Universidade de Lisboa
Faculdade de Ciências
Departamento de Estatística e Investigação Operacional
2012
Modelos Não Lineares de Efeitos Mistos na
Farmacocinética da Ciclosporina em Doentes
Transplantados Renais
Ana Sofia Cunha Cardoso
Dissertação
Universidade de Lisboa
Faculdade de Ciências
Departamento de Estatística e Investigação Operacional
2012
Modelos Não Lineares de Efeitos Mistos na
Farmacocinética da Ciclosporina em Doentes
Transplantados Renais
Ana Sofia Cunha Cardoso
Dissertação orientada pela Prof.ª Doutora Maria Salomé Cabral e
coorientada pela Prof.ª Doutora Ana Paula Carrondo
i
Í
NDICE
Índice...i
Índice de figuras...ii
Índice de tabelas ...iii
Agradecimentos ...v
Resumo...vii
Abstract...ix
Abreviaturas e símbolos ...xi
1. Introdução...1 2. Farmacocinética...3 2.1 LADME ...4 2.1.1 Libertação e absorção ...5 2.1.2 Distribuição ...6 2.1.3 Eliminação ...6 2.2 Estado Estacionário ...8 2.3 Análise Farmacocinética ...9 2.3.1 Análise Compartimental ...10
2.3.2 Análise Não Compartimental ...16
2.4 Análise Populacional ...18
3. Modelo Não Linear de Efeitos Mistos ...21
3.1 O Modelo...22
3.2 Extensões do modelo básico ...27
3.2.1 Efeitos aleatórios e estruturas da matriz D ...27
3.2.2 Variabilidade intra individual. Modelação da matriz de variância–covariância dos erros aleatórios...27
3.2.3 Modelo geral...35
3.2.4 Covariáveis dependentes do tempo...36
3.2.5 Interpretação dos parâmetros do modelo e objetivos da inferência ...38
3.3 Estimação e inferência no modelo não linear hierárquico de efeitos mistos ...39
3.3.1 Função verosimilhança e estimação dos parâmetros...39
3.3.2 Inferência ...44
3.4 Qualidade do ajustamento...45
3.5 Construção do modelo...46
3.6 Abordagens não paramétricas...48
3.7 Programas para farmacocinética populacional...49
4. Modelação da ciclosporina em doentes transplantados renais...51
4.1 Dados...53 4.2 Modelo...57 4.3 Ajustamento do modelo ...59 4.4 Validação: ...69 4.5 Parâmetros farmacocinéticos ...74 4.6 Discussão ...76 4.7 Conclusão...80 Bibliografia...81
ii
Í
NDICE DE FIGURAS
Figura 2.1 - Perfil da concentração sanguínea em função do tempo de um fármaco hipotético
administrado em doses múltiplas por via oral. ...9
Figura 2.2 - Esquema de input e output de um fármaco no organismo na presença de um, dois e três compartimentos. ...11
Figura 3.1 – Gráficos de semivariogramas versus distância para correlações espaciais isotrópicas com ρ=1 e efeito pepita=0.1...34
Figura 3.2 – Principais passos na construção e validação de um modelo ...48
Figura 4.1 – Esquema dos tempos de amostragem do estudo nas duas ocasiões. ...54
Figura 4.2 – Perfil observado da concentração de ciclosporina ao longo do tempo, imediatamente após entrada no estudo (primeira ocasião) e cerca de 6 meses depois (segunda ocasião) no indivíduo 114. ...54
Figura 4.3 – Perfil observado da concentração de ciclosporina versus tempo após entrada no estudo dos 82 indivíduos (primeira ocasião)...55
Figura 4.4 - Excerto dos dados do grupo de modelação, relativo ao doente 101. ...57
Figura 4.5 – Estimativas EB dos efeitos aleatórios de lV e de lCl versus as covariáveis em estudo...61
Figura 4.6 – Gráfico de dispersão dos resíduos padronizados versus valores ajustados...65
Figura 4.7 – Gráfico de dispersão dos resíduos padronizados versus tempo. ...65
Figura 4.8 – Papel de probabilidades da normal dos resíduos padronizados. ...66
Figura 4.9 – Papel de probabilidades da normal das estimativas dos efeitos aleatórios...66
Figura 4.10 – Estimativa do semivariograma amostral dos resíduos padronizados...67
Figura 4.11 – Gráfico das concentrações observadas versus valores preditos com base no ajustamento populacional e individual...70
Figura 4.12 – Gráfico das concentrações observadas e valores preditos, com base no ajustamento populacional (“típico”) e individual, ao longo do tempo, para os indivíduos 114 e 131...73
iii
Í
NDICE DE TABELAS
Tabela 2.1 - Descrição, gráficos e equações dos modelos de um compartimento mais frequentes
em TDM, considerando eliminação a partir do compartimento central, em dose única e em
doses múltiplas...13
Tabela 2.2 – Relação do MRT com o modelo de um compartimento e estimação dos parâmetros farmacocinéticos a partir de dados de dose única...17
Tabela 2.3 - Fatores de variabilidade farmacocinética. ...18
Tabela 3.1 – Funções de variância...30
Tabela 3.2 - Função de autocorrelação para modelação de correlação dos erros aleatórios em dados igualmente espaçados e de natureza inteira. ...32
Tabela 3.3 - Modelos de semivariograma isotrópicos para estruturas de correlação espacial...34
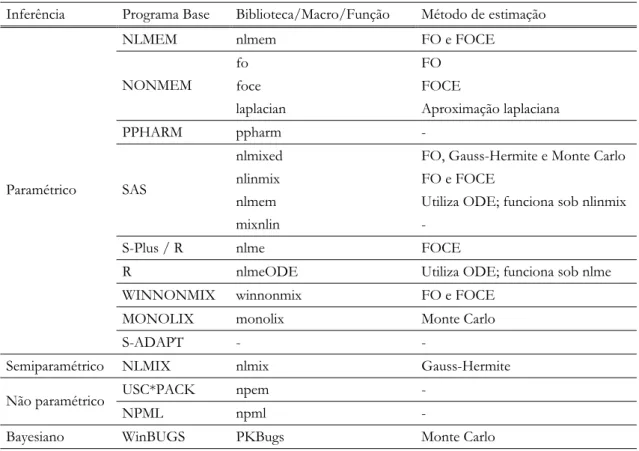
Tabela 3.4 - Programas e respetivo método de estimação de modelos não lineares de efeitos mistos aplicados a farmacocinética...50
Tabela 4.1 - Caracterização dos dados utilizados no desenvolvimento e validação do modelo. ...56
Tabela 4.2 - Estimativas iniciais dos parâmetros. ...59
Tabela 4.3 – Efeitos fixos e aleatórios do modelo base (Modelo 1)...60
Tabela 4.4 - Comparação de modelos com diferente número de efeitos aleatórios. ...60
Tabela 4.5 - Modelos obtidos por introdução sequencial de covariáveis no modelo base (Modelo 1), inicialmente associadas a lCl e depois a lV. ...63
Tabela 4.6 – Comparação de modelos com covariáveis e diferentes efeitos aleatórios. ...64
Tabela 4.7 – Comparação de modelos com diferentes estruturas de variância. ...68
Tabela 4.8 - Parâmetros do modelo final (Modelo 24)...69
Tabela 4.9 - Resultados da validação interna e externa do modelo final (Modelo 24). ...72
v
A
GRADECIMENTOS
No fim desta etapa não posso deixar de expressar a minha gratidão aos familiares, amigos e colegas, que me apoiaram e incentivaram a ir sempre um pouco mais além. Enumerá-los equivale a correr o risco de esquecer algum, por isso, aqui fica uma palavra de reconhecimento para todos quantos, de uma maneira ou doutra, contribuíram para a materialização deste projeto.
O meu profundo agradecimento:
À Professora Doutora Maria Salomé Cabral, pelo conhecimento, trabalho, dedicação e amizade que sempre me dispensou durante a orientação deste trabalho. Graças ao seu empenho e ao seu olhar atento foi possível percorrer este caminho.
À Professora Doutora Ana Paula Carrondo, colega e amiga, pelo constante incitamento à elaboração desta dissertação, bem como, ao seu trabalho como coorientadora.
Ao Doutor José Guerra do Serviço de Nefrologia e Transplantação Renal do Hospital de Santa Maria.
