinical development process at an increased rate. An industry-sponsored White Paper concluded that the reduced timelines for products designated as breakthrough introduce significant chemistry manufacturing and controls (CMC) and good manufacturing practices (GMPs) challenges for sponsors.32 The issue is so important that FDA’s guidance on expedited pathways warns sponsors to “pursue a rapid manufacturing development program to accommodate the accelerated pace” of development and discusses some of the key manufacturing issues to prepare for an expedited program.33 Despite this drawback, the program has remained popular with sponsors as breakthrough requests have reached 441 as of June 2016.34
3. Fast Track Designation
Fast Track was implemented in 1997 as part of the Food and Drug Administration Modernization
Act (FDAMA). While FDA began identifying products as “fast track” as early as 1988, the formal designation and fast track program was not implemented until 1997. As with other expedited programs, fast track designation is designed to facilitate the development and expedite the review of a drug that treats a serious disease or condition and nonclinical or clinical data indicate the potential for the product to address unmet medical need (qualified infectious disease products (QIDPs) also qualify for Fast Track).35 Similar to breakthrough program, the Fast Track
designation provides for more frequent meetings with FDA to discuss drug development and ensure collection of appropriate data needed for approval, more frequent written correspondence from FDA, eligibility for Accelerated Approval and Priority Review, and a rolling review. When it was first implemented, the fast track designation was thought to provide a significant value for patients as new, innovative products would reach the market more quickly.
While the benefits provided by Fast Track were initially met with enthusiasm by industry, interest in the designation waned because increasingly FDA staff: 1) provided regular meetings with companies to discuss development and review issues (e.g., pre-IND, EOP2, pre-NDA, etc.), 2) was willing to provide follow-up meetings (when necessary), and 3) was willing to provide correspondence outside of the formal meeting structures for products that seek to treat life-threatening diseases and for potentially innovative and/or novel drug products. Furthermore, rolling reviews and dispute resolution were also available for products that were not designated as Fast Track, which further diluted the benefits of the designation. The decrease in fast track interest aligns with recent research that found that fast track was the second least utilized expedited program.36 Now, with the implementation and popularity of the Breakthrough Therapy designation, the potential exists that fewer sponsors will seek Fast Track.
4. Priority Review Designation
The Priority Review program was implemented in 1992 as part of the first PDUFA. A Priority Review provides a 6-month review instead of the standard 10-month review1 and can be granted
18- and 1month reviews, respectively, under the PDUFA V NME Program due to FDA’s
by FDA to a drug that treats a serious disease or condition and, if approved, would provide “a significant improvement” in treatment or “provide a treatment where no adequate therapy exists (QIDPs are also entitled to priority review).”37 The criteria to demonstrate “significant improvement” over available therapies include evidence of increased effectiveness, elimination or substantial reduction of a drug reaction, documented enhancement of patient compliance, or evidence of safety or effectiveness in a new patient subpopulation. Priority review has been the most popular expedited program based on recent analysis.38
B. Measuring Expedited Programs
The thesis study will take an unprecedented look into the expedited pathways through the lens of measuring development times. With respect to the purpose of the expedited programs, i.e., to speed the clinical development or regulatory review time of medicines that address unmet
medical need or treat serious or life-threatening diseases, it is important to identify whether these programs have successfully accomplished their intended purpose. Simply counting the number of approvals from each program is not sufficient. As FDA has commented: “measuring innovation based solely on the number of new drugs approved has considerable limitations.”39 Other
research has attempted to identify a definition of innovation. For example, categorizing new drug approvals into categories such as first-in-class, advances-in-class, and additions-to-class40 or even touting definitions for “transformative” products.41 Further, a work by Aronson, Ferner and
Hughes titled “Defining rewardable innovation in drug therapy” defined “highly innovative” products as those that address a new target or a novel mechanism, improve the identification of patients who are likely to benefit or be harmed, or involve the novel use of an existing
compound.42 Given the state of fluidity regarding the definition of pharmaceutical innovation, and the intent of this research project, the thesis project will rely on 1) FDA’s approval of a biopharmaceutical as a new molecular entity (NME) and 2) FDA’s decision to include a product in an expedited program as the guiding definitions of novel and innovative biopharmaceutical product.
Due to the intent of FDA’s expedited programs to speed the identification of novel products and move them through development to the market, the key measurement of the thesis project will be
to assess the amount of time it takes a product in one or more of FDA’s expedited programs to move through clinical development and regulatory review to an approval. As previously noted, the time from IND submission to an NDA decision can take many years. However, for novel drugs, the expedited programs have been put into place to speed the time it takes to move through development to commercialization. The clinical development and review time measurement is an approach that has been utilized in previous research, including Kaitin and DiMasi43, Shulman and Brown44 and Downing et al.45 Further, Moore and Furberg utilized development time as one
measure in their study of FDA approved products in 2008.46 However, these research projects
did not specifically utilize this measure to analyze all four of FDA’s expedited programs during the time periods selected for the thesis project. Therefore, the clinical development and review time (from IND submission to NDA approval) will be used in this research project in order to measure the success of FDA’s expedited programs in speeding the development of novel or innovative drugs.
