UNIVERSIDADE DO ALGARVE
Sistemas Conjugados: Importância na
ação e potenciação de fármacos.
Nuno Alexandre Guerreiro Alves
Dissertação
Mestrado Integrado em Ciências Farmacêuticas
Trabalho efetuado sob a orientação de: Prof.º Dr.º Am adeu Ferna ndes Bri gas
Sistemas Conjugados: Importância na
ação e potenciação de fármacos.
Declaração de autoria de trabalho.
Declaro ser o autor deste trabalho, que é original e inédito. Autores e trabalhos consultados estão devidamente citados no texto e constam na listagem de referências incluída.
_________________________________ (Nuno Alexandre Guerreiro Alves)
Copyright © 2012 por Nuno Alexandre Guerreiro Alves
A Universidade do Algarve tem o direito, perpétuo e sem limites geográficos, de arquivar e publicitar este trabalho através de exemplares impressos reproduzidos em papel ou de forma digital, ou por qualquer outro meio conhecido ou que venha a ser inventado, de o divulgar através de repositórios científicos e de admitir a sua cópia e distribuição com objetivos educacionais ou de investigação, não comerciais, desde que seja dado crédito ao seu autor e editor.
Ao avô Acácio e à avó Margarida, sempre Aos meus pais Ao resto da família
“On bended knees is no way to be free…” Eddie Vedder
i
Agradecimentos
Este espaço é dedicado a todos que, de uma forma ou de outra, contribuíram para que este trabalho fosse projetado e realizado. A todos os meus mais sinceros agradecimentos.
Em primeiro lugar, ao Prof. Dr. Amadeu Brigas, meu orientador, pela total disponibilidade que apresentou para me orientar e ajudar, não só ao longo da realização deste trabalho, mas também ao longo da minha formação académica. Foi, efetivamente, um prazer trabalhar sob a sua orientação.
Ao Prof. Américo Lemos, por ter tido a sorte que comigo partilhasse diversos conhecimentos práticos e teóricos a nível laboratorial.
A todos os restantes professores das áreas de Química, Química Orgânica e, principalmente, Química Orgânica Farmacêutica por me terem ajudado a encontrar o lugar onde melhor me encaixo no seio das Ciências Farmacêuticas.
A todos os meus colegas e amigos, principalmente o Marcos Torrado e o Cesar Costa, por terem marcado e por me terem acompanhado ao longo de todo este percurso.
A toda a minha família, não só pelo apoio que me deram mas também por todos os sacrifícios que fizeram, sendo eles a razão de aqui ter chegado.
Por fim, a toda a Universidade do Algarve tal como a toda a sua estrutura, pelas condições de trabalho e pelo peso e significado que têm tanto na minha formação académica como cívica.
ii
Resumo
Os sistemas conjugados apresentam uma reatividade própria devido à deslocalização das suas ligações. Esta reatividade torna-os importantes não só ao nível da indústria química mas também em questões farmacológicas e toxicológicas. Existem bastantes fármacos que exercem a sua função farmacológica devido à capacidade alquilante inerente aos seus sistemas conjugados, podendo a alquilação dar-se por uma reação tipo-Michael ou por uma substituição nucleofílica assistida por um sistema conjugado, como ocorre com compostos com sistemas alílicos. Muitos fármacos citotóxicos ou antibióticos atuam por um mecanismo de alquilação, seja pela alquilação do DNA ou pela modificação covalente de enzimas e outras proteínas. Estas alquilações interferem com os processos celulares levando à apoptose da célula.
Por outro lado, existem muitos fenómenos de toxicidade que têm vindo a ser associados ao desenvolvimento de diversas doenças, que ocorrem também devido à alquilação de proteínas e do DNA por moléculas com um sistema conjugado na sua estrutura. Estas alquilações podem levar a respostas imunológicas e à diminuição de função de algumas proteínas endógenas, por alterarem a estrutura destas. No caso da alquilação do DNA, podem provocar mutações que podem causar um descontrolo do ciclo celular dando origem ao desenvolvimento de uma neoplasia.
O conhecimento dos mecanismos pelo qual os fármacos e outras moléculas com atividade biológica contendo sistemas conjugados na sua estrutura atuam, a nível endógeno, é bastante relevante tanto na compreensão de determinadas patologias, como na criação de novas moléculas com uma atividade farmacológica melhorada.
Palavras-chave: Sistemas conjugados, Agentes alquilantes, Citotóxicos, Antibióticos, Moléculas tóxicas, Potenciação de fármacos.
iii
Abstract
Conjugated systems show a particular reactivity due to their bond delocalization. This reactivity makes them important not only in chemical industry but also in pharmacological and toxicological issues. There are plenty of drugs that exert their pharmacological activity due to their alkylating ability inherent to their conjugated systems, which can give the alkylation by a Michael-type reaction or a nucleophilic substitution assisted by a conjugated system, as occurs with compounds with an allylic systems. Many cytotoxic drugs or antibiotics act by an alkylation mechanism, being this mechanism the alkylation of DNA or covalent modification of enzymes and other proteins. These alkylations interfere with cellular processes leading to apoptosis of the cell.
Moreover, there are many mechanisms of toxicity that have been associated with the development of various diseases, which occur also due to alkylation of DNA and protein molecules by molecules having a conjugated system in their structure. These alkylations lead to immunologic responses and decrease of the function of some endogenous proteins, by altering their structure. In the case of alkylation of DNA, mutations may occur which may cause an uncontrolled cell cycle, leading to the development of a tumor.
The knowledge of the mechanisms by which drugs, and other biologically active molecules with a conjugated system in its structure, acts at an endogenous level, may become quite important both in understanding certain pathologies, and at the creation of new molecules with an improved pharmacological activity.
Keywords: Conjugated systems, Alkylating agents, Cytotoxics drugs, Antibiotics, Toxic molecules, Drugs potentiation.
iv
Índice
Agradecimentos ... i
Resumo ... ii
Abstract ... iii
I. Introdução aos Sistemas Conjugados ... 1
II. Adições a sistemas alílicos ... 7
II.1. Alilmetanosulfonato e Feniletilmetanosulfonato ... 8
II.2. Isopreno ... 10
II.3. Mitomicina C ... 13
II.4. Tamoxifeno ... 18
III. Adição tipo-Michael ... 21
III.1. Acroleína ... 25
III.2. Derivados da peroxidação de ácidos gordos insaturados ... 33
III.2.1. 4-hidroxi-2-nonenal ... 35
III.2.2. 4-oxo-2-enal ... 39
III.3. cis-2-butene-1,4-dial ... 41
III.4. Terpenóides ... 44
III.4.1. Lactonas diterpénicas ... 45
III.4.1.1. Jolkinolido D ... 46
III.4.2. Lactonas Sesquiterpénicas ... 48
III.4.2.1. Iludina S e Irofulveno ... 49
III.4.2.2. Santamarina ... 51
III.4.2.3. Helenalina ... 52
III.4.2.4. Partenolido ... 53
III.4.2.5. Cnicina e Cinaropicrina ... 54
III.5. Hipotemicina ... 55
III.6. 6-hidroximetil-1H,3H-pirrolo[1,2-c]tiazol ... 56
IV. Outros fármacos com sistemas conjugados ... 57
IV.1. Anfotericina B ... 57
IV.2. Enediínas ... 59
v
V. Discussão/Potenciação e desenvolvimento de fármacos ... 62
V.1. Mitomicina C ... 65 V.2. Bases azotadas ... 66 V.3. Lactonas sesquiterpénicas ... 70 V.3.1. Helenalina ... 73 V.4. Pró-fármaco de enediínas ... 74 V.5. Esteroides ... 76 V.6. Espaçadores multiconjugados ... 78 VI. Conclusão ... 82 VII. Bibliografia ... 83
vi Índice de Estruturas
Estrutura I-1 - Estruturas de ressonância do butadieno. ... 1
Estrutura I-2 - Estrutura molecular da cisplatina 2 e da ciclofosfamida 3. ... 4
Estrutura II-1 - Activação metabólica do Isopreno a IP-1,2-O e IP-3,4-O. ... 11
Estrutura II-2 - Estrutura molécula da mitomicina C. ... 15