A todos os colegas do Serviço Farmacêutico do Hospital de Santa Maria, na pessoa da sua Diretora, Drª Piedade Ferreira, pelo interesse e apoio manifestado.
vii
R
ESUMO
Os parâmetros farmacocinéticos caracterizam o perfil concentração-tempo de um fármaco no organismo sendo, por isso, essenciais na individualização posológica da terapêutica, com vista a maximizar a sua eficácia e reduzir os efeitos adversos.
Os dados necessários para a análise farmacocinética consistem na medição, após a administração, das concentrações sanguíneas do fármaco, obtidas ao longo do tempo para cada indivíduo. Este tipo de dados, aos quais se dá o nome de dados longitudinais, requer particular cuidado na caracterização da variabilidade, uma vez que as observações intra indivíduos tendem a estar correlacionadas. Os modelos mistos, através da incorporação de efeitos fixos (parâmetros associados à população) e efeitos aleatórios (efeitos associados aos indivíduos), permitem modelar esta dependência e acomodar a variabilidade intra e interindividual tendo, por isso, particular interesse nesta área.
Este trabalho teve como objetivo fundamental estimar os parâmetros farmacocinéticos da ciclosporina recorrendo a dados longitudinais obtidos, após administração oral, em doentes transplantados renais, através da aplicação de modelos não lineares de efeitos mistos, também designados por modelos não lineares mistos, e identificar algumas covariáveis responsáveis pela variabilidade intra e interindividual da ciclosporina na subpopulação estudada.
Após uma breve descrição dos conceitos básicos de farmacocinética e do fundamento teórico dos modelos não lineares de efeitos mistos e a sua aplicação em farmacocinética, descreve-se a metodologia de modelação utilizada no tratamento dos dados de ciclosporina. Os dados foram analisados usando a função quinModel da biblioteca nlme do programa S-Plus (versão 6).
A inferência “populacional” é baseada na máxima verosimilhança e as predições individuais são obtidas usando métodos Bayesianos.
Palavras chave: modelos não lineares de efeitos mistos, dados longitudinais,
ix
A
BSTRACT
Pharmacokinetic parameters characterize pharmacological processes within the body that dictate the time-concentration relationship of a drug. These parameters are used to adjust drug dose and serum concentrations in order to produce the desired pharmacological effect and to avoid adverse effects.
The data required to pharmacokinetics analysis consists of drug concentrations, obtained by a serial blood samples collected over time from each subject following cyclosporine dose. This type of data, named longitudinal data, require particular care in treating variability, since the intra-individual observations tend to be correlated.
Mixed effects models that, incorporate both fixed effects, which are parameters associated with an entired population, and random effects, which are associated to individuals randomly selected from a population, allow to model this dependency and accommodate intra and inter variability, making this approach a useful framework in pharmacokinetical data.
The fundamental aim of this work was to estimate pharmacokinetic parameters of oral cyclosporine, in renal transplant recipients, using longitudinal data analysed by nonlinear mixed effects models, also refered to as nonlinear mixed models, and identify possible variables responsible for pharmacokinetics variability.
After a summary of the basic concepts of pharmacokinetics and the theoretical basics of nonlinear mixed effects models and its application to pharmacokinetics, follows the description of modelling methodology of cyclosporine data.
Statistical analyses were made using quinModel function in nlme library of the
S-Plus (version 6). “Population” inference was based on a maximum likelihood method and
individual preditions were based on a Bayesian approach.
Key words: non linear mixed effects models, longitudinal data, pharmacokinetics,
xi
A
BREVIATURAS E SÍMBOLOS
Principais abreviaturas e símbolos utilizados no âmbito da Farmacocinética:
τ intervalo de administração
AUC área sob a curva (area under the curve)
AUC0∞ área sob a curva após dose única
AUCtss área sob a curva em estado estacionário
AUMC área sob a curva do primeiro momento (area under the first-moment curve)
AUMC0∞ área sob a curva do primeiro momento em dose única
C0 concentração do fármaco no tempo zero
Cl clearance
Cmáx concentração máxima do fármaco após administração Cmáxss concentração máxima do fármaco em estado estacionário Cmin concentração mínima do fármaco
Cminss concentração mínima do fármaco em estado estacionário Ct concentração do fármaco no tempo t
CT concentração do fármaco no fim da perfusão (antes do estado estacionário) CTss concentração de fármaco no fim da perfusão em estado estacionário
CYP P450 sistema enzimático citocromo P-450
D dose
dCt/dt alteração da concentração do fármaco ao longo do tempo
dM/dt alteração da quantidade de fármaco ao longo do tempo
F fator de biodisponibilidade IV intravenoso k0 velocidade de perfusão (D/τ) Ka constante de absorção Ke constante de eliminação KM constante de Michaelis-Menten
LADME libertação, absorção, distribuição, metabolismo e excreção MRT tempo médio de residência (mean residence time)
n número de doses administradas SS estado estacionário (steady-state)
xii
t tempo
T tempo de perfusão
t0 tempo de latência (lag-time)
T1/2 tempo de semi-vida
TDM monitorização sérica da terapêutica farmacológica (therapeutic drug monitoring)
tDn tempo após administração ao fim da dose D1, D2, ..., Dn
tmáx tempo pós administração correspondente à concentração máxima Vd volume de distribuição
Vmáx velocidade máxima da capacidade enzimática
(Fitzmaurice, Davidian, Verbeke, & Molenberghs, 2009)
(De Vito, Crass, Blum, Pleasants, & Schentag, 1985; Sheiner, Rosenberg, & Marathe, 1977)
(Pillai, Mentré, & Steimer, 2005)
(Box, Jenkins, & Reinsel, 1994; Diggle, Liang, & Zeger, 1994)
(Medronho, Carvalho, Bloch, Luiz, & Werneck, 2005; Zeger, Liang, & Albert, 1988) (Nothdurft, Kublin, & Lappi, 2006)
(Maitre, Buhrer, Thomson, & Stanski, 1991; Mandema, Verotta, & Sheiner, 1992; Wade, Beal, & Sambol, 1994; Wählby, Jonsson, & Karlsson, 2002)
(R. J. Bauer, Guzy, & Ng, 2007)
(Porta Oltra, Pérez Ruixo, Jiménez Torres, & Pallardó Mateu, 2004; Rui, Zhuo, Jiang, & Chen, 1995; Schädeli, Marti, Frey, & Uehlinger, 2002)
(Savic, Jonker, Kerbusch, & Karlsson, 2007) (Lindstrom & Bates, 1990)
(Brendel et al., 2007; Dartois et al., 2007; Tornøe, Agersø, Nielsen, Madsen, & Jonsson, 2004; Wade, Edholm, & Salmonson, 2005)
(Asberg et al., 2010; Ette, Williams, & Lane, 2004) (Jönsson, Henningsson, Edholm, & Salmonson, 2012)
1. I
NTRODUÇÃO
As ciências biomédicas apoiam-se em outras disciplinas, nomeadamente na estatística, para fazer inferências sobre a magnitude da resposta biológica de interesse. Frequentemente utilizam modelos construídos com base em dados experimentais, que representam o sistema biológico de interesse e permitem explorar a sua estrutura e comportamento. Uma vez que os dados experimentais estão sujeitos a erro, o objetivo principal desses modelos consiste em distinguir a “informação” do sistema, do ruído ou da componente aleatória do sistema (Bonate, 2005a).
O sistema biológico é então descrito por um modelo estatístico composto por uma parte determinística e uma parte aleatória, sendo a resposta biológica representada por uma variável aleatória, contínua ou discreta, cuja distribuição de probabilidade é utilizada para descrever a probabilidade de se observar um determinado valor da resposta de interesse (Davidian, 2007).
Dentro das ciências biomédicas, a farmacocinética é uma especialidade das ciências farmacêuticas, que estuda o percurso de um fármaco no organismo, recorrendo a modelos geralmente não lineares nos parâmetros, designados de modelos farmacocinéticos. Estes modelos, que representam a variação da concentração, ao longo do tempo, de um fármaco após a sua introdução no organismo, derivam da representação do organismo em compartimentos incorporando os pressupostos de como o organismo processa a absorção, a distribuição e a eliminação de um fármaco em cada indivíduo.