II. OBJECTIVES General Objective
The proposed research project seeks to fill a current gap in the literature regarding FDA’s expedited programs by evaluating the success of the expedited programs in speeding innovative products to market. The research project will analyze products approved under FDA’s expedited pathways to identify whether the programs have decreased the amount of development and regulatory review time required for these products to reach the market (i.e., commercialization).
Specific Objectives
1. Catalogue NMEs approved by FDA from 1987-2015 in order to obtain the total study population.
2. For all catalogued products, collect data elements about each product including active ingredient, brand name, IND submission date (if available), NDA approval date, clinical development and review time, expedited program information, orphan drug status, therapeutic area, and sponsor.
3. Based on available data, create a study sample of products approved between the years 1987-2015.
4. Using analysis of variance (ANOVA) and t-test evaluations, analyze and compare products based on the following group descriptors:
Year approved
Time from IND to NDA approval
Number of program designations achieved
Type of program designations
5. Provide a review of the statistical analysis that discusses the differences observed with regard to any changes in the clinical development and regulatory time with respect to the identified group descriptors.
6. Specifically, review any differences between products approved under the standard
development and regulatory review as compared to products that were approved under the expedited review programs with respect to the four group descriptors.
Based on the review of the study, conclusions will be drawn and recommendations made about the ability of legislative and regulatory policies to impact research and development in the biopharmaceutical industry. Additionally, identify any potential future policy decisions that can be identified or any future research projects that can be conducted based on the results of the data.
III. LITERATURE REVIEW
The literature review was conducted across a wide variety of sources including business, medical and scientific journals, academic texts and online libraries from 1930-2016.2 Additionally, FDA’s online database Drugs@FDA, the Washington University at St. Louis (WUSL) Center for Research Innovation in Biotechnology (CRIB) database and other online databases were
extensively searched to generate the study database. Based on the literature review, two key
2 Literature searches conducted included the following: American Medical Association, BioMed Central, Business Source Direct, British Medical Journal, Directory of Open Access Journals (DOAJ), Informa, Ingenta Connect, MedLine/PubMed (NLM), New England Journal of Medicine (NEJM), Oxford Journals, PubMed Central (PMC), Sage Publications, Science Citation Index Expanded, SciVerse Science Direct, Social Sciences Citation Index, SpringerLink, Walter de Gruyter, Wiley Online Library, and Wolters Kluwer – Ovid – Lippincott Williams & Wilkins.
themes were identified: 1) analysis and review of FDA’s regulatory program, specifically expedited programs; and 2) pharmaceutical innovation.
A. FDA and Expedited Programs
A significant amount of research has been conducted about FDA’s regulatory authority, specifically the Agency’s facilitated and expedited pathways. Kesselheim et al.’s “Trends in utilization of FDA expedited drug development and approval programs, 1987-2014: cohort study”47 provides an important starting point for this project because its purpose was to study
whether the utilization and number of approvals for drugs intended to treat serious or life threatening conditions and that address unmet medical need are increasing over time. It
demonstrated that FDA approved 774 drugs from 1987-2014 with the following characteristics:
one third were first-in-class agents;
priority review (43%) was the most prevalent program;
"accelerated approval (9%) the least common; and
“a greater proportion of programs were being applied to drugs that were not the first members of their classes” (i.e., incrementally innovative products).
The study concluded that more new drugs approved by the Agency have been associated with an increasing number of expedited development or review programs. The study also noted that less innovative products utilizing FDA’s expedited programs “divert limited governmental resources” which questions whether FDA’s inclusion criteria for the programs is expanding over time to include less serious conditions or less noteworthy products. While notable, Kesselheim et al. used a simple accounting of FDA’s expedited programs, i.e., whether the programs were
associated with an increase in products approved. Another limitation of the study was that it was not able to include FDA’s breakthrough therapy designation due to the timing of the program’s enactment (2012). Another notable drawback was that the study included FDA’s orphan designation, which is not one of FDA’s expedited programs.
Another key article that forms the basis of this research project is Kinch et al.’s study “An
to catalogue and detail "all legacy drugs and recent NMEs approved for use in the USA…to determine how the enterprise has changed over time, with emphasis on trends and evaluation of the key players responsible for the discovery of new medicines.” The study found 1453 NMEs approved by FDA during the study period. The study also identified interesting statistics about FDA’s annual approval metrics, namely:
FDA approved fewer than 4 per year prior to 1950
the rate of new approvals increased to an avg. of 15/year in the 1950s-1970s
1980s: average NME approvals per year increased to a range of 25-30 where it has remained until today (two years of outliers in the 1990s and 2 years of outliers in the 2010s, which is indicated in their data).
In addition to the annual approval statistics, the data provide deep insight into the organizations that have received NME approvals over the years, but this is not applicable to this research project given the fluidity of product ownership and mergers and acquisitions over the time period studied by the thesis project. The article’s significance is the creation of a single, comprehensive database of all NMEs approved in USA, information from which was used to create this study’s database. While Kinch et al.’s study is one of the key foundations to this project, it represents a linear cataloguing of approvals. Of course this is helpful to provide baseline information about FDA's NME approvals; however, it does not analyze FDA’s expedited pathways.