Estrutura II-3 - Estrutura molecular do toremifeno. ... 20
Estrutura III-1 - Estrutura molecular da acroleína. ... 25
Estrutura III-2 – Estrutura molecular do 2-propen-1-ol. ... 26
Estrutura III-3 – Aductos formados in vitro entre a acroleína e desoxiadenosina. ... 26
Estrutura III-4 – Estrutura dos aductos maioritários da reacção da acroleína com a desoxitimidina. ... 28
Estrutura III-5 – Cross-link formado entre a acroleína e duas desoxiguanosinas. ... 30
Estrutura III-6 – Estrutura molecular do 4-hidroxi-2-nonenal. ... 35
Estrutura III-7 – Cross-link formado pelo 4-hidroxi-2-nonenal e duas desoxiguanosinas. ... 37
Estrutura III-8 – Histidina 33, cisteina 30 e lisina 34. ... 38
Estrutura III-9 –Estrutura do cross-link formado entre o 4-hidroxi-2-nonenal, uma lisina e uma histidina. ... 38
Estrutura III-10 – Estrutura molecular do 4-oxo-2-enal. ... 40
Estrutura III-11 – Estrutura dos aductos exocíclicos formados pelo 4-oxo-2-enal e a desoxiguanosina. ... 40
Estrutura III-12 – 1,N2-biciclo 2´-deoxiguanosina. ... 42
Estrutura III-13 – Cisteína 30 e Lisina 34. ... 44
Estrutura III-14 – Estrutura molecular do isopreno. ... 45
Estrutura III-15 – Estrutura do aducto duplo formado por duas moléculas de jolkinolido D e uma alanina. ... 47
Estrutura III-16 – Estruturas moleculares de α-metileno-γ-lactona 41, butenolido 42, ciclopentenona 43. ... 48
Estrutura III-17 – Éster conjugado, ou alílico. ... 49
Estrutura III-18 – Estrutura molecular do irofulveno. ... 51
Estrutura III-19 – Estrutura molecular da santamarina. ... 52
Estrutura III-20 – Estrutura molecular da helenalina. ... 53
vii
Estrutura III-22 – Cnicina 49 e Cinaropicrina 50. ... 55
Estrutura IV-1 – Estrutura molecular da anfotericina B. ... 58
Estrutura IV-2 – Estrutura molecular do ergosterol 54 e do colesterol 55. ... 58
Estrutura IV-3 – Estrutura molecular do ácido jacárico. ... 61
Estrutura IV-4 – Estrutura geral de um hidroperóxido de um ácido triénico conjugado. ... 61
Estrutura V-1 – Estrutura molecular do 5-fluoruracilo. ... 67
Estrutura V-2 – Análogo da alquilante da timina com nucleofugo em posição alílica. . 68
Estrutura V-3 - Análogo alquilante da guanina com nucleofugo em posição alílica. .... 68
Estrutura V-4 - Análogo alquilante da guanina com um grupo aceitador de Michael exocíclico. ... 69
Estrutura V-5 – Estrutura molecular da timina. ... 69
Estrutura V-6 – Molécula análoga da timina com função alquilante. ... 70
Estrutura V-7 – Alteração do farmacóforo da santamarina 46, uma lactona sesquiterpénica, por aumento das dimensões do sistema conjugado... 71
Estrutura V-8 – Derivado da helenalina. ... 74
Estrutura V-9 – Estrutura molecular de um pró-fármaco de uma enediina. ... 75
Estrutura V-10 – Estrutura do ciclo-pentano-peridro-fenantreno... 76
Estrutura V-11 – Estrutura de um derivado de um esteroide, o colesterol, com capacidade alquilante. ... 77
Estrutura V-12 – Estrutura de um potencial fármaco híbrido de dois alquilantes de DNA unidos por um espaçador multiconjugado. ... 78
Estrutura V-13 - Estrutura de um hidroperóxido de um potencial fármaco híbrido de dois alquilantes de DNA unidos por um espaçador multiconjugado. ... 80
viii
Índice de Mecanismos
Mecanismo II-1 – Reacção (a) SN1 e (b) SN2 de um sistema alílico. ... 7
Mecanismo II-2 – Reacção tipo SN1’ em sistemas alílicos ... 7
Mecanismo II-3 - Reacção tipo SN2’ em sistemas alílicos. ... 8
Mecanismo II-4 - Alquilação pelo alilmetanosulfato por mecanismo Sn2’. ... 9
Mecanismo II-5 – Alquilação pelo alilmetanosulfato por mecanismoSN2. ... 9
Mecanismo II-6 – Alquilação pelo feniletilmetanosulfonato. ... 10
Mecanismo II-7 – Hidrólise do IP-1,2-O. ... 12
Mecanismo II-8 – Formação da desoxiinosina. ... 13
Mecanismo II-9- Mecanismo de alquilação de DNA por parte da mitomicina C. ... 16
Mecanismo II-10 - Mecanismo de formação de aductos do tamoxifeno com o DNA. .. 19
Mecanismo II-11 – Conversão do α-hidroxitamoxifeno. ... 19
Mecanismo III-1 – Adição de Michael. ... 21
Mecanismo III-2 – Reacção tipo-Michael. ... 23
Mecanismo III-3 – Formação de uma base de Schiff. ... 24
Mecanismo III-4 – Mecanismo de formação do aducto 18 da Estrutura (Adaptado de Pawłowicz & Munter, 2006) ... 27
Mecanismo III-5 - Mecanismo de formação dos aductos maioritários entre a acroleína e a desoxitimidina (Adaptado de Pawłowicz & Kronberg, 2008) ... 29
Mecanismo III-6 – Formação do aducto exociclíco entre a acroleína e a desoxiguanosina. ... 29
Mecanismo III-7 – Aductos formados entre a acroleina e a desoxicitidina. ... 31
Mecanismo III-8 – Formação de aducto entre acroleina e desoxicitidina contendo dois exocíclos. ... 31
Mecanismo III-9 – Formação de aducto entre a acroleína e cisteína. ... 32
Mecanismo III-10 – Activação metabólica da ciclofosfamida a fosforamida e acroleína. ... 32
Mecanismo III-11 – Passo de iniciação da peroxidação lípica. ... 33
Mecanismo III-12 – Primeiro passo de propagação da peroxidação lípida. ... 34
Mecanismo III-13 – Segundo passo de propagação da peroxidação lípida. ... 34
Mecanismo III-14 – Formação de um α,β – aldeído insaturado a partir de um hidroperóxido lípidico. ... 34
ix
Mecanismo III-15 – Formação de aducto entre o 4-hidroxi-2-nonenal e a desoxiguanosina. ... 36 Mecanismo III-16 - Oxidação do furano a cis-2-Butene-1,4-dial. ... 41 Mecanismo III-17 – Mecanismo de formação do 1,N2-biciclo 2´-deoxiguanosine (Adaptado de Vu & Peterson, 2005) ... 42 Mecanismo III-18 – Mecanismo de alquilação do jolkinolido D (Adaptado de Sakakura, Takayanagi, & Kigoshi, 2002) ... 47 Mecanismo III-19 – Mecanismo de acção da iludina S (Adaptado de McMorris & Chimmani, 2007) ... 50 Mecanismo III-20 – Mecanismo de inibição das cinases pela hipotemicina. (Adaptado de Barluenga & Jogireddy, 2010) ... 55 Mecanismo III-21 – Mecanismo de adição do 6-hidroximetil-1H,3H-pirrolo[1,2-c]tiazol ao DNA (Adaptado de Soares, Brito, & Laranjo, 2010) ... 56 Mecanismo IV-1 - Mecanismo de acção da C1027, uma enediina (Adaptado San Pedro, Beerman, & Greenberg, 2012) ... 60 Mecanismo V-1 - Mecanismo de acção de uma molécula derivada da mitomicina C. .. 66 Mecanismo V-2 – Mecanismo de acção potencial de uma lactona sesquiterpénica, baseada na estrutura da santamarina 46, dependente da redução de um anel de hidroquinona. ... 72 Mecanismo V-3 – Remoção dos diois protectores dos grupos carbonilo do anel de hidroquinona. ... 72 Mecanismo V-4 – Mecanismo de acção do pró-fármaco de enediina. ... 75 Mecanismo V-5 – Mecanismo de activação de um análogo esteroide a um composto com capacidade alquilante por um mecanismo tipo-Michael. ... 77 Mecanismo V-6 – Alquilação do DNA por um espaçador multiconjugado, através de um mecanismo SN1. ... 79
x
Índice de Figuras
Figura I-1 - Orbital de menor energia do benzeno. ... 2 Figura I-2 – Representação das orbitais de um catião alílico. ... 3 Figura II-1 - Orientação da mitomicina C no seio da dupla hélice de DNA. A seta aponta a desoxiguanosina onde se dá a primeira alquilação. (Adaptado de referência Tomasz, 1995) ... 17 Figura III-1 - Densidade electrónica da 3-butenona. ... 22
1
I. Introdução aos Sistemas Conjugados
Em grande parte das moléculas uma simples estrutura de Lewis permite descrever as ligações existentes na sua estrutura. No entanto existe um grupo restrito de moléculas em que isto não é suficiente. Este grupo é constituído pelas moléculas que apresentam ligações deslocalizadas, ou seja, em que uma ou mais orbitais ligantes não estão confinadas a apenas dois átomos, mas antes deslocalizadas ao longo de três ou mais átomos. Este facto encontra-se associados a moléculas que apresentam uma ou mais ligações π intercaladas com ligações σ, tanto quanto possível planares, podendo as moléculas ser cíclicas ou acíclicas e apresentar heteroátomos na sua constituição.