2 1. Introdução
Nesta área, a resposta de interesse consiste nas concentrações séricas de um fármaco ao longo do tempo para um conjunto de indivíduos, sendo o objetivo de um estudo farmacocinético, caracterizar as alterações dessa variável resposta ao longo do tempo no indivíduo. É igualmente objeto de interesse determinar se essas alterações se relacionam com um conjunto de características (fatores) fisiopatológicas dos indivíduos como, por exemplo, a idade, o peso, a medicação concomitante, as doenças associadas, etc..
Pode-se então dizer que, os dados resultantes dos estudos em farmacocinética são dados longitudinais contínuos com base nos quais se pretende compreender o comportamento “típico” do fármaco na população e em que medida ele varia entre os indivíduos, podendo este conhecimento ser aplicado na recomendação de posologias individualizadas de forma a tirar partido de todo o potencial terapêutico de um fármaco, maximizando a sua eficácia e segurança.
A complexidade da análise de dados longitudinais é aqui acrescida com o facto de os modelos farmacocinéticos serem, em geral, não lineares nos parâmetros. Os modelos não lineares de efeitos mistos são uma escolha natural na execução do objetivo de um estudo farmacocinético.
Neste trabalho o objetivo fundamental é estimar os parâmetros farmacocinéticos da ciclosporina, após administração oral, em doentes transplantados renais, através da aplicação de modelos não lineares de efeitos mistos. A estimação dos parâmetros farmacocinéticos reveste-se de particular interesse na medida em que têm significado fisiológico e caracterizam o comportamento do fármaco no organismo.
No Capítulo 2 deste trabalho são descritos alguns conceitos básicos de farmacocinética que permitem compreender a complexidade dos modelos e, assim, mais facilmente compreender e interpretar os capítulos seguintes. Não se trata de uma descrição exaustiva de todos os fenómenos envolvidos mas de um resumo dos conceitos necessários ao enquadramento da questão e à implementação dos modelos.
No Capítulo 3 apresenta-se o modelo não linear de efeitos mistos, a inferência a ele associada e a sua aplicação em farmacocinética.
No Capítulo 4 é descrita a farmacocinética da ciclosporina e os vários passos efetuados na construção do modelo ajustado aos dados. A validação e discussão do modelo são igualmente apresentadas.
A função quinModel da biblioteca nlme do programa S-Plus (versão 6) foi a usada
2. F
ARMACOCINÉTICA
A farmacocinética é um ramo da farmacologia e uma especialidade na área das ciências farmacêuticas que se desenvolveu significativamente nos últimos 30 anos. A farmacocinética estuda a absorção, a distribuição, o metabolismo e a excreção (ADME) do fármaco e dos seus metabolitos no organismo, assim como os fatores que os modificam, recorrendo a modelos matemáticos que, ao descreverem o trajeto do fármaco no organismo, permitem fazer previsões sobre a quantidade de fármaco disponível para
exercer ação fisiológica1.
A farmacocinética pode então considerar-se como o estudo do percurso, desde o seu input ao seu output, de um fármaco e seus metabolitos no organismo (Matos, 2004).
Habitualmente esse estudo recorre à determinação da concentração do fármaco e/ou dos seus metabolitos no sangue, por ser o líquido biológico de melhor acesso para descrever o perfil da concentração ao longo do tempo (t). Idealmente a concentração devia ser determinada no local de ação (recetores biológicos) mas, a maior parte das vezes, tal não é possível. Assim, utiliza-se a concentração do fármaco em amostras biológicas acessíveis, como o sangue, que estão em equilíbrio, e podem ser relacionadas, com o fármaco associado ao recetor (Boroujerdi, 2001).
Quando a concentração sérica do fármaco tem uma relação estabelecida com o efeito terapêutico e tóxico, as concentrações séricas constituem um indicador da adequabilidade
1 O estudo da relação do efeito biológico do fármaco e a sua concentração no organismo é conhecido por
4 2. Farmacocinética
da terapêutica. Nesse caso, pode ser definido um intervalo de concentrações terapêuticas (margem terapêutica) que, em termos clínicos, deve ser encarado com alguma flexibilidade (Burton, 2006).
A análise farmacocinética é determinante no estabelecimento da posologia recomendada durante o desenvolvimento de novos fármacos mas, na prática clínica, está indicada fundamentalmente na individualização posológica de fármacos com elevada variabilidade interindividual e margem terapêutica estreita (Matos, 2004). Na prática clínica, esta atividade é habitualmente designada de monitorização sérica (TDM – therapeutic drug monitoring).
Os processos farmacocinéticos de input e output de fármaco podem ser caracterizados essencialmente em dois tipos (Boroujerdi, 2001):
• Cinética linear ou de primeira ordem:
A velocidade de absorção, distribuição ou eliminação é proporcional à quantidade ou concentração do fármaco no organismo e é expressa por uma constante de proporcionalidade, K, sendo esta a situação mais comum.
• Cinética não linear, de ordem zero ou ainda cinética de Michaelis-Menten: A velocidade de absorção, distribuição ou eliminação é independente da quantidade ou concentração do fármaco no organismo e é expressa por uma constante. A administração contínua de um fármaco em bomba perfusora (absorção a taxa constante) ou a saturação do sistema enzimático responsável pelo metabolismo (eliminação a taxa constante) são exemplos desta situação.
2.1 LADME
A concentração de um fármaco no organismo é determinada por vários processos, como:
Libertação: corresponde à libertação do fármaco da forma farmacêutica;
Absorção: corresponde à transferência do fármaco do local de absorção para a corrente sanguínea;
Distribuição: resulta da movimentação reversível do fármaco da corrente sanguínea para os tecidos, onde exerce efeito farmacológico;
2. Farmacocinética 5
Metabolismo: consiste na conversão química do fármaco noutras entidades, designadas
de metabolitos, que podem ser ativas ou não;
Excreção: consiste na remoção do fármaco do organismo através de um orgão de excreção (rim, fígado, pulmão ou outro).
O processo conjunto de metabolismo e excreção é habitualmente designado de eliminação. Os fármacos podem ser administrados por via vascular, diretamente na corrente sanguínea, ou extravascular. A absorção só está presente na administração extravascular.
O processo de LADME é condicionado por vários fatores que determinam a variabilidade entre indivíduos (Taylor & Caviness, 1986). Nas secções seguintes descreve-se resumidamente o processo e os parâmetros farmacocinéticos que o caracterizam.
2.1.1 L
IBERTAÇÃO E ABSORÇÃOA libertação do fármaco da forma farmacêutica envolve a desagregação da forma farmacêutica, que lhe serve de veículo, e a sua dissolução nos líquidos biológicos. Esta fase condiciona a absorção que, por sua vez, depende de vários fatores (Boroujerdi, 2001), pelo que, nem todo o fármaco administrado é absorvido e fica disponível na corrente sanguínea. A libertação e a absorção são caracterizadas pelos parâmetros: fator de biodisponibilidade (F) e constante de absorção (Ka). A biodisponibilidade consiste numa medida da velocidade e extensão de absorção da substância ativa que fica disponível para exercer efeito biológico, e em que, o fator de biodisponibilidade representa a percentagem ou a fração de dose (D) administrada que atinge a circulação sistémica.
A constante de absorção traduz a fração da quantidade de fármaco presente no local de absorção que é absorvida em cada momento.
O tempo que medeia entre a administração e o aparecimento de concentrações no sangue
designa-se de tempo de latência (t0) ou, na terminologia inglesa, lag-time. Esse tempo
representa um atraso no efeito terapêutico desejado.
Na fisiologia humana, um fármaco administrado por via oral pode sofrer metabolização no fígado, ou ser excretado no ar expirado, antes de alcançar a circulação sistémica geral. Este circuito é conhecido por metabolismo pré-sistémico ou efeito de primeira passagem.
6 2. Farmacocinética
Para um fármaco administrado por via extravascular, a biodisponibilidade é determinada pela fração absorvida e pela fração que, após absorção, escapa ao efeito de primeira passagem.
A extensão e a velocidade de absorção afetam o perfil de concentração de um fármaco. A área sob a curva concentração-tempo (AUC - area under the curve) é um indicador sensível da quantidade de fármaco que chega à circulação sistémica (extensão de absorção). A sua magnitude é diretamente proporcional à quantidade absorvida.
2.1.2 D
ISTRIBUIÇÃOUma vez atingida a circulação sistémica, o fármaco é distribuído aos tecidos e simultaneamente eliminado através dos orgãos de eliminação.