Kesselheim and Darrow’s “FDA Designations for Therapeutics and Their Impact on Drug Development and Regulatory Review Outcomes” is another cornerstone of the thesis project because it involved an analysis of expedited programs and special pathways to determine their impact on drug development and regulatory review outcomes based on an analysis of 778 NMEs and original biologics approved between 1987-2013. The study found that 45% of priority review drugs, 50% of fast track drugs, and 42% of accelerated approval drugs were first-in-class products."49 Further, the study noted that “drugs receiving special FDA designations have
shortened development and review times” however the study did not evaluate this measurement.50 Additionally, in discussing the idea of “innovation” and pharmaceutical development, they
concluded that “there is no systematic evidence that first-in-class drugs actually offer major advances in treatment as adequate tradeoff for the shortcomings of their safety evaluations.” The
study offered two areas of caution: 1) it is still unknown how well these programs target approval of medications that have high therapeutic value; and 2) it is unknown whether the expedited programs “increase the likelihood that patients will be exposed to drugs that will ultimately be shown harmful or ineffective.” These are important topics covered in Section VIII.
Recommendations.
Another key research that helped to inform the database created as part of this project was Darrow and Kesselheim’s “Drug Development and FDA Approval, 1938-2013.” Specifically, their research helped to provide data regarding FDA’s older expedited programs (specifically, accelerated approval and fast track) that was not available on FDA’s website. One key observation from this database is that "[t]he number of NMEs benefitting from more than one program has increased from under 20% in 1987 to more than 40% in 2013."51 Additionally,
details regarding the use of this research to create the thesis project’s database is detailed in Section V.
Munos’ “Lessons from 60 years of pharmaceutical innovation”52 researched FDA approvals from
1950-2008, where he found 1,222 NMEs were approved, of which 1103 were small molecules and 119 biologics. The study “…investigates the record of pharmaceutical innovation by analyzing data on the companies that introduced the …new drugs that have been approved by FDA since 1950.” Munos concluded that “[i]n the past 60 years, the pharmaceutical industry has delivered over 1220 new drugs that have played an important part in improving public health and extending life expectancy by an average of 2 months per year.” While this work provides
interesting conclusions about the “ownership of NMEs” it does not provide an analysis of FDA’s expedited programs. Due to the fluidity of product ownership (via in- and out-licensing) and at times rapid pace of mergers and acquisitions, an analysis of the product owners was not included in the thesis project.
Lastly, a recent study conducted by the Biotechnology Industry Organization (BIO) measured “clinical development success rates” to enhance clinical development benchmarking metrics.53
While the study was informative about the individual success rates for each phase of clinical development (e.g., Phase I, Phase II), the purpose of the study did not align with the purpose of the thesis research.
B. Pharmaceutical innovation
Another key theme from the literature review is “pharmaceutical innovation.” The bulk of the literature focuses on defining pharmaceutical innovation or identifying ways to create or enhance innovation in biopharmaceutical development. For example, Cohen’s article entitled “Macro trends in pharmaceutical innovation” reviewed and discussed the definition of innovation as it pertains to the pharmaceutical industry. 54
Lanthier et al.’s55 work describes FDA’s current thinking on measuring pharmaceutical innovation. FDA’s longstanding definition of innovation was the number of new molecular entities (NMEs) approved for each year. However, in this work FDA officials questioned the notion that, in fact, the sheer quantity of NMEs “has considerable limitations” i.e., “not all NMEs are equally innovative." The FDA goes on to say that a “new entity could be similar in function and effectiveness to a drug already on the market or a major breakthrough in treatment or technology.” As a proposed solution, Lanthier et al56 put forth three sub-categories to better
identify a product’s “degree of novelty” as follows:
First-in-class - “drugs that are pharmacologically innovative because each represents a new pathway for treating a disease.” These products are “genuinely innovative because each represents a novel approach to drug therapy."
advance-in-class - “drugs that are not first-in-class but received a priority review designation, which is reserved for medicines that potentially offer major advances in treatment.” “Priority review suggests a measurable degree of innovation in the clinical potential of a drug."
addition-to-class - While additions-to-class represent “additional options for patients and clinicians and may possess unique benefits and value for individual patients…they generally represent a lower degree of innovation because at the time of FDA review, they did not distinguish themselves in terms of potential clinical benefit."
Based on FDA’s research of products approved from 1987-2011 (N = 645 products), 203 (32 percent) were first-in-class drugs, 143 (22 percent) were advances-in-class, and 299 (46 percent) were addition-to-class. These nuanced categories provided by FDA help to better articulate
innovation. This work was instrumental for the thesis project in terms of determining to omit a definition of innovation or attempting to redefine innovation given the state of flux in the
literature about the term innovation and how it should be applied to pharmaceutical development. Additionally, this work helped to identify recommendations for future research.
