Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure.
Texto
Imagem




![Table 5 gives the results of an evaluation of the methods’ ability to predict catalytic sites (defined by the Catalytic Site Atlas [66]) in the LigASite apo dataset](https://thumb-eu.123doks.com/thumbv2/123dok_br/18345675.352495/10.918.474.802.91.781/results-evaluation-methods-ability-predict-catalytic-catalytic-ligasite.webp)
Documentos relacionados
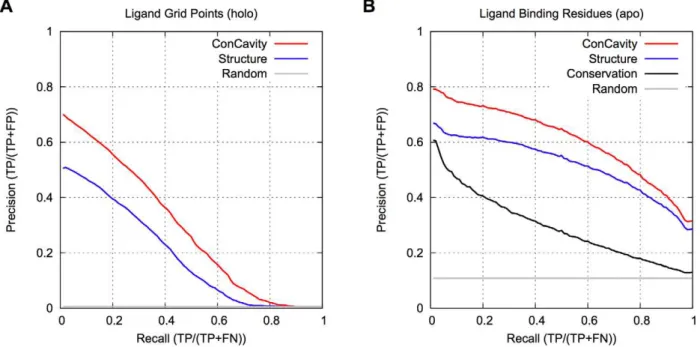
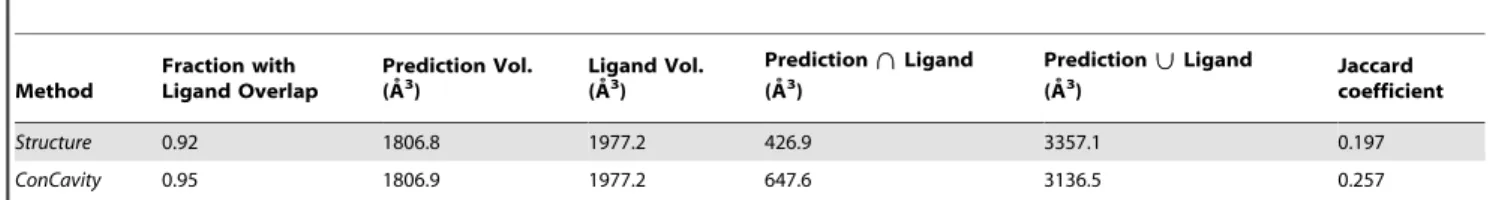
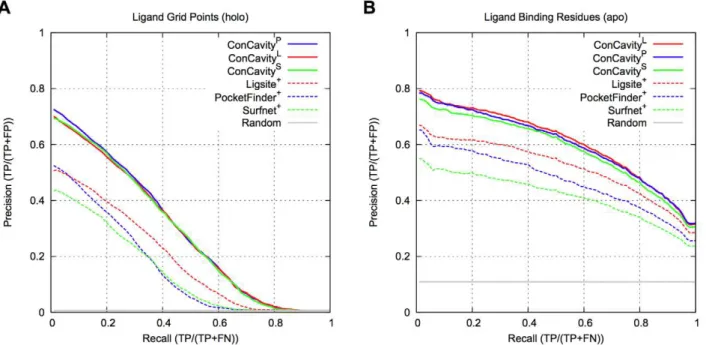
The better performance attained by our approach mainly resulted from two aspects: (i) the HMM-based search was specifically utilized to find the weakly homologous template of
pneumoniae ParC, we have modeled the protein- DNA binding, DNA sequence recognition and cleavage process using a 24-mer duplex corresponding to the strong E binding site of topo IV
Finally, we used alignments produced by our program to study binding site conservation in genome-wide binding data of key transcription factors in the Drosophila blastoderm, with
To take advantage of such cluster information of local sequence fragments for predicting acetylation sites, we took the local sequence around the acetylation site in a query protein
We present a structure-based method for predicting class II epitopes that combines molecular mechanics docking of a fully flexible peptide into the MHC binding cleft followed by
In this work, we demonstrate how a pan-specific HLA-DR prediction method exploiting both peptide and primary HLA sequence can be used to accurately predict quantitative
We tested the enhancer activity of 31 protein-coding exons, which we chose based on strong sequence conservation between zebrafish and human, and occurrence in developmental
Figure S6 Results for Kratchowil set. A comparison of the ROC curves for the Kratchowil set of HSA binders that result from different approaches to prediction of binding affinity