No caso destes compostos, pode entender-se a sua estrutura molecular como uma estrutura híbrida das suas formas canónicas, possíveis estruturas de Lewis da molécula, nunca sendo igual a qualquer uma destas estruturas. Esta representação estrutural da estrutura real como uma média, ponderada em função da estabilidade, das suas formas canónicas é chamada de ressonância. No entanto, é de referir que para escrever as estruturas de ressonância, ou canónicas, há que ter atenção a determinadas regras. (March & Smith, 2007)
O butadieno 1 é um composto com duas ligações π, cujos eletrões π se encontram deslocalizados sobre os quatro átomos de carbono da cadeia, podendo esta molécula ser utilizada para exemplificar a ressonância deste tipo de compostos (Estrutura I-1).
Estrutura I-1 - Estruturas de ressonância do butadieno.
Num cálculo da equação de onda, todas as estruturas de ressonância são contabilizadas conferindo assim uma energia molecular mais baixa, sendo que a diferença entre o resultado desta equação e o valor da estrutura de Lewis de mais baixa energia constitui a energia de ressonância. Este diferencial de energia quantifica a estabilidade extra conferida à molécula pela deslocalização das ligações. (Vollhardt & Schore, 2002)
2
No entanto, esta aproximação torna-se mais correta se for feita com base na teoria das orbitais moleculares. De facto, esta última teoria torna mais fácil a explicação da deslocalização das ligações. Olhando por esta prisma para o benzeno, molécula aromática logo com as ligações deslocalizadas, vemos que todos os átomos de carbono da molécula apresentam uma configuração sp2, usando estas orbitais para estabelecer ligações σ, fazendo com que todos os átomos da molécula se encontrem no mesmo plano. Cada carbono possui ainda uma orbital p, contendo um eletrão. Cada uma destas orbitais estabelece coalescência com as duas orbitais p dos átomos de carbono adjacentes. Esta coalescência forma seis novas orbitais, das quais três são ligantes e ocupam aproximadamente o mesmo espaço. A orbital de menor energia, apresentando apenas um nódulo no plano da molécula, é representada na Figura I-1. (March & Smith, 2007)
Figura I-1 - Orbital de menor energia do benzeno.
A existência desta deslocalização das ligações leva a que, para além de a molécula apresentar uma energia fundamental menor, as ligações apresentem um menor comprimento e que seja necessária mais energia para as romper. (Vollhardt & Schore, 2002)
Existem outros fatores que influenciam a deslocalização de carga, como a conformação da molécula. A deslocalização é tanto mais eficiente quanto mais planar for a molécula, pois esta conformação maximiza a coalescência entre as orbitais p, diminuindo assim a energia fundamental da molécula. Esta maior estabilidade faz com que estas moléculas com capacidade de deslocalização de carga, procurem apresentar uma estrutura planar ou o mais próximo possível disso. Muitas moléculas, por sua vez, vêm a sua deslocalização eletrónicas ser reduzida, ou mesmo impedida de se concretizar por apresentarem átomos que, devido a efeitos conformacionais ou estéreos. (Carey & Sundberg, 2007) (March & Smith, 2007)
3
Os sistemas conjugados, sistemas com ligações duplas ou triplas alternadas com ligações simples, encontram-se estritamente ligados a estes fenómenos de deslocalização das ligações, sendo esta extensamente verificada nestes sistemas. Tendo em conta que a deslocalização de ligações é verificada devido ao overlap entre as orbitais p dos átomos, esta é verificada em sistemas conjugados como o butadieno. Este tipo de deslocalização é verificado ainda em outros sistemas conjugados lineares como carbonilos α,β-insaturados, sistemas conjugados de três ou mais ligações duplas ou triplas, iminas α,β-insaturadas e compostos em que exista uma ligação dupla ou tripla conjugada com um anel aromático. Em termos de reatividades, sabe-se que compostos com extensas conjugações apresentam uma grande reatividade, polimerizando de forma relativamente fácil, especialmente na presença de eletrófilos. Esta reatividade é explicada pela facilidade que estes compostos têm em estabilizar cargas iónicas ou radicais, devido ao seu extenso sistema conjugado, que permite deslocalizar a carga por uma extensa estrutura. (Vollhardt & Schore, 2002)
A deslocalização de ligação ocorre também em sistemas alílicos, em que uma ponte dupla ou tripla se encontra conjugada com a orbital p de um átomo adjacente. Olhando novamente para as orbitais, é fácil perceber que esta conjugação com um átomo adjacente se pode estender por um sistema conjugado maior, caso as ligações múltiplas se encontrem conjugadas entre si. (March & Smith, 2007)
No caso especifico dos sistemas alílicos, o que mais interessa para este trabalho, é a reatividade destes compostos e o facto de estes poderem apresentar um radical, um anião ou um catião alílico, dependendo se a orbital p do átomo adjacente possui um, dois ou nenhum eletrão, bastante estáveis (Figura I-2).
Figura I-2 – Representação das orbitais de um catião alílico.
Existem ainda outros casos específicos de sistemas conjugados em que há uma deslocalização das ligações que por não apresentarem relevância considerável para este trabalho não serão abordados.
4
Os sistemas alílicos, devido à deslocalização eletrónica, para além de apresentarem uma energia mais baixa, apresentam ainda caraterísticas reacionais bastante diferentes dos seus hidrocarbonetos homólogos, nomeadamente ao nível de substituições nucleofílicas. (Vollhardt & Schore, 2002) (March & Smith, 2007)
Os sistemas conjugados são grupos funcionais de grande importância a nível terapêutico, podendo ser olhados por dois prismas diferentes. Se por um lado existem vários fármacos que têm num grupo conjugado o seu farmacóforo que lhe permite bloquear processos celulares através da alquilação do DNA ou enzimas, sendo neste trabalho abordados antibióticos e antineoplásicos, por outro existem vários xenobióticos que exercem a sua toxicidade a nível fisiológico por mecanismos associados também a grupos conjugados. É mostrado neste trabalho que, no caso dos agentes alquilantes do material genético ou de enzimas, devido a este seu mecanismo de ação, tanto podem atuar contra microorganismos infeciosos como contra células tumorais.
Estes compostos conjugados apresentam uma grande capacidade alquilante o que é muito importante em termos de citotoxicidade. De facto e apesar de o seu principal mecanismo de alquilação direta ser diferente, outras moléculas alquilantes como a cisplatina e a ciclofosfamida, uma mostarda nitrogenada (Estrutura I-2), são dos citotóxicos utilizados há mais tempo, devido à sua toxicidade para as células. (Lawley & Phillips, 1996) H3N Pt Cl Cl NH3 O NH P N CH2CH2Cl CH2CH2Cl O 2 3
Estrutura I-2 - Estrutura molecular da cisplatina 2 e da ciclofosfamida 3.
Em boa verdade, os citotóxicos e os xenobióticos tóxicos encontram-se estritamente ligados, não só por atuarem pelos mesmos mecanismos, mas por, muitas vezes, a diferença entre um antineoplásico e um agente mutagénico, se basear na sua concentração. Isto verifica-se no caso de alguns citotóxicos que apresentam atividade mutagénica, e em alguns xenobióticos mutagénicos que desencadeiam processos de apoptose celular, quando se encontram em grandes concentrações. (Hemminki, 1983)
5
(Esterbauer, 1993) Tanto num caso como outro, relaciona-se com um balanço entre as mutações exercidas e a capacidade da célula manter a sua viabilidade. (Hartl & Jones, 2006)
O estudo da ação de compostos tóxicos alquilantes representa um plano importante no estudo da Química Orgânica Farmacêutica. Se o conhecimento dos seus mecanismos permite uma melhor perceção de diversos processos patológicos, indispensável ao avanço da Farmacologia, este mesmo conhecimento também se pode apresentar como uma importante mais-valia no design ou potenciação de novos fármacos que atuem por alquilação de centros ativos de enzimas, proteínas ou material genético.