A study by Cohen reviewed a host of innovation definitions - incremental/sequential innovation vs. pioneering innovations - and discussed the importance of incremental innovations. Cohen found that from 1990-2003, CDER approved 1,171 NDAs; 400 of which were NMEs (34%) and 771 (66%) were non-NMEs. Of the NMEs, 166 (41%) were granted priority review compared with 98 (12%) of non-NMEs granted priority review. He also noted a pronounced decline in recent NME filings in CDER, 50 in 1995 to 24 in 2003. Cohen stated that priority review is a key identifier of innovation because it “provides a sense of the FDA’s judgement of the value of a new drug or use as it relates to previously approved drugs at the time of NDA filing.” Cohen’s research demonstrates the importance of all types of innovation, including incremental
innovation, which lends to the rational to include all NMEs for this thesis research project. Lastly, he noted that “there are no generally accepted measures of innovation that would
conclusively prove” whether a product is innovative or not. Again, this reinforces the decision to omit a specific definition of innovation.
Kaitin and DiMasi’s study “Pharmaceutical Innovation in the 21st Century: New Drug Approvals
for the First Decade, 2000-2009”57 is another key research project that informed this thesis project. The intent of the research was to examine the biopharmaceutical industry’s performance to “assess how companies fared against the myriad economic and political challenges facing the industry” and “measure industry’s future success in bringing new pharmaceutical and
biopharmaceutical products to the market.” 58 Their methodology included an analysis of FDA
approval rates, clinical development durations and regulatory approval phase durations for new drugs and biologics approved in the US from 1980-2009 by reviewing NMEs from original NDAs and therapeutic biologics. The results of their study identified several trends in drug development including changes in clinical and regulatory durations for orphan versus non-orphan designated products, among many others. The research helped to inform the thesis project in terms of the use of clinical development duration and regulatory review as the key measurement.
Additionally, the study helped to inform the methodology and statistical analysis, for example year-to-year comparisons, among others.
The last research article that formed the basis of the thesis project is Kesselheim and Avorn’s “The most transformative drugs of the past 25 years: a survey of physicians.” In this work, the authors discussed the lack of “consensus over what characterizes an innovative drug.” To overcome this, they “conducted an extensive survey of over 180 expert physicians on 646 NMEs and new biological products approved from 1985-2009. The results of their survey indicated that “physicians from many fields appear to hold the greatest regard for highly efficacious new drug classes that address previously unmet medical need.” The study concluded that so-called “transformative drugs” are most often perceived by expert clinicians to be those that “establish new classes and have efficacy that substantially surpasses that of existing alternatives.” In all, this definition of transformative drugs aligns in some ways with two of FDA’s categories of innovation (first-in-class and advance-in-class). Additionally, the research helped to confirm that the thesis project should not focus on defining (or re-defining) innovation per se but utilize FDA’s own thinking regarding innovation such that NME approvals (especially those that utilize an expedited pathway) inherently include at least one of three levels of innovation (first-in-class; advance-in-class; or addition-in-class).
C. Thesis Project
As summarized in Table 2. (next page) there are several important academic works that formed the basis of the design, methodology and statistical analysis of the thesis project. While the thesis builds from the above referenced research in many ways, there are significant differences with the cited literature. For example, the thesis project will utilize the clinical development and
regulatory review time as the key measurement to determine whether FDA’s expedited programs meet their stated objective – to speed drug development for identified drug products. By utilizing this measurement, the research project will be able to definitively measure programs designed to speed these activities. Further, the thesis project will rely on FDA’s decision to approve an NME (especially those that utilize an expedited pathway) to define innovation instead of attempting to re-define innovation. Moreover, the thesis project will focus only on FDA’s expedited programs (accelerated approval, breakthrough therapy, fast track and priority review) rather than broadly
analyzing “facilitated pathways.” Importantly, facilitated pathways are a broader population of regulatory programs.
Table 2. FDA Regulatory Program – Previous Research Statistics
Author(s) Years Studied NMEs Catalogued Measurement
Cohen (2005) 1990-2013 400 Number of approvals;
number of NMEs; number of products in expedited programs
Munos (2009) 1950-2008 1,222 Innovation related to
company/sponsors
Kaitin & DiMasi (2011) 1980-2009 N/A FDA approval rates, clinical
development times, and regulatory approval times
FDA (2013) 1987-2011 645 Number of approvals;
categories of innovation; company/sponsor size and revenue
Darrow & Kesselheim (2014)
1987-2013 778 Frequency of expedited program use
The thesis project is focused on FDA’s expedited programs; orphan designation and QIDP designations are not specifically defined as “expedited programs” by the Agency.59 FDA’s
orphan drug designation is used to identify treatments for rare diseases (as defined by affecting less than 200,000 patients). While orphan drug designation is included in the thesis project as a control in some of the statistical analyses, it is not considered by FDA as an expedited pathway and is thus not evaluated as an expedited pathway. Additionally, the QIDP designation is only available for antibacterial and anti-fungal drug products. Therefore, QIDP-designated products are not included in the study.
Lastly, the thesis project will be the first to include a complete analysis of FDA’s four expedited pathways. Due to the timing of legislative action regarding breakthrough therapy (enacted in 2012), the previous literature did not have the opportunity to review products developed and approved under the breakthrough therapy designation.
IV. METHODOLOGY
The thesis project’s preliminary data set was obtained from a variety of sources, including product labels and summary basis of approval documents available from Drugs@FDA;
information available at FDA T.R.A.C.K. (Transparency, Results, Accountability, Credibility and Knowledge-Sharing) database; information obtained from the Washington University in St. Louis Center for Research Innovation in Biotechnology database60, Darrow and Kesselheim’s database in the New England Medical Journal (NEMJ)61, and other literature articles (notably Nature Reviews Drug Discovery).