Alguns fármacos ligam-se às proteínas plasmáticas (albumina, α-1-glicoproteína ácida) e aos tecidos periféricos. Só o fármaco não ligado, ou livre, está disponível para ser distribuído aos tecidos, eliminado pelos orgãos de excreção e a interagir com o recetor para exercer efeito biológico (Winter, 1994).
O parâmetro farmacocinético utilizado para caracterizar a distribuição de um fármaco no organismo é o volume de distribuição (Vd). Este volume não corresponde necessariamente a um espaço fisiológico. O Vd corresponde a um volume hipotético que relaciona a concentração sérica do fármaco no organismo com a quantidade administrada.
2.1.3 E
LIMINAÇÃOO parâmetro farmacocinético clearance (Cl) descreve a eficiência do processo de eliminação. A clearance de um determinado orgão de eliminação (rim, fígado ou outro) é o volume de sangue, plasma ou soro, que é totalmente “limpo” de fármaco por unidade de tempo.
A maior parte do metabolismo é efetuado no fígado mas, outros orgãos ou tecidos, também podem contribuir, dando origem geralmente a metabolitos mais polares e, portanto, mais fáceis de ser excretados pela bílis, através das fezes, ou pelo rim, através da urina. Habitualmente esses metabolitos são inativos mas também podem ser formas ativas e esse fenómeno ser explorado com fins terapêuticos.
2. Farmacocinética 7
O metabolismo é catalizado principalmente por um sistema enzimático conhecido por citocromo P-450 (CYP P450). O CYP P450 é responsável por diversas interações medicamentosas, uma vez que, alguns fármacos podem competir para o mesmo sistema enzimático, e induzir ou inibir algumas enzimas, afetando assim o seu próprio metabolismo ou o de outros fármacos.
Os fármacos e/ou seus metabolitos excretados pela bílis são armazenados na vesícula biliar. Quando esta é esvaziada para o intestino, parte do fármaco pode ser reabsorvido, completando o processo conhecido como ciclo enterohepático. Como a ingestão de alimentos é um estimulador do esvaziamento da vesícula, podem ser observados picos de concentração secundários, de fármaco ou metabolitos, após as refeições.
O rim é o principal orgão responsável pela excreção de produtos endógenos e xenobióticos. Tal como na absorção, parte deste processo é mediado por transportadores específicos, como a glicoproteína-P, cuja atividade é potencialmente saturável.
A creatinina é um composto endógeno excretado por filtração glomerular. A clearance renal deste composto é considerada um marcador da função renal. Assim, na insuficiência renal, a clearance da creatinina é um parâmetro muito útil no ajuste posológico pois permite estabelecer um paralelismo com a clearance de um fármaco eliminado por via renal.
A eliminação do fármaco ocorre habitualmente por um processo de cinética linear. Neste tipo de cinética, a constante de eliminação (Ke) é um parâmetro, tal como a clearance, independente da concentração, que representa a fração de fármaco que é eliminada por unidade de tempo em cada momento. O Ke pode ser descrito da seguinte forma:
Vd Cl
Ke= (2.1)
O tempo de semi-vida (T1/2), que corresponde ao tempo necessário para que a
concentração do fármaco se reduza a metade, é outro parâmetro importante, pois,
determina o intervalo de administração do fármaco (τ) e o tempo necessário para atingir o
estado estacionário num regime de doses múltiplas (secção 2.2).
Ke
8 2. Farmacocinética
No entanto, alguns fármacos têm cinética de eliminação não linear, uma vez que as enzimas responsáveis pela metabolização (e transporte) ficam saturadas para a gama de concentrações da margem terapêutica. Quando se atinge a capacidade máxima de
metabolização (Vmáx), à medida que a concentração do fármaco aumenta, a capacidade
enzimática de metabolização mantém-se e, por isso, há uma redução da clearance.
O parâmetro KM (constante de Michaelis-Menten), representa a concentração a partir da
qual se observa saturação dos sistemas enzimáticos e é definido como a concentração de
fármaco no organismo correspondente a metade de Vmáx.
Em cinética não linear, o Vd não é afetado. No entanto, como a clearance, e consequentemente o tempo de semi-vida, é dependente da concentração, a utilidade, quer da clearance quer do tempo de semi-vida, é limitada (L. A. Bauer, 2001; Winter, 1994). Existe uma grande variabilidade relativamente à expressão qualitativa e quantitativa dos sistemas enzimáticos, pelo que, fármacos deste tipo são difíceis de monitorizar (Taylor & Caviness, 1986). Além disso, um fármaco com cinética linear, na gama de concentrações terapêuticas, pode apresentar cinética não linear numa situação de intoxicação.
2.2 E
STADOE
STACIONÁRIOHabitualmente uma única administração (dose única) de fármaco não é suficiente para se obter o efeito terapêutico desejado. Assim, o fármaco é administrado repetidamente obedecendo a um determinado intervalo de tempo entre administrações (doses múltiplas). Quando a velocidade de administração ou absorção iguala a velocidade de eliminação é atingido o estado estacionário (SS - steady-state). A disponibilidade do fármaco administrado, num dado intervalo de administração, substitui exatamente a quantidade de fármaco
perdido ou eliminado no intervalo anterior. As concentrações máximas (Cmáx) e mínimas
(Cmin) tornam-se constantes de intervalo para intervalo, e tendo uma dose e um intervalo de
administração adequados, oscilam na margem terapêutica (Figura 2.1). Na Figura 2.1,
CminSS, CmáxSS, tmáx, AUC0∞ e AUCtSS representam, respetivamente, a concentração mínima e
máxima em estado estacionário, o tempo correspondente à concentração máxima e a área sob a curva em dose única e em estado estacionário.
2. Farmacocinética 9
Num fármaco com cinética de eliminação linear, geralmente considera-se que o estado
estacionário é alcançado ao fim de cinco semi-vidas, 5 T× 1/2(Boroujerdi, 2001; Shargel &
Yu, 1999; Taylor & Caviness, 1986).
Para um fármaco com cinética não linear, se a taxa de administração for maior que a capacidade metabólica, o estado estacionário nunca é alcançado e o fármaco acumula-se indefinidamente (Winter, 1994).
Figura 2.1 - Perfil da concentração sanguínea em função do tempo de um fármaco hipotético
administrado em doses múltiplas por via oral.
2.3 A
NÁLISEF
ARMACOCINÉTICAExistem várias abordagens no estudo da farmacocinética, sendo as mais comuns a análise compartimental e a análise não compartimental.
A análise compartimental assenta na descrição matemática do declínio da concentração do fármaco no organismo ao longo do tempo através da utilização de uma amostra biológica como indicador. Em contrapartida, a análise não compartimental utiliza a área da curva concentração-tempo (AUC) como base para a estimação dos parâmetros farmacocinéticos. Estas duas últimas abordagens estão resumidas brevemente nas secções seguintes.
0 0 Tempo Conc ent ra ç ão t0 tmá x Cmá x SS Margem Terapêutica A U C0∞ A UCtSS CminS S Cm á xS S
10 2. Farmacocinética
2.3.1 A
NÁLISEC
OMPARTIMENTALNesta teoria, o organismo é representado como uma série de sistemas ou compartimentos que comunicam reversivelmente uns com os outros (Boroujerdi, 2001).
Um compartimento não é uma região fisiológica ou anatómica real, mas é considerado um tecido ou conjunto de tecidos, com fluxo sanguíneo e afinidade semelhantes para o fármaco, no qual este se distribui de forma uniforme e homogénea.
O fármaco move-se dinamicamente entre compartimentos, de tal forma que, cada molécula tem igual probabilidade de abandonar o compartimento. Este processo de troca de massas entre compartimentos é traduzido por constantes de velocidade (Boroujerdi, 2001; Rowland & Tucker, 1986). Desta forma, o modelo matemático envolvido é um sistema de equações diferenciais que expressam a velocidade a que a quantidade de fármaco, ou a sua concentração, ou o seu efeito farmacológico, é afetado ao longo do tempo em cada compartimento.
Os modelos podem ser mono ou multicompartimentais. Os modelos multicompartimentais são descritos por um compartimento central e um ou vários compartimentos periféricos. Geralmente assume-se que a eliminação ocorre a partir do compartimento central mas pode ser feita a partir dos compartimentos periféricos.