Sendo o objetivo deste trabalho explorar os mecanismos de reação entre fármacos e outros xenobióticos com grupos conjugados, levando em conta o paralelismo de ação entre estes e visando a potenciação e design de fármacos, no trabalho as moléculas serão apresentadas de acordo com o seu mecanismo de alquilação.
Não descurando a importância do desenvolvimento de novos e mais potentes antibióticos dado o crescente aparecimento de estirpes de microorganismos resistentes às terapias atualmente disponíveis, é o cancro que constitui uma das maiores, se não a maior, preocupação a nível de investigação científica. Dada a expressão desta patologia, da sua progressão e considerando também a relativa resistência destes casos a terapia medicamentosa, assim como a elevada taxa de mortalidade associada a determinados tipos de neoplasia, torna-se imperativo aumentar a eficiência terapêutica contra esta patologia.
Apesar da reconhecida importância e potencialidade ao nível da alquilação por compostos conjugados, existem também outros fármacos e moléculas conjugadas que, apesar de não atuarem por meio de uma alquilação mediada por este sistema, devem direta ou indiretamente a sua ação à existência de um sistema conjugado na sua estrutura molecular, e como tal merecem também referência.
É na perspetiva de alcançar um futuro com um maior e melhor leque de opções no combate ao cancro, que os sistemas conjugados, assim como a Química Orgânica Farmacêutica, podem assumir um papel de relevância. A adição ou modificação de compostos conjugados em moléculas com atividade farmacológica contra o cancro associado ao conhecimento das características das células tumorais, podem assim
6
conferir não só uma maior citotoxicidade e uma maior versatilidade da ação dos fármacos, que permita superar a capacidade que as células tumorais têm em desenvolver resistência aos agentes quimioterapêuticos, mas também conferir uma maior seletividade aos citotóxicos que permita diminuir as suas reações adversas, através do aproveitamento de algumas características específicas que diferenciam as células tumorais das saudáveis.
7
II. Adições a sistemas alílicos
Os sistemas alílicos são sistemas que apresentam uma conjugação entre uma ligação π e a orbital p do átomo adjacente. (March & Smith, 2007) (Vollhardt & Schore, 2002) Os sistemas alílicos podem ser alvos de reações nucleófilicas, podendo reagir, tal como as outras moléculas, por um mecanismo SN1 e SN2 (Mecanismo II-1).
X X Nuc Nuc Nuc X Nuc Nuc -X -X (a) (b)
Mecanismo II-1 – Reação (a) SN1 e (b) SN2 de um sistema alílico.
No entanto, devido à sua estrutura conjugada que permite uma deslocalização das ligações π, estes grupos funcionais podem reagir por outros mecanismos chamados de SN1’ e SN2’, que são mecanismos variantes dos já mencionados SN1 e SN2. (March &
Smith, 2007)
O mecanismo SN1’ é um mecanismo em que, tal como no SN1, se forma um carbocatião
após a quebra da ligação com o nucleofugo. Esta carga positiva é deslocalizada ao longo do sistema alílico, dando-se o ataque nucleofílico no carbono 3 do sistema alílico posicionado na margem oposta do sistema, em relação ao carbono 1 anteriormente ligado ao nucleofugo, ao contrário do que ocorre nos mecanismos SN1 (Mecanismo
II-2). Olhando para a estrutura de um sistema alílico é fácil de perceber que o produto deste tipo de reação nucleofílica apenas se distingue do produto de uma reação SN1, se o
sistema alílico não for simétrico.
X
Nuc -X
Nuc Mecanismo II-2 – Reação tipo SN1’ em sistemas alílicos
8
Quanto ao mecanismo SN2’, este ocorre por um ataque nucleofílico ao carbono 3 do
sistema alílico. No mesmo passo da reação deste ataque dá-se uma deslocalização concertada da ligação dupla e o abandono do nucleofugo (Mecanismo II-3). Tal como na reação SN1’, o produto desta reação SN2’ também não apresenta qualquer diferença
do produto de uma reação SN2 em sistemas alílicos simétricos.
Nuc
X
Nuc -X
Mecanismo II-3 - Reação tipo SN2’ em sistemas alílicos.
Em termos de reatividade os compostos alílicos apresentam também uma velocidade de reação relativa superior aos seus alcanos correspondentes. Esta maior reatividade ocorre em todos os tipos de substituição nucleofílica. No caso das reações tipo SN1, este facto
prende-se com a capacidade que o sistema alílico tem em estabilizar o catião formado, por ressonância. Em relação a reações do tipo SN2, a maior velocidade de reação está
relacionada com a estabilização, também por ressonância, do estado de transição da reação. (March & Smith, 2007)
Esta velocidade de reação dos sistemas alílicos é tanto maior quanto o número de possíveis estruturas de ressonância estáveis. A adição de grupos alquílicos ao sistema também aumenta a sua reatividade desde que não interfira com a reação por motivos de impedimento estéreo. Isto ocorre devido a um fenómeno chamado hiperconjugação, que se baseia numa sobreposição entre a orbital p vazia do carbocatião e uma orbital ligante vizinha. Resumidamente, o grupo alquílico aumenta a estabilidade do carbonato ou do estado estacionário da reação pela doação de carga eletrónica. (Ochran & Uggerud, 2007) (Streitwieser, Jayasree, Leung, & Choy, 2005)
II.1. Alilmetanosulfonato e Feniletilmetanosulfonato
Os ésteres alilmetanosulfonato 4 e feniletilmetanosulfonato 5 são compostos que apresentam um potencial mutagénico e carcinogénico na medida em que tem a capacidade de estabelecer aducto com desoxiguanosinas existentes no DNA. Tanto o feniletilmetanosulfonato como o alilmetanosulfonato funcionam como agentes alquilantes do átomo de oxigénio existente na posição 6 do nucleótido de guanina das desoxiguanosinas. Esta alquilação pode levar a mutações pontuais por uma transversão9
de bases, nomeadamente uma transversão G→A, já que uma desoxiguanosina alquilada no Oxigénio na posição 6, vai emparelhar com uma timidina em vez de uma citidina. (Eder, Dornbusch, & Fischer, 1987) (Eder, Kutt, 2001)
Apesar de ambos os compostos serem alquilantes e terem um átomo-alvo em comum, cada um estabelece a ligação covalente por um mecanismo diferente.
A estrutura do metanosulfonato constitui uma das estruturas mais básicas de um composto alílico exercendo genotoxicidade de forma direta. (Eder, Dornbusch, & Fischer, 1987) Devido ao impedimento estéreo, dadas as dimensões do grupo metanosulfonato, a reação de alquilação do alilmetanosulfonato dá-se preferencialmente por via de uma adição conjugada. O ataque nucleofílico do oxigénio dá-se no átomo de carbono na extremidade oposta ao grupo abandonante, por um mecanismo do tipo SN2’,
já que este carbono apresenta um menor impedimento estéreo do que o carbono diretamente ligado ao grupo metanosulfonato (Mecanismo II-4).
S O O O DNA DNA 4
Mecanismo II-4 - Alquilação pelo alilmetanosulfato por mecanismo Sn2’.
No entanto apesar da reação apresentada atrás se dar com maior facilidade ocorrendo assim de forma preferencial, o alilmetanosulfonato também pode reagir com a base azotada por via de uma reação do tipo SN2 simples, em que a substituição nucleofílica é
assistida por uma ponte dupla, e facilitada por o nucleofugo se encontrar ligado a um carbono alílico (Mecanismo II-5).
S O O O DNA DNA 4
Mecanismo II-5 – Alquilação pelo alilmetanosulfato por mecanismoSN2.
Ambas as reações de alquilação do alilmetanosulfonato com o DNA celular dão o mesmo aducto, como produto. (Eder, Kutt, & Deininger, 2001)
10
Por sua vez, o feniletilmetanosulfonato reage por um mecanismo SN1, em que após a
dissociação do grupo abandonante metanosulfonato, se dá um rearranjo da molécula que dá origem a um ião fenónio, em que o carbonato é altamente estabilizado por ressonância, já que se encontra em posição alílica em relação a duas ligações duplas. Após a formação deste ião, dá-se o ataque nucleofílico por parte do Oxigénio, estabelecendo-se assim o aducto (Mecanismo II-6).
S O O O DNA DNA 5
Mecanismo II-6 – Alquilação pelo feniletilmetanosulfonato.