The process for collecting data from each database and literature articles began with searching for each of product approved by FDA from 1987-2015 using the following three databases:
1. U.S. Food and Drug Administration maintains Drugs@FDA: a database that is publicly available and stores a variety of information about drugs and biological products approved by the Agency. For example, the database includes the drug’s commercial and non-proprietary name, its sponsor/manufacturer, FDA application number, date of approval, the chemical type, and its review classification. The database also includes links to its label information, its approval history, correspondence (between FDA and sponsor), and FDA review
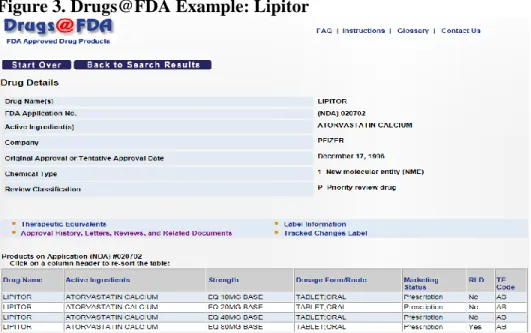
documents (e.g., summary basis of approval). Figure 3 (next page) contains an example from Drugs@FDA.
2. Washington University in St. Louis Center for Research Innovation in Biotechnology
(WUSTL CRIB): based on Dr. Michael Kinch’s work to catalogue biopharmaceuticals in the
United States (including approvals prior to the creation of FDA). The database stores over 1,724 drug products from over 1,232 biopharmaceutical companies. The database includes several meta data for each product, including commercial and nonproprietary name, chemical type, indication, target, approval date, patent information, and development milestones (if available). See Figure 4 for an example of the available information for Lipitor.
Figure 3. Drugs@FDA Example: Lipitor
Figure 4. WUSTL CRIB Example: Lipitor
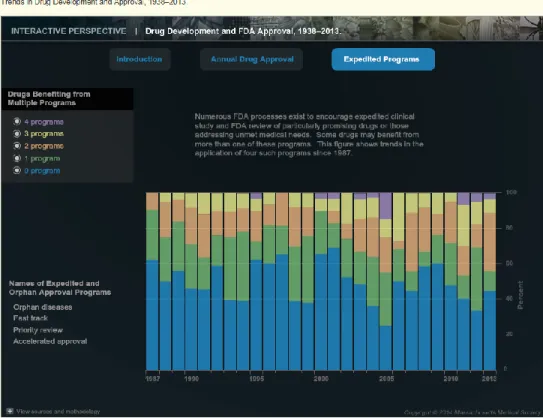
3. Darrow and Kesselheim’s Trends in Drug Development and Approval, 1938-2013: The
research conducted by Darrow and Kesselheim resulted in a useful database that captures FDA approvals from 1938-2013 (See Figure 5 for a representation of the database). The database also captures approvals for each of the following expedited and special FDA pathways: accelerated approval, fast track, orphan drugs and priority reviews. The database was used mainly to capture approvals for accelerated approval and fast track that were not captured in Drugs@FDA.
Information from the above referenced data sources and other literature articles was combined into a database that identified the following key attributes for each product: active ingredient,
brand name, IND submission date (if available), NDA approval date, therapy area, sponsor name orphan designation, and the expedited pathway(s) (if applicable). For this project, the sponsor name identified was the company that was most involved in the development and
commercialization of the product (as denoted in FDA summary basis of approvals and product labels). The development time was calculated from the date of the IND submission and the NDA approval date.
Figure 5. Darrow and Kesselheim Interactive Graphic
A. Descriptive Categories
The following coding systems were used to identify descriptive categories to facilitate the statistical analyses described in Subsection B:
1. Approval Year
A number 1-4 was denoted for each product based on the year of approval per the following groupings:
1 - 1987- 2001 2 –2002-2007 3 - 2008-2012 4 – 2013-2015
These year groupings were chosen to align with the enactment of PDUFA laws, as follows: 1987-2001 (pre-PDUFA, PDUFA I, and PDUFA II); 2002-2007 (PDUFA III); and 2008-2012
(PDUFA IV); and 2013-2015 (PDUFA V). The purpose of grouping the approvals in alignment with PDUFA enactment dates is to potentially derive analyses that may identify with specific policies and/or processes implemented as part of the PDUFAs.
2. Expedited Program
A number 1 was inputted for each expedited pathway along with a column to identify the total number of expedited pathways for each product. For example, a product that was approved under the standard development process and standard review would receive a “0” while a product approved under all four expedited programs would receive a “4.”