Na Figura 2.2, as constantes K representam a transferência de massa entre compartimentos. O compartimento central foi identificado como 1, e os compartimentos periféricos como 2 e 3. O valor 0 foi atribuído ao exterior do sistema. A constante Ka, que representa a constante de absorção, só está presente se se considerar uma administração extravascular. Os tecidos altamente perfundidos (fígado, coração, pulmão e rim) devido ao seu alto fluxo sanguíneo e ao rápido equilíbrio com a concentração do fármaco no sangue, são geralmente considerados como fazendo parte do compartimento central. Assim, o compartimento central, em muitos dos modelos, é composto pela circulação sistémica e pelos tecidos altamente perfundidos. O compartimento central corresponde habitualmente ao compartimento de amostragem.
Para incluir outras regiões do organismo que alcançam o equilíbrio com a concentração do fármaco na circulação sistémica de forma mais lenta, ou quando um fármaco tem determinada afinidade para um dado orgão ou região, são necessários modelos mais complexos com dois, três ou quatro compartimentos.
2. Farmacocinética 11
No entanto, a regra de ouro é incluir sempre o menor número possível de compartimentos consistente com o comportamento do fármaco, com a realidade fisiológica, e a facilidade de determinação dos parâmetros relevantes (Boroujerdi, 2001).
Figura 2.2 - Esquema de input e output de um fármaco no organismo na presença de um, dois e três
compartimentos.
MODELOS FARMACOCINÉTICOS
Os modelos farmacocinéticos procedem da integração analítica ou numérica das equações diferenciais (Rowland & Tucker, 1986)., sendo caracterizados pelos parâmetros farmacocinéticos referidos anteriormente (secção 2.1).
A distinção entre os vários modelos farmacocinéticos é feita, essencialmente, pelo número de compartimentos, pela cinética do seu input e output e pelo tipo de administração.
1 1 1 2 2 2 K12 K21 K 12 K21 K21 K12 K20 K10 K10 K20 1 compartimento 2 compartimentos 3 1 2 K12 K21 3 compartimentos 1 2 2 3 1 3 K31 K13 k20 K12 K21 K31 K13 k10 K12 K21 K31 K13 K30 3 1 1 K12 K21 1 2 2 3 2 3 k31 k13 k20 K12 K21 K31 K13 K20 K21 K12 K32 K23 K10 K30 K10 3 2 1 K21 2 1 1 3 2 3 K32 K21 K12 K32 K21 K12 K32 k10 K30 k20 3 2 1 K21 2 1 1 3 2 3 K32 k30 K21 K12 K32 K10 K21 K12 K32 K10 K30 k20 K23 K23 K23 K20 K12 K23 K23 K23 K20 K30 1 3 2 K12 K21 K31 K20 K30 K13 K10 Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka K10 1 Ka I II III IV V VI VII VIII IX X XI XII XIII XIV XV XVI XVII K12 1 1 1 2 2 2 K12 K21 K 12 K21 K21 K12 K20 K10 K10 K20 1 compartimento 2 compartimentos 3 1 2 K12 K21 3 compartimentos 1 2 2 3 1 3 K31 K13 k20 K12 K21 K31 K13 k10 K12 K21 K31 K13 K30 3 1 1 K12 K21 1 2 2 3 2 3 k31 k13 k20 K12 K21 K31 K13 K20 K21 K12 K32 K23 K10 K30 K10 3 2 1 K21 2 1 1 3 2 3 K32 K21 K12 K32 K21 K12 K32 k10 K30 k20 3 2 1 K21 2 1 1 3 2 3 K32 k30 K21 K12 K32 K10 K21 K12 K32 K10 K30 k20 K23 K23 K23 K20 K12 K23 K23 K23 K20 K30 1 3 2 K12 K21 K31 K20 K30 K13 K10 Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka Ka K10 1 Ka I II III IV V VI VII VIII IX X XI XII XIII XIV XV XVI XVII K12
12 2. Farmacocinética
Na Tabela 2.1 estão indicadas, para o modelo de um compartimento, com eliminação a partir do compartimento central, as equações diferenciais e as respetivas equações integradas para os modelos farmacocinéticos mais utilizados (Boroujerdi, 2001; Shargel & Yu, 1999).
Podem ser descritos outros modelos que acomodem, por exemplo, uma dose de carga seguida de doses de manutenção, eliminação mista (eliminação simultaneamente linear e não linear feita por diferentes orgãos de eliminação), etc., que tornam as expressões mais complexas.
Em modelos multicompartimentais, as equações têm, além disso, de acomodar as constantes de velocidade entre os compartimentos e, potencialmente, admitir eliminação também a partir dos compartimentos periféricos.
Um modelo de dois ou mais compartimentos pode ser mais realista na descrição do comportamento do fármaco no organismo, mas introduz dificuldades na interpretação fisiológica dos parâmetros obtidos (Boroujerdi, 2001).
Em análise compartimental, os métodos clássicos de estimação dos parâmetros farmacocinéticos, recorrem, sob determinados pressupostos, à análise gráfica e a linearizações dos modelos (Boroujerdi, 2001; Rowland & Tucker, 1986).
Nas últimas décadas, a existência de computadores com uma velocidade de cálculo cada vez maior tem levado ao desenvolvimento de métodos computacionais que permitem estimar os parâmetros farmacocinéticos diretamente por regressão não linear.
A biodisponibilidade (F), no entanto, dado o seu significado fisiológico, continua a ser estimada por análise não compartimental (secção 2.3.2), a partir da razão entre a AUC após administração oral e a AUC após administração endovenosa.
Os parâmetros farmacocinéticos obtidos no desconhecimento de F, não são os reais e designam-se de aparentes (Cl/F, Vd/F) (Shargel & Yu, 1999).
2. Farmacocinética 13
Tabela 2.1 - Descrição, gráficos e equações dos modelos de um compartimento mais frequentes em TDM, considerando eliminação a partir do compartimento
central, em dose única e em doses múltiplas.
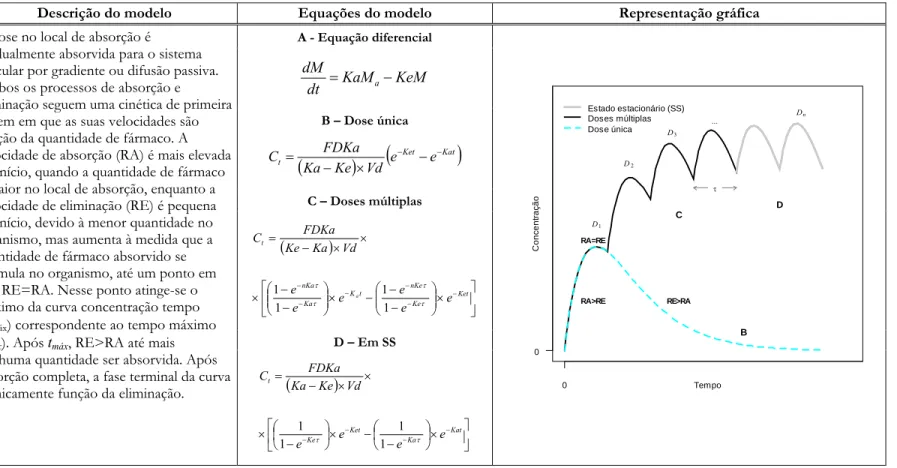
Tipo de modelo Descrição do modelo Equações do modelo Representação gráfica A - Equação diferencial KeM KaM dt dM a− = B – Dose única ( )
(
Ket Kat)
t e e Vd Ke Ka FDKa C − − − × − = C – Doses múltiplas ( ) ⎥ ⎦ ⎤ ⎢ ⎣ ⎡ × ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − − × ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − × × × − = − − − − − − Ket Ke nKe t K Ka nKa t e e e e e e Vd Ka Ke FDKa C a τ τ τ τ 1 1 1 1 D – Em SS I input e output de primeira ordem Ex.: administração oralA dose no local de absorção é
gradualmente absorvida para o sistema vascular por gradiente ou difusão passiva. Ambos os processos de absorção e
eliminação seguem uma cinética de primeira ordem em que as suas velocidades são função da quantidade de fármaco. A velocidade de absorção (RA) é mais elevada no início, quando a quantidade de fármaco é maior no local de absorção, enquanto a velocidade de eliminação (RE) é pequena no início, devido à menor quantidade no organismo, mas aumenta à medida que a quantidade de fármaco absorvido se acumula no organismo, até um ponto em que RE=RA. Nesse ponto atinge-se o máximo da curva concentração tempo (Cmáx) correspondente ao tempo máximo
(tmáx). Após tmáx, RE>RA até mais nenhuma quantidade ser absorvida. Após absorção completa, a fase terminal da curva
é unicamente função da eliminação. ( )
⎥ ⎦ ⎤ ⎢ ⎣ ⎡ × ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ − − × ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ − × × × − = − − − − Kat Ka Ket Ke t e e e e Vd Ke Ka FDKa C τ τ 1 1 1 1 0 0 Tempo Conc ent raç ão Estado estacionário (SS) Doses múltiplas Dose única B D C RA>RE RA=RE RE>RA D1 D2 ... D3 Dn τ 2. Farmacocinética 13