II.2. Isopreno
O isopreno 6 foi classificado como um possível carcinogénico para as células humanas pela Internacional Agency for Research on Cancer. Este análogo, metilado na posição 2, do 1,3-butadieno é bastante abundante no ambiente, sendo proveniente tanto de fontes antropológicas, como combustões, como de fontes naturais, já que o isopreno é um produto de vários processos biológicos, principalmente, em plantas. (Begeman, Boysen, & Georgieva, 2011) (de las Heras & Rodriguez, 2003)
Apesar de as emissões de isopreno para a atmosfera através de fontes naturais excederem a emissão por fontes antropológicas em cerca de 300 vezes, é pelo do fumo do tabaco que há uma maior exposição do organismo humano ao isopreno.
Este composto orgânico conjugado demonstrou ser carcinogénico em roedores, causando neoplasias nos pulmões e na glândula de Harder, uma glândula lacrimal profunda (Delgado, 2005). Estes tumores encontram-se associados a mutações pontuais, mais concretamente transversões A→T, no proto-oncogene K-ras, no codão 61. O
11
isopreno também demonstrou induzir quebras de cadeia de DNA e outras transversões, predominantemente A→G, em alguns estudos in vitro com outras linhas de células que não as de roedores. (Begeman, Boysen, & Georgieva, 2011)
No entanto, o isopreno por si é inofensivo, sendo que a sua atividade carcinogénica é atribuída aos seus metabolitos. A molécula de isopreno é metabolizada pelo CYP2E1, uma enzima do citocromo P450. A oxidação pela ação do CYP2E1 vai produzir dois tipos de epóxidos: o IP-1,2-O 7, ou 2-metil-2-viniloxirano, e o IP-3,4-O 8, ou propen-2-iloxirano (Estrutura II-1).
CYP450
+
O O
6 7 8
Estrutura II-1 - Ativação metabólica do Isopreno a IP-1,2-O e IP-3,4-O.
Os monoepóxidos podem sofrer uma epoxidação subsequente formando-se um diepóxido que, tal como os monoepóxidos, será alvo de hidrólise ou conjugação com glutationa, de modo a ser excretado do organismo. No entanto este fenómeno não se dá de forma imediata, permitindo assim aos metabolitos do isopreno exercerem uma atividade nociva no organismo.
Estes epóxidos têm uma elevada capacidade alquilante, não só pela tensão do anel oxirano e eletronegatividade do carbono interno deste, o que é inerente a esta classe de compostos, mas também devido ao facto de um dos carbonos do anel ser um carbono em posição alílica, o que aumenta a reatividade deste, facilitando uma reação de adição nucleofílica. Esta particularidade dos metabolitos do isopreno permite que estes atuem como agente alquilantes de diversas macromoléculas biológicas. A alquilação de nucleósidos pelos epóxidos derivados da oxidação do isopreno é tida como a causa que desencadeia a mutagénese e carcinogénese associada a este composto.
Apesar do carácter alquilante e ambos pode dizer-se que existe uma diferença de toxicidade entre os dois epóxidos já que o IP-3,4-O forma uma maior quantidade de aducto com o DNA, que o IP-1,2-O. Isto é explicado com base na estrutura deste último, já que, para além da eletrofílicidade do carbono interno do anel oxirano, este carbono é também um carbono terciário em posição alílica. Assim este carbono apresenta uma grande facilidade em reagir por um mecanismoSN1, já que, o anel reage
12
de forma espontânea dando-se uma rutura do anel. Esta rutura leva ao alívio da tensão do anel, que é energeticamente favorável, como também forma um carbocatião alílico terciário que é estabilizado por ressonância, constituindo um catião bastante estável em ambiente intracelular. Este carbocatião vai ser atacado por moléculas de água (Mecanismo II-7). Assim o IP-1,2-O constitui uma molécula bastante instável, cuja rápida hidrólise não lhe permite formar aducto com os nucleosídeos de forma considerável, em relação ao IP-3,4-O.
O O H O H OH OH
Mecanismo II-7 – Hidrólise do IP-1,2-O.
Apesar de a alquilação do nucleósido se poder dar pelo estabelecimento de uma ligação covalente entre um átomo de azoto existente na sua base azotada, e um qualquer dos carbonos integrantes do anel oxirano do IP-3,4-O, a reação vai dar-se preferencialmente com o carbono interno do anel, já que se encontra numa posição alílica, reagindo assim mais facilmente.
Não obstante estabelecer aducto também com a desoxiguanosina, são os aducto estabelecidos com a desoxiadenosina que se apresentam como os mais importantes na genotoxicidade dos metabolitos do isopreno, já que as principais mutações são as transversões de uma base de adenina para outra base azotada sendo que as alquilações dão-se, de forma quase exclusiva, nos átomos de azoto existentes nas posições 1 e 3 do anel de purina por um mecanismo SN2. (Begeman, Boysen, & Georgieva, 2011)
Os aducto considerados mais importantes na genotoxicidade do isopreno são os estabelecidos pelo IP-3,4-O com o átomo de azoto na posição 1 da purina. Estes aducto reagem de forma intramolecular formando-se uma desoxiinosina, que se origina por uma desaminação na posição 6 da adenina da desoxiadenosina. O mecanismo de formação deste composto é apresentado no Mecanismo II-8. Esta desoxiinosina apresenta assim uma hipoxantina como base azotada, molécula esta que apresenta uma grande similaridade com a guanina e estabelece assim par com uma citosina. Este facto pode levar a incorporações de bases azotadas incorretas aquando da replicação do DNA
13
o que justificaria algumas mutações pontuais, nomeadamente transversões A→G. (Begeman, Boysen, & Georgieva, 2011)
N N NH2 N N O dR N N NH N N dR HO N N H2N N N dR O H O H N N O N N OH dR N N HO N N dR O -NH2
Mecanismo II-8 – Formação da desoxiinosina.
A compreensão total dos mecanismos de interação dos metabolitos do isopreno com o DNA, pode assim ser um passo em frente na avaliação da genotoxicidade deste composto conjugado.
II.3. Mitomicina C
A mitomicina C 9 é um potente fármaco, com propriedades anticancerígenas e antibióticas, descoberto em 1950, e obtido em culturas de Streptomyces caespitosus. Desde ai têm sido desenvolvidos e descobertos diversos análogos da mitomicina C, sendo incorporados no grupo das mitomicinas. (Tomasz, 1995)
Apesar de apresentar também propriedades antibacterianas, a mitomicina C tem vindo a ser utilizada desde 1960 em quimioterapia devido à sua atividade contra tumores sólidos, mantendo-se atualmente como um importante citotóxico usado em quimioterapia combinada em casos de cancro da mama, do pulmão, da próstata, estômago, pâncreas e cancro coloretal. Mais recentemente têm vindo a ser
14
demonstrados alguns efeitos da mitomicina C a nível da expressão de alguns genes, no organismo humano. (Tomasz & Palom, 1997) (Lown, 1983)
Nos microorganismos bacterianos, a mitomicina C tem como efeitos a inibição da síntese e recombinação do DNA, e causa também a quebra dos cromossomas e troca de segmentos de DNA entre cromatídeos irmãos, apresentando atividade tanto contra bactérias Gram -, como contra Gram +. (Lown, 1983) (Tomasz & Palom, 1997)
Os efeitos da mitomicina C podem ser entendidos, de forma relativamente fácil, à luz do seu mecanismo de ação intermolecular. A mitomicina C funciona como um agente alquilante bifuncional do DNA, estabelecendo cross-links entre as duas cadeias complementares da dupla hélice. Esta ação é de tal modo tóxica, que apenas um cross-link pode causar a morte de uma célula bacteriana. Em certos casos, esta pode apenas estabelecer monoaductos com o material genético da célula ou, em casos ainda mais raros, estabelecer um aducto com duas ligações à mesma cadeia de DNA. Ambos estes tipos de alquilação são menos tóxicos para a célula que os cross-links. A forma de atuar da mitomicina C foi descoberta em 1963, e em 1995 ainda não era conhecido nenhum outro antibiótico obtido de forma natural, que apresentasse um mecanismo de ação semelhante a este. Apesar de a mitomicina C reagir com as moléculas do DNA, a síntese de RNA e proteínas não sofre uma alteração que possa ser considerada relevante. (Tomasz, 1995)
A mitomicina C, sendo citotóxica, apresenta muitos efeitos adversos como leucopenia, trombocitopénia, vómitos, náuseas e febre. Estes efeitos de toxicidade devem-se à ação da molécula sobre os tecidos proliferativos da matriz óssea e epitélio do sistema gastrointestinal. (Lown, 1983)
No entanto a capacidade desta mitomicina em estabelecer aducto com o DNA, sejam eles monoaductos ou cross-links, é dependente da redução do anel de quinona presente na sua estrutura (Estrutura II-2). Esta redução, que pode ser enzimática, pela ação da redutases endógenas, ou química, vai desencadear uma serie de reações espontâneas na molécula, ficando esta com a capacidade de reagir com nucleófilos do DNA. (Denny, 2001)
15 O O N NH O H2N NH2 O O 9
Estrutura II-2 - Estrutura molécula da mitomicina C.