A separate coding system was used to identify each combination of expedited programs a product could achieve, as follows:
1. Accelerated Approval (A) 2. Breakthrough Therapy (B) 3. Fast Track (F)
4. Priority Review (P) 5. Accelerated approval and
Breakthrough Therapy (AB)
6. Accelerated Approval and Fast Track (AF)
7. Accelerated Approval and Priority Review (AP)
8. Breakthrough Therapy and Fast Track (BF)
9. Breakthrough Therapy and Priority Review (BP)
10. Fast Track and Priority Review (FP) 11. Accelerated Approval, Breakthrough
Therapy and Fast Track (ABF) 12. Accelerated Approval, Breakthrough
Therapy and Priority Review (ABP) 13. Breakthrough Therapy, Fast Track
and Priority Review (BFP) 14. Accelerated Approval, Fast Track
and Priority Review (AFP)
15. Accelerated approval, Breakthrough Therapy, Fast Track and Priority Review (ABFP)
3. Therapeutic Area
1 – Analgesics 2 – Anesthetics 3 – Bone 4 – Cardiovascular 5 – Dermatology 6 – Digestive 7 – Endocrine 8 – Immunologic 9 – Infectious 10 – Metabolic 11 – Neurologic 12 – Oncology 13 – Psychiatry 14 – Ophthalmology 15 – Reproduction 16 – Respiratory 17 – Rheumatology 18 – Sleeping 19 - Urological
To provide an even more granular review of the data, statistical analyses will also be run that will include two or more descriptive groups. For example, analyses that look at expedited program approval time across the chose time periods will be examined. Additionally, specific therapeutic areas will be analyzed to determine if expedited programs perform differently in infectious disease versus bone (for example).
B. Study Data Universe and Sample Size
Based on the identified methodology, 796 drug products were initially catalogued as NMEs approved from 1987-2015. The data used for the study included only novel drugs and therapeutic biologics approved by FDA’s CDER from 1987-2014, excluding imaging and contrasting agents, radiopharmaceuticals, generic drugs, or supplemental applications for drugs or biologics.
Additionally, other products under FDA’s authority were considered out of scope and not included in the study such as dietary supplements, vaccines, diagnostic products, and blood and blood components. Lastly, products were removed where IND approval dates and NDA approval dates did not align to the same initial approval indication.
The sample population was derived from the broader dataset by choosing those products with IND submission and approval date information. As such, a total of 303 products were chosen for further analysis. Of these, the products with the fastest and longest study durations were removed from the sample population to omit potential outliers. Further, single-item categories derived from the descriptive categories for Approval Year, Therapeutic Category, and Expedited Program were eliminated due to statistical software limitations.
V. STATISTICAL ANALYSIS
For the samples identified in each descriptive category (as previously defined), the thesis project compared the sample populations to one another via t-test, ANOVA, and subsequent comparative analyses to obtain an answer to the thesis question.
All statistical analyses referenced in the review of analysis and conclusions are listed in tables in Section IX. Appendix, which depict the results of the t-test, ANOVA and/or comparative
analyses of the expedited programs with respect to the following:
Subsection 1: Years of approval
Subsection 2: Expedited programs direct comparison
Subsection 3: Combinations of expedited programs
Subsection 4: Expedited programs and therapeutic areas
VI. REVIEW OF ANALYSIS A. Years of Approval
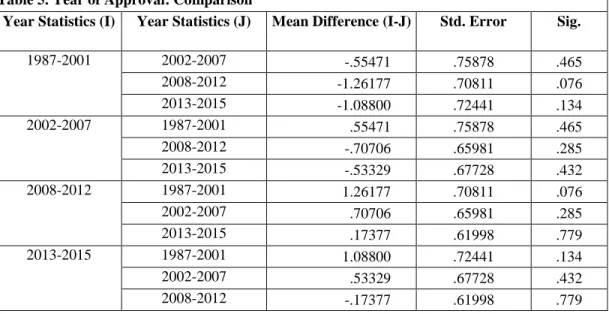
On average, little differences were observed between the different year grouping in terms of review times when looking at both standard and expedited programs in the sample. As indicated in Table 4, the range of development and regulatory review times is from 6.90-8.16 years. The variance in terms of means was not statistically significant. Interestingly, the development times were lower in the first year grouping (1987-2001), which are aligned to pre-PDUFA, PDUFA I, and PDUFA II. When one-to-one analyses were conducted, group one (1987-2001) was 1.26 years faster than year group three (2008-2012), with statistical significance of 0.076.
In order to take a refined review of the year periods, specific expedited programs were isolated and reviewed within each period to attempt to identify any trends. For time period one (1987-2001), the results were not statistically significant but were aligned to what was seen in the total program grouping where those programs approved under the expedited program were observed to have shorter development and review times (See Table 7). In time period two (2002-2007), some variance in the mean years begin to emerge with accelerated approval mimicking its shorter
development and review duration when compared to other programs (See Table 9), which is discussed further in subsection C. Moreover, year grouping two also begins to demonstrate the longer duration of times associated with fast track combinations (even in combination with other expedited programs) that are also discussed in more detail in subsection B (See Tables 27-28), although these figures are marginally significant (significance = 0.054).