14 2. Farmacocinética
Tipo de modelo Descrição do modelo Equações do modelo Representação gráfica A - Equação diferencial KeM k dt dM = − 0 B – Antes de atingir o SS
(
Ket)
t e Vd Ke k C − − × = 0 1 C – Após de atingir o SS Vd Ke k Ct = ×0D– Fim de perfusão antes de atingir o SS
(t T)
Ke T t C e C = − −
E – Fim de perfusão após atingir o SS II input de ordem zero e output de primeira ordem Ex.: administração IV contínua, transdérmica ou oral de libertação prolongada Ket T t C e C = ss − 0 0 T e m p o Co nc ent ra ç ã o fi m d e p e r fu s ã o e m S S F i m d e p e r fu s ã o a n te s d e S S B D C E S S 0 T 0 T s s CT CT s s F - Dose única
(
KeT)
Ke(tT) t e e Vd Ke k C − − × − − × = 0 1 G – Doses múltiplas(
)
(t T) Ke Ke nKe KeT t e e e e Vd Ke k C − − − − − × ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − × × − × = τ τ 1 1 1 0 H –Em SS III input intermitente de ordem zero e output de primeira ordem Ex.: administração IV intermitenteO fármaco é introduzido na circulação sistémica a uma velocidade constante e eliminado do organismo a uma velocidade que depende da concentração no
organismo. No início a quantidade no compartimento central é pequena e a velocidade de eliminação também é pequena e menor que a velocidade de administração. À medida que a
administração continua, a quantidade no organismo acumula-se e a velocidade de eliminação aumenta gradualmente. Até se atingir um nível em que a quantidade administrada é semelhante à quantidade eliminada por unidade de tempo e a concentração no organismo permanece constante. Assim, a acumulação do fármaco no organismo pode ser vista como a diferença entre a velocidade de input e a velocidade de output. No estado estacionário deixa de haver acumulação e dM/dt=0, pelo que k0=KeM. Quando se pára a
administração do fármaco, a concentração decai a uma velocidade que é dependente da concentração.
(
)
(t T) Ke Ke KeT t e e e Vd Ke k C − − − − × ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ − × × − × = τ 1 1 1 0 0 0 T e m p o Con c ent ra ç ã o D o s e ú n i c a D o s e s m ú l ti p l a s E m e s ta d o e s ta c i o n á r i o T e m p o d e p e r fu s ã o (T) B F H B B G 0 T D1 D2 . .. D3 Dn τ 14 2. Farmacocinética2. Farmacocinética 15
Tipo de modelo Descrição do modelo Equações do modelo Representação gráfica A - Equação diferencial: KeM dt dM − = B - Dose única: Ket t e Vd D C = × − C – Doses múltiplas: Ket Ke nKe t e e e Vd D C − − − × ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − = ττ 1 1 D - Em SS IV input instantâneo e output de primeira ordem E.x: bólus IV Assume que:
• input no organismo e a transferência do fármaco do plasma para os tecidos é rápida; • a quantidade de fármaco no organismo no instante zero (t=0) é igual à dose (D) administrada;
• a velocidade de eliminação do fármaco (dM/dt) no tempo t é proporcional à quantidade do fármaco no organismo. Assim, dM/dt é variável e diminui à medida que a quantidade (M) de fármaco no organismo diminui, mas a constante de eliminação Ke é constante em todos os
tempos t. t Ke e Ket e Vd D C − − ⎟⎠× ⎞ ⎜ ⎝ ⎛ − = τ 1 1 0 0 T e m p o C onc ent raç ã o E s t a d o e s ta c i o n á r i o ( S S ) D o s e s m ú l ti p l a s D o s e ú n i c a B D C D1 D2 . . . D3 Dn τ A – Equação diferencial (equação de Michaelis-Menten) M t t máx t K C C V dt dC + × − = B – Dose única t M máx t C C t K V t C C0 0 ln − = − C - Doses múltiplas V input instantâneo e output de ordem zero Ex.: administração em bólus IV
Quando Vmáx diminui, aumenta o tempo necessário para eliminar uma dada quantidade de fármaco no organismo. Um aumento de KM, com Vmáx inalterado, aumenta o tempo de eliminação do fármaco do organismo. O KM não é uma constante de eliminação.
( )
Vd D t C Ct= (1) D1 = 1 Vd D t C t C C n D n D n t = n = n + − ( ) ) ( ( 1) ) ( 0 0 Te m p o Con c en tr aç ão B C D o s e s m ú ltip la s D o s e ú n ica D1 D2 ... D3 Dn τC0 – concentração do fármaco no tempo zero; CTss – concentração de fármaco no fim da perfusão após atingir o SS; Ct – concentração do fármaco ao fim do tempo t; CT – concentração do fármaco no fim da perfusão antes de atingir o SS; dCt/dt – alteração da concentração do fármaco ao longo do tempo; dM/dt – alteração da quantidade de fármaco ao longo do tempo; IV – intravenoso; k0 – velocidade de perfusão; n- número de doses administradas; t – tempo; T – tempo de perfusão; tDn– tempo após administração ao fim da dose D1, D2, ..., Dn. 2. Farmacocinética 15
16 2. Farmacocinética
2.3.2 A
NÁLISEN
ÃOC
OMPARTIMENTALA análise não compartimental faz uma análise direta das concentrações, independente dos pressupostos compartimentais e de transferência de massa, baseando-se nos momentos de uma variável aleatória.
A ideia chave é a de que a passagem de um fármaco pelo organismo pode ser considerada um processo estocástico sujeito a algumas flutuações aleatórias. Por exemplo, se o que se está a medir é a alteração da concentração de um determinado fármaco no organismo com o tempo, a alteração é função da variável independente “tempo” e a variável associada à alteração é uma variável aleatória (Boroujerdi, 2001).
Em teoria, um conjunto de observações concentração-tempo pode ser considerado realização de uma variável aleatória. Assim, considerando que, a variável aleatória associada às curvas concentração-tempo pode ser definida por uma função densidade de
probabilidade dada pela concentração do fármaco (Ct) no tempo (t), tem-se:
∫
+∞ ∞ = = 0 0 0 Ctdt AUC μ (2.3)∫
+∞ ∞ = = 0 0 1 tCtdt AUMC μ (2.4)A equação dada por (2.3) representa a área sob a curva da concentração vs tempo (AUC) em dose única e a equação (2.4) representa a área sob a curva do primeiro momento (AUMC – area under the first-moment curve) na mesma situação.
A determinação da AUC e da AUMC pode ser feita por métodos de integração ou mais simplesmente pela regra dos trapézios (Boroujerdi, 2001; Rowland & Tucker, 1986).
O parâmetro farmacocinético determinante na análise não compartimental é o tempo médio de residência (MRT – mean residence time). O MRT, que caracteriza o processo de eliminação do fármaco, corresponde à média do tempo em que o conjunto total de moléculas permanece no organismo e pode ser descrito como:
2. Farmacocinética 17 ∞ ∞ = 0 0 AUC AUMC MRT (2.5)
O MRT calculado desta forma relaciona-se com a teoria compartimental de tal forma que, para um compartimento, o MRT é equivalente às expressões indicadas na Tabela 2.2, donde é possível estimar os parâmetros farmacocinéticos habituais (Boroujerdi, 2001). Para mais compartimentos considera-se que o MRT total corresponde ao somatório do MRT de cada compartimento (Shargel & Yu, 1999).
Tabela 2.2 – Relação do MRT com o modelo de um compartimento e estimação dos parâmetros
farmacocinéticos a partir de dados de dose única.