A necessidade de redução do anel quinona para que a mitomicina C se torne ativa, constitui um mecanismo de seletividade por parte deste fármaco, visando apenas tumores sólidos. Este fenómeno está relacionado com o ambiente de hipoxia que a molécula encontra no interior deste tipo de tumores, que contrasta com o ambiente existente noutros tipos de neoplasias em que a maior concentração de Oxigénio molecular, inibe a redução da mitomicina C à sua forma ativa. Esta seletividade própria da mitomicina C é bastante importante já que, normalmente, os tumores sólidos apresentam uma elevada resistência a tratamentos que impliquem a utilização de oxigénio molecular ou radiação, dificultando assim a terapêutica. (Tomasz & Palom, 1997)
Em termos de interações moleculares, a mitomicina C estabelece ligações covalentes apenas com as desoxiguanosinas presentes no DNA podendo estas constituir monoalquilações ou cross-links, como já tinha sido referido. O estabelecimento de ligações intermoleculares é assistido por um sistema conjugado, na medida em que os ataques nucleofílicos pelos grupos amina da desoxiguanosina, se dão em carbonos alílicos da molécula de mitomicina C. Esta reação de adição, maioritariamente feita à posição N2 da desoxiguanosina, é assim facilitada pelo sistema conjugado, o que vai de encontro à importância da redução do anel de quinona, para a atividade alquilante da mitomicina C. (Yang & Wang, 1999)
O mecanismo de alquilação do DNA celular por parte da mitomicina C é apresentado no Mecanismo II-9.
16 O O N NH O H2N NH2 O O OH OH N NH O H2N NH2 O O OH OH N NH H2N NH2 O O OH OH N NH H2N NH2 O O DNA OH OH N NH2 H2N NH2 O O DNA OH OH N NH2 H2N DNA Redução - MeOH - NH2COOH
Mecanismo II-9- Mecanismo de alquilação de DNA por parte da mitomicina C.
A formação de cross-links e de monoaductos com o DNA por parte da mitomicina C apresenta seletividade para sequências do tipo CpG, apesar de ser possível a ocorrência de monoaductos fora deste tipo de sequência, mas numa quantidade muito menor. A rigidez da molécula de mitomicina C, a distância entre as duas funções alílicas alquilantes, e as interações não-covalentes estabelecidas pela mitomicina C com determinadas bases azotadas do DNA quando este citotóxico se encontra entre as duas cadeias da dupla hélice, ajudam a compreender esta seletividade por parte do fármaco para as sequências 5´-CpG-3´, em que p representa a ponte fosfato entre a desoxiguanosina e a desoxicitidina, com esta ultima na extremidade 5´.
A seletividade da mitomicina C é explicada por a sua primeira alquilação requerer, em circunstâncias normais, não só uma desoxiguanosina na cadeia onde se vai ligar primariamente, mas também uma desoxicitidina ligada à extremidade 5´ desta desoxiguanosina. Esta sequência implica que na cadeia oposta exista uma desoxiguanosina complementar à desoxicitidina, nucleosídeo este que apresenta uma grande importância no posicionamento, e consequentemente na ação, da mitomicina C em relação à dupla cadeia de DNA. Esta desoxiguanosina estabelece uma ponte de hidrogénio através do seu grupo amina na posição N2, com o grupo carbamato do
17
fármaco, ficando este grupo funcional direcionado para o terminal 5´ da cadeia em que se dá a primeira alquilação (Figura II-1). Esta interação não-covalente intermolecular leva a uma orientação da molécula de mitomicina C que faz com que esta se aproxima da desoxiguanosina onde se vai dar a primeira alquilação, facilitando este primeiro passo da alquilação bifuncional. A formação do cross-link é consumada quando o grupo amina N2, na cadeia complementar à cadeia onde se deu a primeira alquilação, devido à proximidade do grupo carbamato por via da ponte de hidrogénio anteriormente estabelecida, ataca o carbono electrofílico que se encontra ligado ao carbamato, e que também se encontra numa posição alílica. Assim se estabelece a segunda alquilação do cross-link entre as duas cadeias complementares de DNA. (Yang & Wang, 1999) (Tomasz, 1995)
Figura II-1 - Orientação da mitomicina C no seio da dupla hélice de DNA. A seta aponta a desoxiguanosina onde se dá a primeira alquilação. (Adaptado de referência Tomasz, 1995) Esta seletividade é entendida como uma potente arma desenvolvida pelos microorganismos produtores de mitomicina C contra outros microorganismos seus competidores, já que estas interações não-covalentes garantem que se formem preferencialmente cross-links, que são mais tóxicos que os monoaductos.
No entanto, devido ao tamanho e rigidez da molécula do fármaco, a sua ação de agente alquilante bifuncional fica confinada a esta sequência específica. Isto constitui uma desvantagem para a ação citotóxica da molécula no tratamento de neoplasias, pois a sequência 5´-CpG-3´ é relativamente pouco abundante no genoma humano. De facto, apenas 5% das desoxiguanosinas presentes no genoma das células dos mamíferos se encontra nestas sequências, ao contrário do que acontece nos microorganismos bacterianos onde esta percentagem pode chegar aos 70%. (Tomasz, 1995)
18
Este “calcanhar de Aquiles” da mitomicina C deixa, no entanto, algumas portas abertas no que diz respeito à modulação e desenvolvimento de moléculas análogas mais potentes e eficientes que a mitomicina C.
II.4. Tamoxifeno
O tamoxifeno 10, um trifeniletileno, é um composto com atividade antiestrogénica, sendo amplamente utilizado como adjuvante em quimioterapia da neoplasia da mama, assim como na profilaxia desta doença em mulheres que apresentem um alto risco de a vir a desenvolver. (Kuramochi, 1996) (Surh, 1998)
Apesar de ser benéfico no tratamento e profilaxia do cancro da mama, o tratamento com tamoxifeno aumenta o risco de cancro do endométrio, assim como o risco de desenvolvimento de eventos tromboembólicos em mulheres. O mecanismo de indução de cancro do endométrio ainda não está completamente compreendido, pensando-se que estarão envolvidos fatores hormonais, assim como fatores relacionados com genotoxicidade. (Gamboa da Costa & Pereira, 2007)
Esta mesma genotoxicidade já foi verificada também em roedores tratados com tamoxifeno, sendo que este fármaco exibe uma ação hepatocarcinogénica, tendo sido detetados aducto de tamoxifeno com o material genético dos hepatócitos. Este facto sugere que seja este o mecanismo inerente à genotoxicidade e carcinogénese associada à utilização de tamoxifeno, tanto em roedores como em humanos.
O tamoxifeno forma aducto com o DNA, após ser metabolicamente ativado por enzimas do citocromo P450, nomeadamente o CYP2E1 e o CYP3A1. Esta ativação metabólica torna o tamoxifeno num agente electrofílico com grande capacidade alquilante, devido a uma oxidação da posição α do grupo etil, pela remoção de um hidreto, levando assim à formação de um intermediário que possui um carbocatião alílico na posição onde se deu a oxidação. Este carbocatião apresenta uma reatividade elevada, reagindo com nucleófilos existentes no DNA, e estabelecendo ligações covalentes, permitindo assim a formação de diversos aducto (Mecanismo II-10). (Okubo, Nagai, & Ushiyama, 1998) As ligações covalentes entre o tamoxifeno e o DNA estabelecem-se preferencialmente com o grupo amina exocíclico da base azotada guanina, ou seja, o azoto na posição 7, do nucleósido desoxiguanosina.
19 N O N O N O DNA P450 Oxidação DNA 10
Mecanismo II-10 - Mecanismo de formação de aducto do tamoxifeno com o DNA.