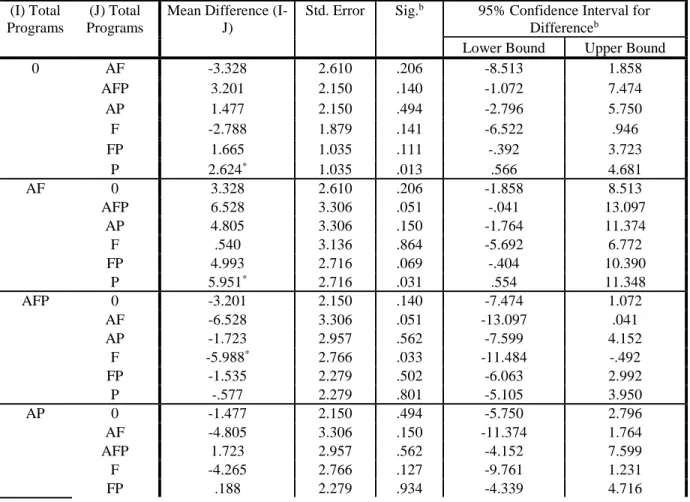
Year group three (2008-2012) demonstrated similar results where programs with accelerated approval programs (AFP and AP) demonstrated shorter development and review times as compared to other programs (statistical significance = 0.025; See Tables 11-12). Products approved with priority review alone were 2.6 years faster than standard development and review programs (statistical significance = 0.013; See Table 13). Fast Track programs (F) were 2.8 mean years slower than programs approved under standard development and review. The results are aligned with other delays observed with products approved with the fast track designation in this time period and other observations that are discussed in subsection B.
Lastly, time period four (2013-2015) demonstrated similar mean differences as observed in other year groupings such as faster development and regulatory review for products approved with accelerated approval (and in combinations ABFP, ABP, and AFP) as well as products developed and approved with the breakthrough therapy designation (e.g., BP) (reminder: this is the only time period where breakthrough is available) (See Table 14-15). Notably, products approved under all four expedited programs (ABFP) were 7.2 years faster than products with breakthrough, fast track and priority review (BFP) (significance: 0.02) and products with accelerated approval, breakthrough and priority (ABP) were 4.8 years faster than BFP products (significance: 0.04). However, most of the statistics for this year grouping were marginally significant.
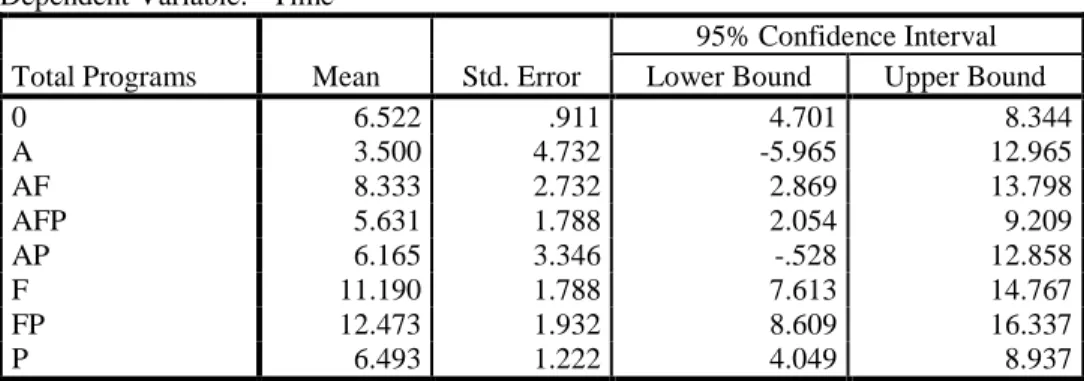
B. Number of Expedited Programs
The next set of descriptives used to analyze the sample population was the number of expedited programs for each product. With regard to the number of expedited programs per product, products with three expedited program designations were identified as having a 1.5 year faster development and regulatory review time that having no expedited program at all (significance of 0.081; See Table 18).
While not statistically significant, there is a clear difference in the mean average in terms of the number of years it takes to move through clinical development and regulatory review for products that do not have any expedited program designations versus those with one, two, three or four designations (See Table 18). This would be expected given the increased touchpoints, FDA oversight, and communication between sponsors and FDA when a product receives a designation for an expedited pathway.
C. Expedited Programs
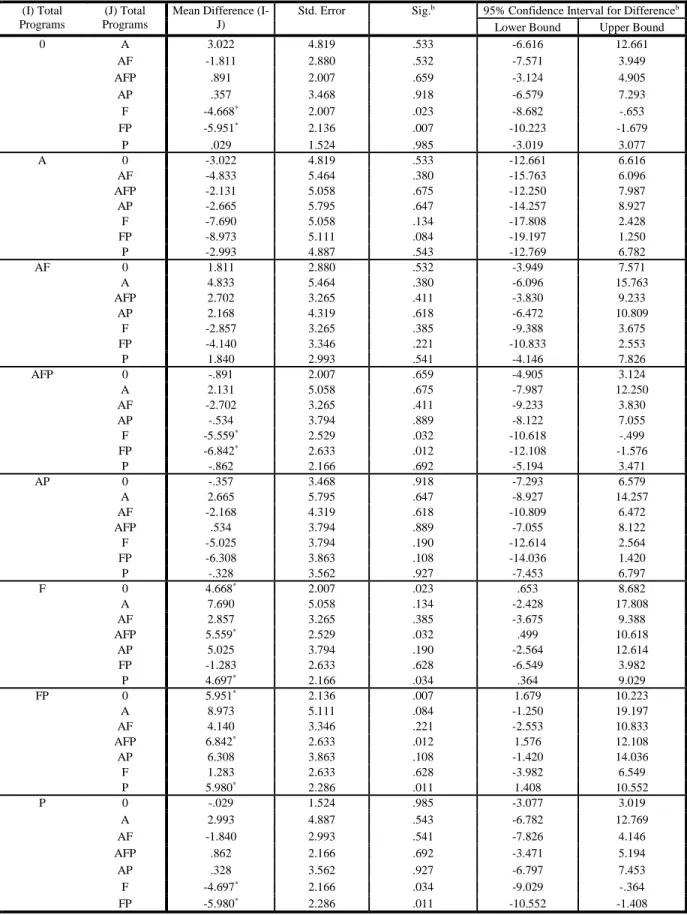
In this study, products that receive combinations of expedited programs fared much better in terms of shorter development and regulatory review time than standard reviews. Product
programs that received all four expedited pathway designations (N=2) were associated with 6.24 year decrease in development and review time than products that receive Fast Track designation alone (significance = 0.046; See Table 29c).