1 compartimento Dose única Parâmetros farmacocinéticos
input instantâneo e output de
primeira ordem
(ex: bólus IV) Ke
MRTbólus = 1 AUMC MRT D AUC D Cl bólus total × = = 2 AUC AUMC D MRT Cl Vd total bólus × = × =
input ordem zero intermitente e output de primeira ordem
(ex: IV intermitente) 2 1 T Ke MRTperf = + AUMC MRT D AUC D Cl perf total × = = AUC T k AUC AUMC T k Vd 2 2 0 2 0 × − =
input de primeira ordem e output
de primeira ordem (ex: oral) Ke Ka MRTPO = 1 + 1 AUMC MRT FD AUC FD Cl PO total × = =
(
)
oral bólus bólus AUC Ka FD AUC MRT D Vd= × + ×1/k0 – velocidade de perfusão; T – tempo de perfusão.
A determinação de MRT, a partir de dados obtidos após doses múltiplas ou em estado estacionário, é mais complexa sendo rara a sua aplicação prática (Boroujerdi, 2001; De Vito et al., 1985; Perrier & Mayersohn, 1982; Smith & Schentag, 1984).
A análise não compartimental não é habitualmente utilizada, de forma independente, na estimação de parâmetros farmacocinéticos, mas em complementariedade com a análise compartimental.
18 2. Farmacocinética
2.4 A
NÁLISEP
OPULACIONALOs métodos de análise compartimental e não compartimental, descritos nas secções anteriores (2.3.1 e 2.3.2) exigem um elevado número de observações por indivíduo para descrever corretamente o perfil da curva concentração-tempo. Tal só é possível no ambiente controlado de um estudo farmacocinético.
Os estudos farmacocinéticos tradicionais apresentam rigorosos critérios de inclusão, em que os indivíduos incluídos são selecionados de modo a constituir um grupo homogéneo de doentes, uma vez que, as metodologias de análise envolvidas não permitem caracterizar a variabilidade interindividual (Tabela 2.3), sendo esta considerada uma fonte de ruído que deve ser eliminada (Ette & Williams, 2004a). Estes estudos são conduzidos, por isso, de forma artificial, não traduzindo a utilização normal do fármaco na prática clínica.
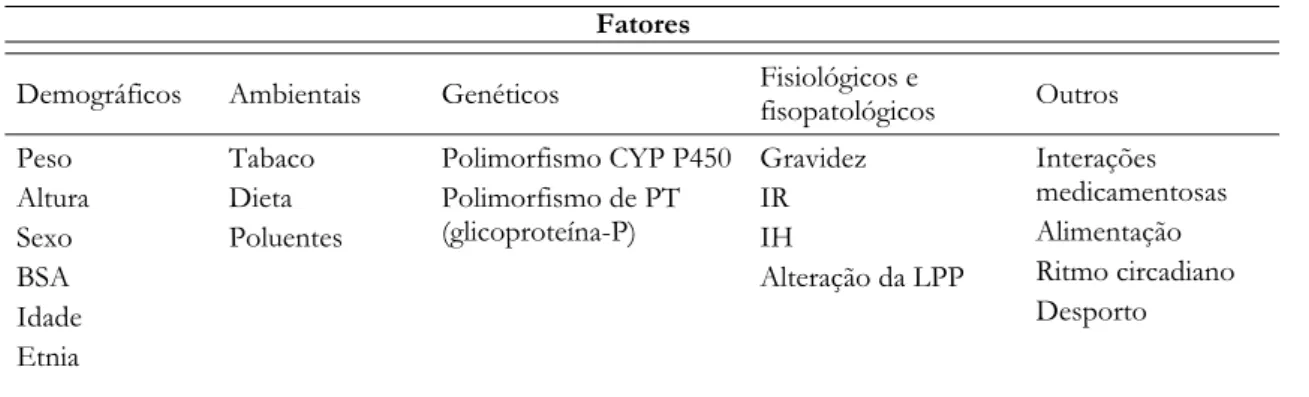
Tabela 2.3 - Fatores de variabilidade farmacocinética.
Fatores
Demográficos Ambientais Genéticos Fisiológicos e fisopatológicos Outros Peso Altura Sexo BSA Idade Etnia Tabaco Dieta Poluentes Polimorfismo CYP P450 Polimorfismo de PT (glicoproteína-P) Gravidez IR IH Alteração da LPP Interações medicamentosas Alimentação Ritmo circadiano Desporto
BSA – área de superfície corporal; IR – insuficiência renal; IH – insuficiência hepática; LPP – ligação às proteínas plasmáticas; PT – proteínas de transporte
No fim da década de 70, início da década de 80, o grupo de investigação liderado por Sheiner e Beal, publicou uma série de artigos (Sheiner & Beal, 1980, 1981a, 1983; Sheiner et al., 1977) que descrevem uma nova abordagem na análise farmacocinética, que mais tarde veio a ser designada por farmacocinética populacional, e um software, NONMEM, que permite implementar a análise estatística envolvida (modelos não lineares de efeitos mistos) (Bonate, 2005b), embora atualmente já existam outras alternativas informáticas (secção 3.7).
Nesta nova metodologia procura-se avaliar e quantificar fontes de variabilidade em vez de tentar eliminá-las, por isso, a utilização de um grupo de indivíduos da rotina clínica, que
2. Farmacocinética 19
representam a utilização normal do fármaco, é vantajosa. Conhecer a variabilidade e a sua magnitude é importante para estabelecer regimes posológicos adequados (Ette & Williams, 2004a).
Esta metodologia tem ainda a vantagem de poder ser aplicada a indivíduos com um pequeno número de observações (amostragem reduzida), pois, permite “emprestar” informação de indivíduos semelhantes, admitindo que o comportamento farmacocinético de um indivíduo deve ser semelhante ao de indivíduos com idênticas características.
Por este motivo, esta metodologia pode ser aplicada em populações especiais que, por razões éticas, não poderiam ser estudadas de outra forma: recém nascidos, idosos, imunodeprimidos, doentes críticos e oncológicos (Ette & Williams, 2004a), entre outros. Apesar de inicialmente a amostragem reduzida ter sido encarada com alguma reserva (Pillai et al., 2005), esta metodologia rapidamente ganhou adeptos sendo atualmente aprovada pela
FDA (Food and Drug Administration)2 e pela EMA (European Medicines Agency)3, quer para
dados da rotina, quer para desenvolvimento de novos fármacos (Committee for Medicinal Products for Human Use [CHMP], 2007; US FDA Center for Drug Evaluation and Research [CDER], 1999).
A análise estatística desta abordagem farmacocinética será o tema do próximo capítulo.
2 Agência americana do medicamento.
3 Agência europeia do medicamento anteriormente designada por European Agency for the Evaluation of Medicinal
3. M
ODELO
N
ÃO
L
INEAR DE
E
FEITOS
M
ISTOS
Os modelos não lineares de efeitos mistos para dados contínuos são uma metodologia de análise cada vez mais utilizada, quando os dados resultam de medições repetidas sobre os indivíduos, em particular, quando essas repetições são a consequência da medição da característica em estudo ao longo do tempo, isto é, quando se têm dados longitudinais, e em que a expressão matemática, que relaciona a variável resposta com as variáveis preditoras, é não linear nos parâmetros.
Essa relação não linear entre a variável resposta e, pelo menos, um dos parâmetros do modelo é muitas vezes baseada em modelos do mecanismo de produção da resposta, tendo geralmente uma interpretação física ou biológica (Pinheiro & Bates, 2000), como é o caso dos modelos farmacocinéticos discutidos no Capítulo 2.
A existência no modelo quer de efeitos fixos, parâmetros associados à população, quer de efeitos aleatórios, associados aos indivíduos, leva a que o modelo se designe de efeitos mistos (também designado por modelo não linear misto ou com efeitos mistos).
Os modelos não lineares de efeitos mistos são, por isso, modelos mistos em que um ou mais efeitos fixos e aleatórios ocorrem de forma não linear no modelo (Pinheiro & Bates, 2000).
Estes modelos têm particular interesse em farmacocinética onde, a partir de dados longitudinais, se pretende fazer inferência sobre as características subjacentes ao perfil dos indivíduos selecionados da população.
22 3. Modelo Não Linear de Efeitos Mistos
Em dados longitudinais, as observações sobre um indivíduo têm tendência a estar correlacionadas. Por outro lado, em dados que seguem um modelo não linear é frequente que a variância associada à resposta varie sistematicamente com a magnitude da resposta (heterocedastecidade). A incorporação de efeitos aleatórios vai permitir acomodar a dependência das observações no mesmo indivíduo e reconhecer e quantificar fontes de variabilidade.