Sabe-se que o α-hidroxitamoxifeno 11, um metabolito do tamoxifeno, também apresenta capacidade de estabelecer aducto com o DNA, pois, devido à sua estrutura apresenta a capacidade de se converter no intermediário da metabolização do tamoxifeno, que possui o carbocatião alílico (Mecanismo II-11). Assim, a α-hidroxilação do tamoxifeno também pode ser entendida como uma ativação metabólica. (Okubo, Nagai, & Ushiyama, 1998)
N O OH -OH +OH N O 11
Mecanismo II-11 – Conversão do α-hidroxitamoxifeno.
Por outro lado, alguns análogos do tamoxifeno, como o toremifeno 12 (Estrutura II-3), apresentam uma toxicidade muito inferior à do tamoxifeno. Isto explica-se devido aos substituintes eletrotractores que destabilizam a formação do carbocatião. (Kuramochi,
20
1996) Este facto pode ser a chave no desenvolvimento de análogos do tamoxifeno, que apresentem uma menor toxicidade.
N O
Cl
12
21
III. Adição tipo-Michael
Antes de definir uma adição tipo-Michael, é importante compreender o conceito inerente à reação de Michael.
Uma reação de Michael, assim chamada devido a Arthur Michael, químico orgânico norte-americano que a descobriu e estudou, consiste num ataque nucleofílico por parte de um ião enolato, um dador de Michael cuja formação se dá por catálise básica, a um aceitador de Michael, que pode ser um carbonilo α,β-insaturado, como uma cetona ou
um aldeído, ou um nitrilo. (March & Smith, 2007)
Na reação de Michael, o nucleófilo também pode ser um anião derivado de um dicarbonilo β ou um qualquer derivado de um destes compostos. A adição de Michael aparenta ser uma adição conjugada 1,2, devido à manutenção do grupo carbonilo no produto final, no entanto esta é uma adição conjugada 1,4, em que o grupo carbonilo se mantém devido à tautomerização do produto final, um enol, para a sua forma ceto (Mecanismo III-1). (Vollhardt & Schore, 2002)
O O H B - HB + O O O H H O O
Mecanismo III-1 – Adição de Michael.
Apesar de uma adição 1,2 ser cinéticamente favorável, ocorrendo mais rapidamente devido à maior deficiência eletrónica do carbono do grupo carbonilo por estar diretamente ligado a um átomo de Oxigénio, o produto da adição 1,4 é predominante na reação de Michael. O efeito da deslocalização da densidade eletrónica também vai diminuir a eletrofílicidade do carbono do grupo carbonilo participando, de certo modo, na preferência com que se verifica uma adição 1,4, em detrimento de uma 1,2. (Cai & Bhatnagar, 2009)
22
O produto da adição conjugada 1,4 é termodinamicamente mais estável, devido à manutenção do grupo carbonilo, que é constituído por uma ligação dupla mais forte, sendo assim mais estável que os produtos formados na reação conjugada de adição 1,2, como são o caso dos hidratos e hemiacetais. Além disso, a adição 1,4 produz um anião enolato, que é estabilizado por ressonância. (Carey & Sundberg, 2007)
No entanto, é de referir que existem nucleófilos que reagem preferencialmente por uma adição conjugada 1,2 como é o caso da hidroxilaminas, semicarbazidas e hidrazinas. Apesar do produto apresentar uma energia fundamental mais alta do que numa adição 1,4, este tipo de reação leva à formação de uma imina cuja precipitação irá, eventualmente, levar ao estabelecimento de um equilíbrio de reação direcionado para a formação destes produtos. (Vollhardt & Schore, 2002)
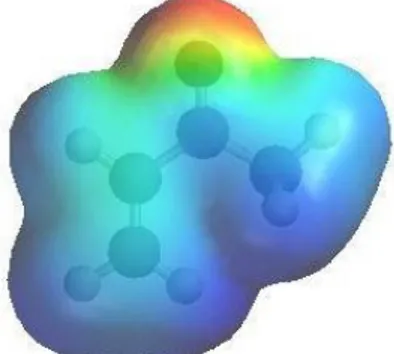
Olhando para o mecanismo geral de uma reação de Michael, vemos que a reação ocorre devido à ação nucleofílica do carbono α do enolato que vai atacar o carbono β do composto carbonilo α,β- insaturado. O carácter eletrotrator do grupo carbonilo vai roubar densidade eletrónica, tornando assim o carbono β do carbonilo α,β- insaturado mais suscetível a um ataque nucleofílico, como se poder observar pela densidade eletrónica da molécula de 3-butenona, na Figura III-1.
Figura III-1 - Densidade eletrónica da 3-butenona.
Em jeito de sumário, uma reação de adição de Michael resume-se a uma adição conjugada de um enolato, para formar um composto com dois grupos carbonilo.
Pode entender-se uma reação tipo-Michael como uma derivação da reação de adição de Michael, ocorrendo por um mecanismo em tudo semelhante a esta, excetuando o facto de a adição a um aceitador de Michael, um carbonilo α,β- insaturado, ocorrer através do estabelecimento de uma ligação covalente por parte de um outro nucleófilo que não um
23
enolato, ou outra molécula análoga, dispensando-se assim, a catálise básica Mecanismo III-2. O Nuc O Nuc O Nuc R1 R2 R1 R2 R1 R2 HO Nuc R1 R2
Mecanismo III-2 – Reação tipo-Michael.
Em determinados casos em que o sistema π conjugado do aceitador de Michael apresente mais que duas ligações duplas conjugadas, a ligação pode dar-se na posição 1 e noutra posição do sistema conjugado, que não a posição 4, desde que esta mesma posição seja par.
Os grupos nucleofílicos mais importantes que atuam na reação tipo-Michael, a nível fisiológico, são os grupos amina e os grupos tiol. Este facto apresenta uma grande relevância pois muitos fármacos apresentam um aceitador de Michael no seu farmacóforo, existindo ainda outras moléculas que exercem a sua atividade no organismo, também por possuírem um carbonilo α,β- insaturado na sua estrutura molecular. Ao contrário do que acontece em laboratório onde este tipo de reações é facilmente reversível através de aquecimento, uma vez no organismo humano os aceitadores de Michael formam aducto estáveis com diversas moléculas endógenas. Assim estas reações em meio orgânico representam um foco de elevada importância tanto a nível farmacológico, podendo muitas delas apresentar uma atividade citotóxica, citostática, antibacteriana ou atuarem na modulação da resposta inflamatória, devido à capacidade de estabelecerem ligações com proteínas, enzimas ou com o material genético das células. (Gersch & Kreuzer, 2012)
Grande parte dos efeitos nocivos associados a compostos alílicos devem-se à sua ativação metabólica a um carbonilo α,β- insaturado. Composto alílicos com bons grupos abandonantes são hidrolisados a álcoois alílicos, que são posteriormente metabolizados pelo álcool desidrogenase a um carbonilo α,β- insaturado, que, como já se viu, poderá
24
atuar como um aceitador de Michael. (Toennes, Schmidt, & Fandiffo, 2002) (Eder, Dornbusch, & Fischer, 1987)
Devido a os compostos carbonilo α,β- insaturado manterem o grupo carbonilo após a adição tipo-Michael, ainda podem ser alvos de um outro ataque nucleofílico. Em meio orgânico, este ataque é normalmente feito por um grupo amina, estabelecendo-se uma base de Schiff (Mecanismo III-3) Este mecanismo ocorre no estabelecimento de cross-links entre este tipo de compostos e bases azotadas ou aminoácidos.
R1 O H2N R2 R1 OH N R2 H H R1 N R2 - H2O
Mecanismo III-3 – Formação de uma base de Schiff.
Apesar de ser possível o estabelecimento da base de Schiff antes da adição tipo-Michael, esta reação normalmente ocorre numa muito menor extensão, já que de certo modo o carbono do grupo carbonilo se encontra reacionalmente desativado relativamente ao carbono β, pois a deslocalização de densidade eletrónica do sistema conjugado confere-lhe uma menor eletrofílicidade devido à eletronegatividade do grupo funcional do qual faz parte. Após a adição de tipo-Michael a ligação dupla entre os dois carbonos é perdida, mas o grupo carbonilo mantém-se intacto. Assim a perda de contributo eletrónico por parte da ponte dupla, aumenta a reatividade do grupo carbonilo, reagindo este com grupos amina existentes na vizinhança, formando-se assim a base de Schiff. (Cai & Bhatnagar, 2009)
As reações tipo-Michael também apresentam um papel importante no desenvolvimento de algumas patologias, nomeadamente na carcinogénese e aterosclerose, já que alguns aducto formados podem causar mutações, que levem ao descontrolo do ciclo celular, mas que não causem a apoptose celular.
25
III.1. Acroleína
A acroleína 13, ou propen-2-enal, é o aldeído α,β- insaturado de estrutura mais simples, constituindo a estrutura básica de todos os carbonilos α,β- insaturados (Estrutura III-1). Este composto é formado a nível endógeno, como um produto secundário da degradação lipídica por peroxidação e pela oxidação de outras moléculas como aminoácidos ou poliaminas. No entanto a acroleína constitui também um poluente ambiental, podendo ser produzida por fontes exógenas, nomeadamente em diversas combustões, como de madeira, combustíveis fósseis e outros compostos orgânicos. A acroleína é ainda encontrada em grande concentração no fumo do tabaco. Este composto constitui também uma importante molécula a nível da indústria química. (Thompson & Burcham, 2008)
O
13
Estrutura III-1 - Estrutura molecular da acroleína.
A acroleína é uma das molécula que apresenta o maior carácter electrofílico de toda a série de aldeídos α,β- insaturados, podendo reagir com diversos nucleófilos, por uma reação tipo-Michael ou ainda pelo estabelecimento de uma base de Schiff. As propriedades tóxicas deste composto devem-se assim à sua capacidade de reagir com várias moléculas endógenas, causando depleção de glutationa, formando aducto com proteínas, DNA, RNA e ainda cross-links inter- ou intra-moleculares. (Cai & Bhatnagar, 2009) Estas reações podem levar a mutações, locais abásicos no DNA e quebras de cadeia deste, perdas de função de proteínas, e, dependendo da extensão dos danos causados, morte celular, sendo que estes factos vão ao encontro dos efeitos provocados por uma exposição aguda à acroleína que compreendem uma desregulação de diversos mecanismos celulares envolvidos na apoptose, regulação do ciclo celular, transcrição e biossíntese de proteínas. (Thompson & Burcham, 2008) (Wang, Prorok, & Vaughan, 1993)
A toxicidade do 2-propen-1-ol 14 (Estrutura III-2), um álcool alílico utilizado como herbicida e intermediário em algumas sínteses industriais, deve-se à sua ativação metabólica a acroleína. (Eder, Dornbusch, & Fischer, 1987) (Toennes, Schmidt, & Fandiffo, 2002)
26
HO
14
Estrutura III-2 – Estrutura molecular do 2-propen-1-ol.
Constituindo a estrutura reacional base de todos os restantes α,β–aldeído insaturados que constituem produtos secundários da peroxidação lipídica, os mecanismos de ação da acroleína, podem de certa forma ser extrapolados para os restantes produtos secundários da oxidação lipídica.
Em termos de interações com o material genético, a acroleína apresenta a capacidade de estabelecer aducto com todos os nucleósidos que fazem parte das cadeias de DNA ou RNA. Apesar de se conhecer a estrutura de alguns dos aducto formados, existe a noção que devido às características estruturais e reacionais da acroleína, ainda existem muitos outros tipos de aducto cuja estrutura ainda se encontra por elucidar. (Pawłowicz & Munter, 2006)
Em ratinhos expostos a acroleína foi encontrada uma acumulação de aducto de acroleína com desoxiadenosina, no núcleo os seus hepatócitos. Em reação in vitro entre a acroleína e o DNA de células do timo de vitelo verificou-se que uma mesma desoxiadenosina pode estabelecer aducto com mais que uma molécula de acroleína. Destas reações obtiveram-se quatro principais monoaductos de acroleína com desoxiadenosina (Estrutura III-3).
Estrutura III-3 – Aducto formados in vitro entre a acroleína e desoxiadenosina.
Os aducto exocíclicos 15 e 16 são os que se formam em maior quantidade. O aducto 15 é formado por uma reação tipo-Michael entre a acroleína e o azoto N6 do nucleótido. Após esta adição dá-se uma reação de substituição intramolecular que tem como produto o aducto 15. Quanto ao aducto 16, é formado por uma reação entre o aducto 15
27
e uma outra molécula de acroleína por uma reação tipo-Michael e uma adição nucleofílica intramolecular.
Em relação aos aducto formados em menor extensão, o aducto 17 forma-se por um rearranjo de Dimroth do aducto 15 e uma subsequente abertura do anel propano. Por sua vez, o aducto 18 é produto de uma reação de adição tipo-Michael do aducto 17 com outra molécula de acroleína e uma posterior ciclização (Mecanismo III-4).
Mecanismo III-4 – Mecanismo de formação do aducto 18 da Estrutura (Adaptado de Pawłowicz & Munter, 2006)
Seria de esperar que se apresentasse como pouco provável a reação de duas moléculas de acroleína com a mesma molécula de desoxiadenosina no DNA, no entanto uma desoxiguanosina após uma primeira reação tipo-Michael apresenta-se como mais reativa perante uma acroleína, reagindo três vezes mais rápido, do que uma desoxiadenosina não alquilada. Esta superior reatividade explica não só a formação de aducto com duas acroleínas mas também que o aducto mais abundante seja o aducto 16. (Pawłowicz & Munter, 2006)
A acroleína também reage com a desoxitimidina 19, formando-se aducto estáveis. Tal como no caso da desoxiadenosina, também aqui se formam aducto com mais que uma molécula de acroleína, o que permite uma panóplia imensa de aducto possíveis. Existem, no entanto, cinco aducto maioritários (Estrutura III-4). (Pawłowicz & Kronberg, 2008)
28 Estrutura III-4 – Estrutura dos aducto maioritários da reação da acroleína com a desoxitimidina. A formação do aducto 19 dá-se através de uma reação tipo-Michael entre o azoto N3 da base azotada e a acroleína. Todos os restantes aducto têm o aducto 19 como percursor, sendo que este reage posteriormente com outra molécula de acroleína, na sua forma enol 25, seguido de uma reação de desidratação, no caso dos aducto 21, 22 e 23, ou de uma reação de adição nucleofílica intramolecular no caso do aducto 20 (Mecanismo III-5). (Pawłowicz & Kronberg, 2008)
29 Mecanismo III-5 - Mecanismo de formação dos aducto maioritários entre a acroleína e a desoxitimidina
(Adaptado de Pawłowicz & Kronberg, 2008)
Os aducto formados entre a acroleína e a desoxiguanosina 26 serão talvez os mais estudados, ocorrendo por uma adição nucleofílica tipo-Michael entre o azoto na posição N2 do nucleósido, e a acroleína, que funciona como aceitador de Michael. Após esta reação ocorre uma ciclização devido a um ataque nucleofílico intramolecular por parte do azoto N1 ao carbono electrofílico do grupo aldeído, resultante da adição tipo-Michael (Mecanismo III-6). (Kozekov, Turesky, & Alas, 2010)
NH N N O NH2 N dR O NH N N O N H N dR O N N N O N H N dR OH 26
30
Esta reação entre a acroleína e a desoxiguanosina, à imagem do que acontece também com o 4-hidroxi-2-nonenal 32 (III.2.1.), é mutagénica, dando lugar a transversões G→T. Para além destas mutações pontuais a acroleína tem a capacidade de formar cross-links entre duas desoxiguanosinas, em sequências 5’-CpG-3’. A mitomicina C 9 (Estrutura II-2) também apresenta seletividade para esta sequência formando também aducto entre duas desoxiguanosinas, sabendo-se que a razão se prende com interações intermoleculares, e sendo a sua ação farmacológica dependente destes cross-links. Neste caso específico, a formação destes bis-adutos ocorre por um mecanismo diferente do que normalmente acontece com os α,β–aldeídos insaturado, já que devido à reação intramolecular que ocorre aquando da formação do aducto exocíclico, não há manutenção do grupo carbonilo, necessário à formação de uma base de Schiff. Assim a formação destes cross-links ocorre pelo encadeamento de, primeiro, a reação que tem como produto o aducto exocíclico entre uma desoxiguanosina e uma molécula de acroleína e um subsequente ataque nucleofílico simples ao carbono do aducto exocíclico ligado ao hidroxilo, por parte do azoto N2 de uma outra desoxiguanosina emparelhada com a desoxicitidina 27 ligada à extremidade 5´ da desoxiguanosina onde se deu a primeira alquilação, estabelecendo-se assim uma ligação covalente (Estrutura III-5). (Kozekov, Turesky, & Alas, 2010)
N N N O N H N dR HN N N O HN N dR
Estrutura III-5 – Cross-link formado entre a acroleína e duas desoxiguanosinas.
Por fim, na reação entre a acroleína e a desoxicitidina 27 também são formados dois tipos de aducto, um em que se forma um exociclo e outro em que se formam dois exociclos (Mecanismo III-7). (Pawłowicz & Klika, 2007)