As expected, products developed and reviewed under combinations of expedited programs fared well against standard development and review (SDR). For example, product programs that received accelerated approval, fast track designation and priority review were associated with a statistically significant decrease in development and regulatory review time of 2.42 years (significance = 0.03) versus a standard review program and 4.54 years faster than products with Fast Track (See Table 29c).
Nearly as fast were those products that received breakthrough therapy and priority review (BP), which were 2.37 years shorter than SDR products (significance = 0.168; See Table 29b). Notably, programs with breakthrough therapy and priority review were 4.48 years faster than those programs with fast track (significance = 0.026; statistically significant) and 2.58 years faster than products with fast track and priority review (significance = 0.163; not statistically significant; See Table 29b).
The study demonstrated that products developed under the accelerated approval pathway will decrease development and review time by 1.4 years (significance of 0.045; See Tables 19-20). While the use of the accelerated approval pathway has waned over time62, this study demonstrates
that it is still a viable regulatory pathway that decreased development and regulatory review by nearly one and half years.
In similar fashion to the accelerated approval pathway, the study demonstrated that the products with breakthrough therapy designation were associated with a 1.3 year decrease in development and regulatory review time as compared to a product with no expedited designations (0.197 significance; See Tables 21-22). While this figure is not statistically significant, it does demonstrate some evidence that breakthrough designation offers some utility versus a product with no expedited designations. This is expected given the increased senior management involvement with products designated as breakthrough therapies.
Product programs that receive priority review alone (See Table 29a) demonstrated a decrease in development and review time of 2.59 years versus Fast Track designation alone (significance = 0.034).
On the other side of the spectrum, Fast Track designation is associated with a slower clinical development and regulatory review time versus a product with standard review (8.11 years vs. 7.61 years; 0.389 significance; See Tables 23-24). While the figure is not statistically significant, it is still remarkable that a program designed to decrease development time would be even
minimally associated with increased times.
D. Therapeutic Areas
The final descriptive grouping was based on therapeutic area. The mean development and regulatory review time for therapy areas ranged from 5.42 years to 11.20 years (See Table 30). Of note, products in psychiatry (5.62 years), infectious disease (6.69 years), and cardiovascular (6.80 years) and immunology (6.90 years) were notably faster than other therapy areas.
One-to-one therapy area analyses draw out more concrete analysis (See Table 31).3 For example, infectious disease products were 2.1 years faster than neurology products (significance = 0.038), and 3.18 years faster than ophthalmology products (significance = 0.047). On the other side of the spectrum, bone drugs were notably slower in terms of development and regulatory review
times when compared to cardiovascular (+4.39 years; significance = 0.028), immunology (+4.29 years; significance = 0.039), infectious disease (+4.5 years; significance = 0.022) and psychiatry (+5.57 years; significance = 0.028).
Additional analyses were conducted that isolated the specific expedited programs used to approve products within therapy areas. For example, two products for bone indications were approved under the standard development and review pathway while two were approved using fast track and priority review and one was approved under priority review. Of these, the products approved under the fast track and priority review programs were much slower (18.96 years; See Tables 32-33). While these are not statistically significant, it does offer some insight into the delays
associated with fast track that could be evaluated further.
In terms of statistically significant findings with regard to expedited programs approved for specific therapy areas, the following were observed in the infectious disease therapy area (See Table 34):
Products with ABFP were associated with a 13.16 year shorter development and review time when compared to products approved with BFP (significance = 0.008)
Products with AFP were associated with a 10.94 year shorter development and review time versus products approved with BFP (significance = 0.013).
Products with AP were associated with a 10.5 year shorter development and review time versus products with BFP (significance = 0.004)
VII. CONCLUSIONS A. Thesis Conclusions
The thesis project sought to prove the success of FDA’s expedited pathways based on the
measurement of development and regulatory review time. The thesis project demonstrated mixed results with respect to the four groupings chosen to analyze the expedited programs: year of approval, number of expedited programs, type of expedited program and therapeutic area. The years of approval grouping demonstrated a notable finding in that the development and regulatory review time actually increased since 1987, important because the FDA and industry have worked to refine the development and regulatory review processes since 1992, with Congress