Os modelos não lineares de efeitos mistos mereceram grande atenção por parte da comunidade científica nos finais dos anos 80. Durante os anos 90 assistiu-se a um grande desenvolvimento destes modelos com a criação de novas metodologias e técnicas computacionais para a sua análise. Hoje em dia são uma “ferramenta” de trabalho em várias áreas científicas existindo já programas estatísticos específicos para a sua aplicação. Neste capítulo apresentam-se os modelos não lineares de efeitos mistos, para a análise de dados contínuos com distribuição normal ou aproximadamente normal (Davidian, 2007), tendo em vista a sua aplicação a farmacocinética.
3.1
O M
ODELOConsidere-se uma amostra de N indivíduos i
(
i=1,..,N)
da população em estudo. Sejaij
y o valor da j-ésima resposta ou observação
(
j=1,...,ni)
do indivíduo i, obtida notempo tij, realização da variável aleatória (v.a.) Yij, a que se dá o nome de variável resposta.
Ao vetor Yi =
(
Y ,...,i1 Yini)
Tdas variáveis resposta dá-se o nome de perfil do indivíduo i,sendo o número total de observações dado por
∑
= = N i i n M 1 . Em dados longitudinais ao conjunto de medições repetidas sobre o mesmo indíviduo dá-se o nome de grupo.
O modelo não linear de efeitos mistos pode ser descrito em duas fases. A esta descrição em duas fases dá-se o nome de formulação hierárquica do modelo não linear de efeitos mistos (Davidian & Giltinan, 1995):
Fase 1 - Modelo individual (variação intra indivíduos)
(
ij i)
ijij f e
3. Modelo Não Linear de Efeitos Mistos 23
onde f é uma função não linear de um vetor de covariáveis xij, que sumarizam o
conjunto de condições a que o indivíduo i estava sujeito na observação j, e θ é um vetor i
1 ×
r de parâmetros, específico de cada indivíduo i . O termo eij é um erro aleatório que dá
a variabilidade intra individual e que se assume satisfazer a igualdadeE
(
eij|θi)
=0, o queimplica E
(
Yij |θi) (
= f xij,θi)
para cada j. Assume-se habitualmente a homocedastecidade eindependência do erro aleatório de modo que, cov
(
eij θi)
cov( )
eij Ini2
| = =σ , sendo I a ni
matriz identidade de ordem ni. A distribuição normal é a hipótese geralmente considerada
para a distribuição dos eij condicional a θ , tal que, i ei |θi ~ N
(
0,σ2Ini)
(Davidian &Giltinan, 1995, n.d.).
No âmbito da farmacocinética, a função não linear f é representada por um modelo
farmacocinético, sendo Yij a v.a. associada à concentração sérica do fármaco e xij um
vetor de covariáveis como o tempo, a dose, etc..
Fase 2 - Modelo populacional (variação entre indivíduos)
) , , ( i i i d a β b θ = i=1,..,N (3.2)
onde d é uma função r dimensional, β é um vetor p×1 de parâmetros fixos, ou efeitos
fixos, b é um vetor i q×1 de efeitos aleatórios e a é um vetor i a×1 de covariáveis
correspondendo aos atributos do indivíduo i. Cada elemento de d está associado com o
correspondente elemento de θ , de modo que a relação funcional pode ser diferente para i
cada elemento. A relação entre os efeitos fixos e aleatórios pode não ser linear (Davidian & Giltinan, 1995, n.d.; Fitzmaurice et al., 2009)
O vetor θ , caracteriza a variabilidade entre indivíduos. Esta variabilidade é atribuída a duas i
componentes, uma sistemática e outra aleatória. A componente sistemática contém a variabilidade atribuída à dependência sistemática do parâmetro relativamente às características do indivíduo (e.g., covariáveis como a idade, o peso, o sexo, etc.). A restante variabilidade, que não se consegue explicar, mas que deve ser incluída no modelo, é atribuída à componente aleatória.
24 3. Modelo Não Linear de Efeitos Mistos
O modelo populacional envolve assim a especificação de cada elemento de θ e portanto i
de d, de modo a modelar a dependência deste relativamente às componentes sistemática e
aleatória através da incorporação no modelo de covariáveis explicativas a e de efeitos i
aleatórios b , respetivamente (Davidian & Giltinan, 1995, n.d.). i
Geralmente assume-se que os b são independentes entre si e independentes dos i
(
)
T in 1 i i = e ,...,e i e e dos ai com: E(
bi|ai)
=E( )
bi =0 e cov(
bi|ai)
=cov( )
bi =D (3.3)onde D é uma matriz de variância-covariância definida positiva e idêntica para todos os
indivíduos, que caracteriza a magnitude da variabilidade não explicada pelas covariáveis nos
elementos de θ e as associações entre eles. Outra hipótese em geral considerada é a de i
( )
0 Dbi ~ N , (Davidian & Giltinan, n.d.).
No contexto da farmacocinética é frequente d ser uma função não linear das componentes
sistemática e aleatória. Considere-se como exemplo o modelo de um compartimento com input e output de primeira ordem, em dose única, apresentado no Capítulo 2 (Tabela 2.1).
(
)
(
Ket Kat)
t Ka Ke Vd e e FDKa C − − − × − = (3.4)Considerando que Ke=Cl/Vd (2.1), o modelo pode ser escrito da seguinte forma:
⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ − × ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ − = −Vdt −Kat Cl t e e Vd Vd Cl Ka FDKa C (3.5) sendo θ =i
(
)
T i i i Vd Cl Ka , ,(
r = ; admitindo que 3)
(
)
T i i i i = w,g ,Clcr a , com wi =peso, gi=idade e Clcri =clearance da creatinina, uma variável binária que assume o valor 1 se > 50
ml/min e 0 caso contrário, vai considerar-se a Cli a depender de wi e de Clcri, e Vdi a
depender de gi.
Tendo em atenção a relação acabada de indicar e, com base no facto de se saber que os parâmetros farmacocinéticos (Cl, Ka, Vd) são positivos e exibirem habitualmente uma
3. Modelo Não Linear de Efeitos Mistos 25
distribuição assimétrica com coeficiente de variação (CV) constante e, como tal, permitirem assumir uma distribuição log-normal (Fitzmaurice et al., 2009), o modelo sugerido para as componentes de d é dado por:
(
)
(
)
(
)
(
)
(
i i)
(
i i i)
i i i i i i i i i i i i i b Clcr w d Cl b g d Vd b d Ka 3 6 5 3 3 3 2 4 2 2 2 1 1 1 1 exp b , β , a exp b , β , a exp b , β , a + + + = = = + + = = = + = = = β β β θ β β θ β θ (3.6)onde bi = (b1i, b2i, b3i)T
(
q=3)
, β=(
β1,...,β6)
T(
p=6)
(Davidian & Giltinan, 1995). Neste modelo foi associado um efeito aleatório a todos os parâmetros, pois, do ponto de vista biológico, é improvável obter um parâmetro sem variabilidade ou em que a sua variabilidade esteja completamente explicada pelas covariáveis. No entanto, em alguns casos, o efeito aleatório pode ser retirado. A opção de remover o efeito aleatório é então uma opção estatística para obter a parcimónia do modelo (Davidian & Giltinan, n.d.). Com base em (3.6) garante-se que os parâmetros farmacocinéticos são positivos; alémdisso, se bi segue uma distribuição normal, então, os θ seguem uma distribuição log-i
normal (e como tal, assimétrica).
Em geral, a forma de d é baseada no conhecimento do problema combinado com considerações de ordem empírica e objetivos da inferência (Fitzmaurice et al., 2009). Embora a relação não linear entre d e os parâmetros seja frequente, alguns autores como Lindstrom & Bates (1990) restringem d à relação linear:
i i i
i Aβ Bb
θ = + (3.7)
onde Ai é uma matriz r×p dependente de covariáveis (cujas linhas dependem dos
elementos de a ) e Bi i é uma matriz r×q de zeros e uns que permite que alguns elementos
de θi não tenham efeito aleatório associado (Fitzmaurice et al., 2009).
No caso anterior, esta linearização pode ser facilmente obtida fazendo a reparametrização4: