Universidade Nova de Lisboa
Faculdade de Ciências e Tecnologia
Departamento de Química
Preparação de carvões activados a partir de biomassa e de
matrizes zeolíticas
Por
Margarida Simões Ferreira Proença
Dissertação apresentada na Faculdade de Ciências e Tecnologia da Universidade Nova de Lisboa para obtenção do grau de Mestre em Engenharia Química e Bioquímica
Orientadores: Professora Doutora Ana Paula Carvalho Professora Doutora Isabel Fonseca
Lisboa
i
COPYRIGHT
Autorizo os direitos de copyright da presente tese de mestrado, denominada “Preparação de carvões activados a partir de biomassa e de matrizes zeolíticas”.
iii
Agradecimentos
Quero agradecer às minhas orientadoras a Professora Doutora Ana Paula Carvalho e a Professora Doutora Isabel Fonseca por me terem aceite como aluna de mestrado e pelo seu entusiasmo, apoio, disponibilidade, partilha de conhecimentos, partilha de vida, foi um privilégio ter oportunidade de as conhecer e trabalhar com ambas. O facto de o meu trabalho de investigação poder ser desenvolvido em paralelo com uma componente humana maravilhosa, contribuiu grandemente para que, para além do gosto pelo trabalho científico desenvolvido, este fosse desenvolvido com vontade e com um sorriso e boa disposição. Para isso contribuíram também todas as pessoas que conheci na FCUL, em especial as minhas colegas de laboratório, Terese, Martinha, Ana Sofia, Cristina, Susana. Fizeram dos meus dias, dias melhores. Foi um privilégio conhecer-vos, e agradeço toda a atenção, disponibilidade, brincadeiras, tudo! Nunca poderia imaginar que seria tão bem recebida, num local onde não conhecia ninguém. Muito Obrigada por me fazerem sentir em casa. À Terese e à Martinha, um abraço mais que especial, estiveram lá desde o inicio e a vocês devo o meu sorriso ao chegar de manhã ao
laboratório e dar de caras com o sol a entrar por aquela JANELA ☺. Não posso deixar de
referir e agradecer em particular também à Sara Fernandes pois embora nos tenhamos cruzado durante um curto espaço de tempo, eu a chegar e ela a partir, foi uma querida e sempre disponível para qualquer dúvida, e ao Nuno Neng que me ajudou com essa bela ferramenta que é o Endnote. À Ana Sofia, que conheci mais tarde, um Obrigada, pela partilha de vida, alguns conselhos e um bom dia sempre bem-disposto. À Cristina, sempre atenta e que se preocupa e se lembra de tudo o que falamos, muito obrigada pela companhia e pela partilha de vida.
O momento em que agora me encontro é o culminar de vários anos de trabalho, de estudo, de aprendizagem… Tem sido um longo percurso académico. Todas as pessoas que se cruzaram comigo ao longo de todo este tempo e com quem aprendi, com quem estudei, com quem sorri, com quem partilhei vitórias e fracassos, os meus, os deles, os nossos. Todos influenciaram a pessoa que sou e que agora chega finalmente a esta meta. Todos os meus colegas e amigos da FCT. Há tanta gente a quem quero expressar o meu MUITO OBRIGADA, é uma gratidão conhecer-vos a todos, e ter a vossa amizade. Um agradecimento especial à Rachel, pelas nossas conversas intermináveis, pelos longos dias de estudo “pó binte!”, pelos conselhos e desabafos; ao Fábio, pela amizade de que já perdi a conta dos anos, as conversas sem fim e sobre todo e qualquer assunto, as palhaçadas e gargalhadas, e apoio, e o sorriso boa energia que tens sempre e que tiveste sempre que precisei; à Telma, e à sua força e todos os momentos que partilhamos desde o momento que entramos na FCT. A juntar a todas estas pessoas mais que especiais, todos os meus amigos de Linda-a-Velha, eternas amizades, à Joana, à Jessica, à Raquel. À Vânia, não há palavras para descrever todo o apoio que me deu, a minha gratidão transcende-me Amiga! Quero também, formalmente agradecer ao meu amigo Ruben Freire, que antes e depois da FCUL sempre foi um companheiraço, um amigo verdadeiro e sincero, obrigada!
disponível para ajudar e a quem agradeço a realização de todos os ensaios de caracterização e de catálise efectuados na FCT.
Um agradecimento também à Doutora Isabel Nogueira, do IST, pelas imagens TEM das amostras preparadas. À Engenheira Ângela Nunes, do ISEL, pela realização da calcinação na preparação do zeólito HY.
Um agradecimento muito especial ao Nuno Figueiredo, pelo apoio e incentivo que demonstrou nestes anos, a nunca desistir, pela alegria com que coloriu os momentos de cansaço ou de estudo e por partilhar profundamente das minhas alegrias e vitórias.
v
Sumário
Este trabalho foi desenvolvido em duas vertentes. Numa primeira fase, foram preparados carvões activados a partir de biomassa. Foi estudada a utilização de borras de café como precursor e recorreu-se ao carbonato de potássio como agente activante.
Numa segunda fase foram preparados carvões activados a partir de matrizes zeolíticas. Seguindo os procedimentos da literatura preparam-se dois tipos de amostras: usando a estrutura NaY como molde e o álcool furfurílico como precursor de carvão e usando a estrutura HY como molde e um polímero fenol-formaldeído como precursor de carvão
As amostras foram caracterizadas por adsorção de N2 e de CO2, difracção de raios X, espectroscopia de infravermelho por reflectância difusa, microscopia electrónica de transmissão, pHPZC, dessorção a temperatura programada e microanálise elementar.
Os carvões obtidos a partir da biomassa, revelaram uma natureza microporosa, e áreas
específicas aparentes que rondam os 1500 m2/g. Algumas amostras foram usadas como
catalisadores na reacção de abertura de anel do óxido de estireno, mostrando boas propriedades catalíticas, isto é conversões e selectividade elevadas.
Relativamente aos carvões obtidos a partir de matrizes zeolíticas, de um modo geral reproduziram-se os resultados da literatura, tendo-se em alguns casos obtido materiais
com áreas superficiais aparentes mais elevadas que as descritas.
Abstract
Carbon materials were prepared using as precursor biomass and using Y zeolite as template. In the former process, spent coffee grounds were used as precursor and K2CO3 as the chemical activation agent. In the latter, two experimental procedures, selected from the literature, were followed. One is based on the use of poly-furfuryl alcohol as precursor and NaY as matrix and in the other, phenol formaldehyde polymer and HY zeolite were the precursor and the template, respectively.
The characterization of the carbons materials samples included N2 and CO2 adsorption, measurement: X-ray Diffraction, Infrared Spectroscopy, Transmition Electron Microscopy, Elemental Analysis, pH at point zero charge and Temperature Programmed Desorption.
It was found that activated carbons prepared from biomass have a high amount of
micropores with BET surface area around 1500 m2/g. A sample which the surface was
functionalized was tested in ring open reactions of epoxides, and excellent results of conversion and selectivity, both close to 100%, were obtained.
Carbon materials prepared through Y zeolite templates have in general properties comparable to those found in the literature. A sample with BET surface area larger than the published data was obtained.
vii
Lista de abreviaturas e Acrónimos
AF - Álcool Furfurílico
BDDT - Brunauer, Deming, Deming, Teller
BET - Brunauer, Emmett, Teller
DR - Dubinin-Raduskhevich
DRX - Difracção de Raios X
FAU – Estrutura faujasítica
I.U.P.A.C. - International Union of Pure and Applied Chemistry
TEM - Microscopia Electrónica de Transmissão
Lista de Símbolos
αs - Valor correspondente a nads/n0,4 β - Coeficiente de afinidade
λ - Comprimento de onda
θ - Ângulo de Bragg
ABET - Área superficial aparente, determinada pelo método BET
am - Área média ocupada por uma molécula de adsorvato, na monocamada
B - Constante independente da temperatura e característica da estrutura porosa do adsorvente, obtida pela equação DR
b - Ordenada na origem
c - Constante da equação BET
d - Distância interplanas
E0 - energia característica de adsorção obtida pela equação de DR
E1 - Calor de adsorção relativo à primeira camada adsorvida
EL - Calor de condensação de vapor
m - Declive
NA - Número de Avogadro
nads - quantidade adsorvida à pressão p
a m
n - Quantidade adsorvida numa monocamada
p - Pressão
p/p°°°° - Pressão relativa
p°°°° - pressão de saturação do gás ou vapor R - Constante dos gases ideais
ix
VDR(N2) - volume microporoso obtido pela equação DR aplicada aos dados de adsorção de N2 a – 196 °C
VDR(CO2) - volume microporoso obtido pela equação DR aplicada aos dados de adsorção de CO2 a 0 °C
VMeso - Volume mesoporoso
VTotal - Volume poroso total
VαSuper - Volume supermicroporoso
VαTotal - Volume microporoso total
VαUltra - Volume ultramicroporoso
w0 - Volume microporoso obtido pela representação gráfica da equação DR
Índice
AGRADECIMENTOS... III
SUMÁRIO... V
LISTA DE ABREVIATURAS E ACRÓNIMOS... VII
LISTA DE SÍMBOLOS...VIII
ÍNDICE... X
ÍNDICE DE FIGURAS... XII
ÍNDICE DE TABELAS... XV
I. ESTUDO BIBLIOGRÁFICO... 1
I.1.CARVÕES ACTIVADOS... 1
I.1.1.Estrutura... 1
I.1.2. Preparação de carvões activados... 3
I.1.1.2. Carvões activados preparados a partir de biomassa...5
I.1.3. Carvões activados preparados a partir de uma matriz... 5
I.1.4. Catálise... 7
I.1.4.1. Reacções de abertura de anel de epóxidos...9
I.2.TÉCNICAS DE CARACTERIZAÇÃO DOS CARVÕES ACTIVADOS... 12
I.2.1. Adsorção de gases... 12
I.2.1.1. Isotérmicas de adsorção...14
I.2.1.2. Histerese e condensação capilar...16
I.2.1.3. Métodos de análise das isotérmicas de adsorção...18
I.2.1.3.1. Modelo e equação de Brunauer-Emmet-Teller (BET)...18
I.2.1.3.2. Método αs...20
I.2.1.3.3. Equação de Dubinin-Radushkevich (DR)...22
I.2.2. pH no ponto de carga zero (pHpzc)... 23
I.2.3. Dessorção a temperatura programada... 24
I.2.4. Análise elementar... 26
I.2.5. Difracção de raios X... 27
I.2.6. Espectroscopia de infravermelho por reflectância difusa... 28
I.2.7. Microscopia electrónica de transmissão... 28
II. PREPARAÇÃO E TÉCNICAS DE CARACTERIZAÇÃO DE CARVÕES ACTIVADOS A PARTIR DE BIOMASSA... 30
II.1.PREPARAÇÃO DAS AMOSTRAS... 30
xi
II.2.1. Adsorção de gases... 33
II.2.1.1. Adsorção de N2 a -196 °C...33
II.2.1.2. Adsorção de CO2 a 0 °C...34
II.2.2. pH no ponto de carga zero (pHPZC)... 35
II.2.3. Dessorção a Temperatura Programada... 35
II.2.4. Microanálise Elementar... 36
II.2.5. Ensaios catalíticos... 36
III. PREPARAÇÃO E TÉCNICAS DE CARACTERIZAÇÃO DE CARVÕES ACTIVADOS USANDO COMO MATRIZ O ZEÓLITO Y... 38
III.1.CARVÕES ACTIVADOS PREPARADOS A PARTIR DO ZEÓLITO NAY... 38
III.1.1. Impregnação e polimerização do precursor de carvão... 38
III.1.2. Carbonização... 40
III.1.3. Remoção da matriz zeolítica e lavagem do carvão... 40
III.2.CARVÕES ACTIVADOS PREPARADOS A PARTIR DO ZEÓLITO HY... 42
III.2.1. Preparação do zeólito HY... 42
III.2.2. Preparação do compósito zeólito-polímero... 42
III.2.3. Cross-link do polímero e calcinação... 43
III.2.4. Remoção da matriz zeolítica e lavagem do carvão... 44
III.3.TÉCNICAS DE CARACTERIZAÇÃO... 44
III.3.1. Difracção de raios X... 44
III.3.2. Espectroscopia de infravermelho por reflectância difusa... 45
III.3.3. Microscopia electrónica de transmissão... 45
IV. APRESENTAÇÃO E DISCUSSÃO DOS RESULTADOS... 46
IV.1.APLICAÇÃO DOS MÉTODOS DE ANÁLISE DAS ISOTÉRMICAS... 46
IV.2.CARACTERIZAÇÃO DOS CARVÕES PREPARADOS A PARTIR DE BIOMASSA... 51
IV.2.1. Ensaios de adsorção... 51
IV.2.2. Análise Elementar... 58
IV.2.3. pHPZC... 59
IV.2.4. Dessorção a Temperatura Programada (TPD)... 59
IV.2.5. Ensaios catalíticos... 61
IV.3.CARACTERIZAÇÃO DOS CARVÕES PREPARADOS A PARTIR DE UMA MATRIZ DE ZEÓLITO Y... 65
IV.3.1. Caracterização dos carvões preparados a partir do zeólito NaY... 65
IV.3.2. Caracterização dos carvões preparados a partir do zeólito HY... 71
CONCLUSÕES E PERSPECTIVAS FUTURAS... 78
BIBLIOGRAFIA... 81
ANEXOS...I
Índice de Figuras
Figura I. 1 – Representação da secção transversal de um hipotético material poroso com (a) poro fechado; (b) poro aberto numa extremidade, poro cego, poro em forma de “tinteiro” ou “garrafa”; (c) poro aberto em forma de cilindro; (d) poro aberto em forma de fenda; (e) poro aberto que atravessa o material; (f) poro aberto numa extremidade ou poro cego; (g)
rugosidade da superfície externa do material [2].... 2
Figura I. 2 – Representação da estrutura porosa de um carvão activado [3].... 3
Figura I. 3 - (a) Representação esquemática da supercavidade da estrutura porosa do zeólito Y (FAU), (b) representação dos canais interligados do zeólito Y.... 6
Figura I. 4 – Representação do processo de preparação de carvão activado a partir de uma matriz zeolítica.... 7
Figura I. 5 – Esquema da reacção genérica da alcoólise do óxido de estireno.... 10
Figura I. 6 – Mecanismos de reacção de abertura de anel do óxido de estireno utilizando um catalisador básico.... 10
Figura I. 7 – Mecanismo reaccional de abertura do anel de epóxido utilizando um catalisador ácido. ... 11
Figura I. 8 – Classificação da IUPAC para as isotérmicas de adsorção de gases (adaptada de [30]).. 15
Figura I. 9 – Classificação dos ciclos de histerese (processos de adsorção-dessorção irreversíveis), de acordo com a IUPAC (adaptado de [30]).... 17
Figura I. 10 – Representação αs hipotética para adsorventes microporosos.... 21
Figura I. 11 – Esquema da representação αs.... 21
Figura I. 12 - Grupos funcionais que podem ser encontrados na superfície dos carvões activados e libertados por dessorção, a temperatura programada, na forma de CO ou CO2.... 26
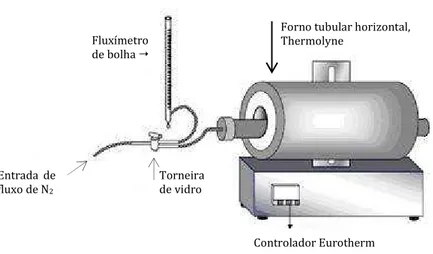
Figura II. 1 – Representação esquemática da montagem para carbonizações sob fluxo de azoto... 31
Figura II. 2 – Perfil térmico das activações. ... 31
Figura II. 3 – Equipamento ASAP 2010. ... 33
Figura II. 4 – Instalação volumétrica para estudos de adsorção (FCUL), onde foram realizados os ensaios de adsorção de CO2 a 0 °°°°C.... 34
Figura III. 1 –Preparação da amostra (a) NaY_AF_A e (b) NaY_AF_B, sob agitação.... 39
Figura III. 2 – Amostra (a) NaY_AF_A e (b) NaY-AF_B, após lavagem com tolueno e secagem em estufa a 100 °°°°C.... 39
Figura III. 3 - Perfil térmico da polimerização do álcool furfurílico no zeólito NaY.... 40
Figura III. 4 - Perfil térmico de carbonização para a obtenção do carvão a partir dos compósitos zeólito NaY/álcool furfurílico.... 40
xiii
Figura III. 7 - Perfil térmico da calcinação do polímero.... 44
Figura IV. 1 - Isotérmica de adsorção de N2 a -196 °°°°C da amostra NL/1:1/700. Os pontos de adsorção e
dessorção são representados, respectivamente, a vazio ( ) e a cheio ( )... 46
Figura IV. 2 - Representação gráfica da aplicação da equação de BET para a amostra NL/1:1/700.47
Figura IV. 3 – Representação gráfica da curva αs da amostra NL/1:1/700.... 49
Figura IV. 4 – Representações DR correspondentes à análise das isotérmicas de adsorção de (a) azoto a -196 °°°°C e (b) CO2 a 0 °°°°C na amostra NL/1:1/700.... 50
Figura IV. 5 - Isotérmicas de adsorção de azoto a -196 °°°°C (a) efeito do tipo de borra de café utilizada; (b) efeito da temperatura de activação; (c) efeito da variação da razão
K2CO3:biomassa. Os pontos de adsorção e dessorção são representados, respectivamente, a
vazio e cheio.... 51
Figura IV. 6 - Isotérmica de adsorção de azoto a -196 °°°°C da amostras NL/1:1/700(HNO3). Os pontos
de adsorção e dessorção são representados, respectivamente, a vazio (□) e cheio(■).... 55
Figura IV. 7 - Isotérmicas de adsorção de CO2 a 0 °°°°C das amostras preparadas a partir de biomassa.
... 56
Figura IV. 8 - Quantidade de grupos funcionais contendo oxigénio, determinada pela integração das curvas de CO2 e CO obtidas no espectro de TPD, usando uma função Gaussiana múltipla
[34, 37].... 61
Figura IV. 9 - Comparação da utilização de diferentes massas de catalisador, aplicadas à reacção de abertura de anel do óxido de estireno, utilizando como álcool o etanol.... 62
Figura IV. 10 - Comparação da utilização do etanol e do butanol como reagentes da reacção,
utilizando a mesma massa de catalisador (0,050 g).... 62
Figura IV. 11 – Isotérmicas de adsorção de azoto a -196 °°°°C das amostras NaY_AF_A e NaY_AF_B. Os pontos de adsorção e dessorção são representados, respectivamente, a vazio ( e ) e cheio( e ).... 66
Figura IV. 12 – Distribuição de tamanho de poros das amostras NaY_AF_A e NaY_AF_B, obtida pelo método DFT aplicado às isotérmicas de adsorção de N2 a -196 °°°°C.... 68
Figura IV. 13 – Difractograma de raios X do zeólito NaY, dos carvões NaY_AF_A e NaY_AF_B e da amostra NaY_AF_A antes de ser carbonizada.... 69 Figura IV. 14 - Espectros de Infravermelho do zeólito NaY, do carvão NaY_AF_B e da amostra
NaY_AF_B antes de ser carbonizada.... 70 Figura IV. 15 – Imagens obtidas por TEM dos carvões preparados a partir do zeólito NaY (a) amostra
NaY_AF_A (b) pormenor da amostra NaY_AF_A (c) amostra NaY_AF_B (d) pormenor da amostra
NaY_AF_B... 71
Figura IV. 16 - Isotérmicas de adsorção de azoto a -196 °°°°C das amostras HY_FF_500 e HY_FF_900. Os pontos de adsorção e dessorção são representados, respectivamente, a vazio ( e ) e cheio( e ).... 72
Figura IV. 18 - Difractograma de raios X do zeólito HY, dos carvões HY_FF_500 e HY_FF_900 e do compósito HY_FF.... 75
Figura IV. 19 - Espectros de infravermelho do zeólito HY e do carvão HY_FF_900... 76
xv
Índice de Tabelas
Tabela I. 1 – Grupos funcionais e respectiva temperatura de decomposição por dessorção a
temperatura programada e gás libertado [34].... 25
Tabela II. 1 – Designação das amostras de carvão preparadas a partir de biomassa por activação química com carbonato de potássio.... 32
Tabela III. 1 – Parâmetros de preparação das amostras feitas a partir de zeólito NaY e álcool furfurílico.... 41
Tabela IV. 1. - Parâmetros texturais das amostras obtidas a partir de biomassa.... 54
Tabela IV. 2 – Volumes porosos obtidos a partir da aplicação da equação DR às isotérmicas de adsorção de CO2 a 0 °°°°C e às isotérmicas de N2 a -196 °°°°C.... 58
Tabela IV. 3 – Resultados obtidos na análise elementar (% em massa) das amostras NL/1:1/700 e NL/1:1/700(HNO3).... 59
Tabela IV. 4 – Resultados obtidos nos ensaios de TPD das amostras NL/1:1/700 e
NL/1:1/700(HNO3).... 60
Tabela IV. 5 – Conversão e selectividade para a reacção de abertura de anel do óxido de estireno, para os diferentes ensaios realizados.... 63
Tabela IV. 6 – Características texturais das amostras NaY_AF_A e NaY_AF_B... 67
1
I. Estudo Bibliográfico
I.1. Carvões Activados
A síntese de carvões activados é um tema de investigação muito importante devido às características que estes materiais apresentam, relacionadas com a sua estrutura porosa e a sua química superficial.
As diversas aplicações deste tipo de materiais dependem das propriedades físicas e químicas da sua superfície nomeadamente, da distribuição de tamanho de poros, área superficial específica e volume poroso.
Estas propriedades, conjugadas com a crescente preocupação ambiental, resultam na aplicação industrial destes materiais em processos de purificação, separação e recuperação. Nestes processos os carvões activados são usados como adsorventes, podendo também em outras aplicações ser empregues como suporte de catalisadores.
No entanto, o seu uso como catalisador tem vindo a adquirir uma relevância cada vez maior, devido a serem materiais muito porosos, apresentando áreas específicas elevadas, serem muito estáveis termicamente e poderem ser obtidos a partir de diversos resíduos, nomeadamente resíduos lignocelulósicos.
I.1.1.Estrutura
podem ou não dar origem a grupos funcionais, e a sua presença ou ausência na superfície do carvão conferem-lhe propriedades químicas relevantes.
Por serem materiais com diversas características e propriedades é essencial que a sua caracterização seja exaustiva e a mais completa possível. Para tal deverão ser caracterizados texturalmente tendo em vista a determinação da área específica, volume poroso e distribuição de tamanho de poros. Devido à formação dos grupos funcionais na superfície dos carvões activados, estes deverão ser caracterizados utilizando técnicas adequadas à identificação desses grupos, tais como TPD, XPS, análise elementar, entre outras.
Na Figura I.1 apresentam-se os vários tipos de poros que, segundo a IUPAC [1], podem ser encontrados na estrutura dos carvões activados.
Figura I. 1 – Representação da secção transversal de um hipotético material poroso com (a) poro fechado; (b) poro aberto numa extremidade, poro cego, poro em forma de “tinteiro” ou “garrafa”; (c) poro aberto em forma de cilindro; (d) poro aberto em forma de fenda; (e) poro aberto que atravessa o material; (f) poro aberto numa extremidade ou poro cego; (g) rugosidade da superfície externa do material [2].
3
Figura I. 2 – Representação da estrutura porosa de um carvão activado [3].
Os microporos constituem a parte principal da área interna de um carvão activado e, em ensaios de adsorção, são preenchidos a pressões relativas baixas. Os microporos dividem-se em duas categorias, ultramicroporos, para poros com dimensões menores que 0,7 nm e supermicroporos, onde se enquadram os poros de dimensão 0,7-2 nm.
I.1.2. Preparação de carvões activados
Os carvões activados podem ser produzidos a partir de um material sólido, de origem natural ou sintética, desde que apresente um elevado teor de carbono. Este material é sujeito a tratamentos de modificação, a altas temperaturas, que permitem realizar uma decomposição controlada do sólido, promovendo o desenvolvimento de porosidade onde inicialmente se encontrava a massa de sólido [3].
As propriedades e características dos carvões activados vão depender do tipo de material utilizado e dos processos químicos e de transformação a que este for sujeito.
Os métodos de preparação de carvões activados podem dividir-se em duas categorias: a activação física e a activação química.
vapor de água, ar, oxigénio), numa gama de temperaturas entre 800-1000 °C, levando a um acentuado desenvolvimento da porosidade.
A activação química implica a carbonização de uma mistura de matéria-prima com um agente activante, sob atmosfera inerte, a temperaturas entre os 400 e os
1000 °C, seguida da remoção de compostos químicos por lavagem minuciosa do
material obtido.
A vantagem da activação química relativamente à activação física, está relacionada com a obtenção de rendimentos superiores, resultantes do facto de a activação química se processar numa única etapa. Além disso realiza-se a temperaturas de activação mais baixas, o que permite o desenvolvimento de porosidade a temperaturas inferiores do que no processo da activação física [1]. Por outro lado, o processo de activação química exige que seja feita uma lavagem após a activação para que o agente activante seja eliminado assim como os produtos resultantes da degradação.
O agente activante usado na activação química pode ser o cloreto de zinco [4], o ácido fosfórico [5-7], o hidróxido de potássio [8-10] ou o carbonato de potássio [11, 12], entre outros. A utilização do ZnCl2 ou do H3PO4 como agente activante, apesar de permitir a obtenção de carvões activados com uma porosidade bem desenvolvida e área superficial específica elevada, tem vindo a ser descontinuada devido a problemas de contaminação ambiental derivados aos resíduos de zinco e da eutrofização provocada pelos compostos de fósforo. O KOH, apesar de permitir obter carvões com elevada capacidade de adsorção, é uma substância perigosa e corrosiva, o que não contribui para a massificação da sua utilização actualmente.
5
I.1.1.2. Carvões activados preparados a partir de biomassa
Tal como foi referido anteriormente, os carvões activados podem ser preparados a partir de qualquer material com uma composição rica em átomos de carbono. É conveniente que a matéria-prima escolhida tenha um custo extremamente reduzido ou que seja um resíduo que possa ser reaproveitado.
Existe uma vasta gama de materiais que têm vindo a ser utilizados como precursores na síntese de carvões activados, entre eles, sub-produtos agrícolas [13-15], caroços de azeitona [16-18], de pêssego [13, 14, 17, 19-21], cascas de amêndoa [16], de arroz [14] ou resíduos de café [22].
O processo de preparação dos carvões activados é dispendioso, uma vez que decorre a temperaturas elevadas, encarecendo a exigência energética, e o rendimento obtido é baixo, derivado da perda de massa significativa que ocorre durante o processo de activação. Por este motivo o material de partida deve ser barato, sendo uma vantagem usar como precursores desperdícios agrícolas ou industriais.
A valorização de um resíduo sólido tem impacto do ponto de vista ambiental e económico, pois permite obter um material de alto valor acrescentado.
Neste estudo usou-se como precursor de carvão activado borras de café, uma vez que é um material orgânico, rico em carbono e de fácil obtenção.
I.1.3. Carvões activados preparados a partir de uma matriz
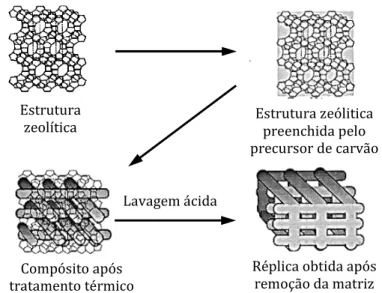
Esta metodologia de preparação dos carvões activados implica que o espaço livre existente na matriz zeolítica seja preenchido com um composto rico em átomos de carbono, o qual dá origem ao carvão activado após a etapa de carbozinação. Depois da remoção da matriz com ácido, cria-se uma estrutura porosa que reproduz a estrutura espacial inicialmente observada no zeólito [23].
O uso de zeólitos para a produção de novos materiais de carbono pode dar origem a materiais microporosos ou mesoporosos, dependendo do “template” utilizado. Na
literatura encontram-se vários estudos sobre o uso do zeólito Y [23-25], zeólito β [23,
26], mordenite [26] , zeólito L [23, 26] e ZSM-5 [26] para a preparação de carvões
activados e as diferenças estruturais que se observam consoante o zeólito usado como “template”.
No presente trabalho, foi utilizado o zeólito NaY, assim como a sua fórmula ácida, HY para a preparação de carvões activados.
O zeólito Y, também denominado por zeólito FAU, apresenta uma estrutura porosa constituída por supercavidades de diâmetro interno de 13 Å, que se encontram ligadas a quatro outras cavidades por passagens circulares com diâmetro de 7,4 Å (Figura I.3.(a)) (o que vai permitir o acesso das moléculas das espécies que vulgarmente se utilizam como precursores de carvão). A estrutura porosa cria uma complexa rede de canais interconectando as cavidades entre si (Figura I.3.(b)) sendo esta a razão pela qual este zeólito assume uma posição de destaque na síntese de carvões porosos.
Figura I. 3 - (a) Representação esquemática da supercavidade da estrutura porosa do zeólito Y (FAU), (b) representação dos canais interligados do zeólito Y.
7
Ao preencher a matriz zeolítica com uma fonte de carbono, e após o processo de calcinação e lavagem desse compósito é possível obter uma estrutura carbonácea estruturada de acordo com a complexa rede criada pela porosidade do zeólito, tal como ilustrado pela Figura I.4.
Figura I. 4 – Representação do processo de preparação de carvão activado a partir de uma matriz zeolítica.
I.1.4. Catálise
O objectivo primordial de cada processo químico é a transformação de matérias-primas em produtos, recorrendo a processos de separação e de reacção. No entanto, o produto pretendido pode não ser o único a ser obtido a partir de uma dada combinação de reagentes. A utilização de catalisadores numa reacção permite melhorar a selectividade e aumentar a velocidade de reacção. Optimizando a selectividade dos catalisadores reduz-se a produção de resíduos, sendo o processo mais limpo. Uma vez que o catalisador aumenta a velocidade de reacção, diminui a exigência energética assim como o consumo de matérias-primas [27].
As vantagens referidas acima são extremamente importantes quer na indústria química e farmacêutica, como no sector da energia e transportes, na produção de combustíveis e no controlo da poluição, revelando-se essenciais para um desenvolvimento sustentável.
Lavagem ácida Estrutura
zeolítica Estrutura zeólitica preenchida pelo precursor de carvão
Réplica obtida após remoção da matriz Compósito após
De acordo com a IUPAC, o termo “Catálise” define o fenómeno pelo qual uma quantidade relativamente pequena de um material estranho à estequiometria -o catalisador-aumenta a velocidade de uma reacção química, não sendo consumido no processo [27].
Ao longo do tempo, ocorre a desactivação dos catalisadores, verificando-se uma diminuição de velocidade de reacção e por vezes também um decréscimo da selectividade.
Dentro da catálise podemos distinguir:
Catálise homogénea ou catálise molecular, que ocorre quando os reagentes e o catalisador se encontram todos na mesma fase.
Catálise heterogénea, que se verifica quando os reagentes e o catalisador se encontram em fases diferentes. Neste tipo de catálise são possíveis diferentes combinações de fases, o que faz com que a reacção ocorra na interface das mesmas. Além disso envolve a existência de centros activos na superfície do catalisador. Uma das vantagens relativamente à catálise homogénea é o facto de o processo de separação do catalisador ser substancialmente mais fácil e daí mais favorável do ponto de vista ambiental e económico.
Catálise enzimática ou biocatálise, utiliza uma enzima como catalisador, o que a coloca num carácter intermédio entre os dois tipos de catálise anteriores.
O comportamento que determinadas substâncias apresentam, ao serem usadas como catalisadores varia, apresentando actividades diferentes assim como selectividades distintas. A actividade de um catalisador relaciona-se com o efeito sobre a velocidade da reacção e podem ser comparados catalisadores diferentes, tendo em conta os seguintes parâmetros [27]:
- temperatura isocinética, ou seja, a temperatura à qual se atinge uma determinada velocidade de reacção
9
- conversão em condições pré-fixadas das variáveis processuais - constantes cinéticas
- tempo de contacto necessário para obter uma conversão pré-fixada, utilizando um reactor específico
No decorrer da reacção, a acção de um catalisador poroso pressupõe uma sequência de processos, que vão constituir o mecanismo da reacção catalítica, e que envolvem (no caso de se tratar de catálise heterogénea) o transporte dos reagentes da fase fluida até à superfície da partícula de catalisador, o transporte dos reagentes no interior dos poros da partícula de catalisador, a adsorção de reagentes nos centros activos, a ocorrência da reacção química entre as espécies adsorvidas, a dessorção dos produtos adsorvidos, o transporte dos produtos através dos poros até à superfície externa de catalisador e, por fim, o transporte dos produtos da superfície externa da partícula de catalisador para a fase fluida [27].
Se se tratar de um catalisador não poroso ou de um catalisador cujos centros activos se encontram na periferia das partículas o mecanismo reaccional será mais simplificado do que o descrito anteriormente, ocorrendo as etapas de transporte no interior da partícula de catalisador.
I.1.4.1.
Reacções de abertura de anel de epóxidos
No decorrer deste trabalho foi escolhida uma reacção de abertura de anel de um epóxido para estudar as propriedades catalíticas dos carvões activados preparados.
Os epóxidos são compostos muito versáteis e muito usados em síntese orgânica e em
química fina para a produção de β-hidroxiéteres, solventes orgânicos de extrema
importância e intermediários na síntese de α-alcóxiacetonas e α-alcóxiacidos [28].
O
+
R OHO OH
R
+
OH O R
Óxido de Estireno Álcool (P1) (P2)
Figura I. 5 – Esquema da reacção genérica da alcoólise do óxido de estireno.
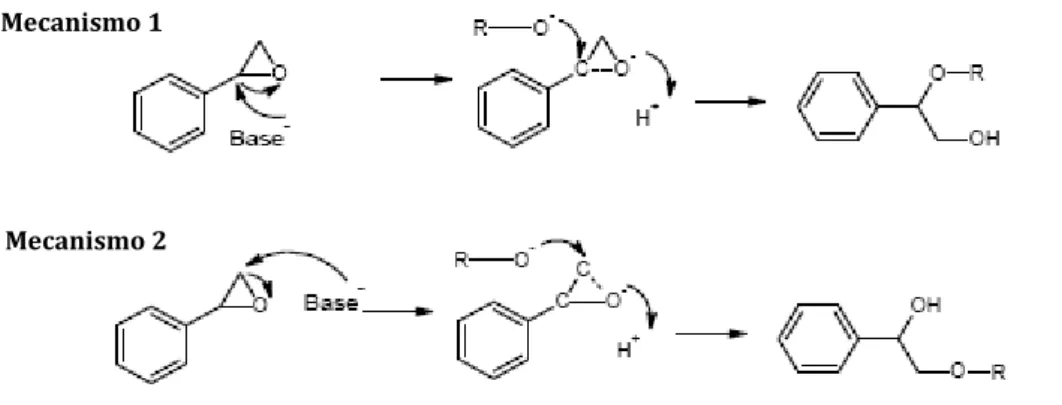
Se for utilizado um catalisador a selectividade para cada um dos produtos, P1 e P2, varia.
Se se utilizar um catalisador básico, espera-se que o produto obtido seja o P1, de acordo com um dos mecanismos indicados na Figura I.6. O mecanismo mais provável é o mecanismo 2, pois a abertura do anel é feita pelo carbono menos substituído, onde o impedimento esteroquímico provocado pelo anel é menor do que no caso do mecanismo 1, onde o ataque é feito ao carbono mais substituído e consequentemente sob um efeito esteroquímico muito mais elevado.
Figura I. 6 – Mecanismos de reacção de abertura de anel do óxido de estireno utilizando um catalisador básico.
Ao ser utilizado um catalisador ácido, espera-se que o produto obtido seja o P2, de acordo com um dos mecanismos descritos na Figura I.7, consoante a reacção ocorra pelo ataque do álcool ao carbono substituído ou não. Esta reacção é do tipo SN2 e o mais provável é a reacção ocorrer segundo o mecanismo 1, uma vez que o intermediário que se forma é mais estável que no caso do mecanismo 2.
Mecanismo 1
11
Mecanismo 1
Mecanismo 2
I.2. Técnicas de caracterização dos carvões activados
As propriedades texturais e químicas têm um papel relevante no comportamento dos carvões activados como adsorventes ou suporte de catalisadores.
Estas propriedades foram estudadas nos carvões activados preparados no decorrer deste trabalho, recorrendo às técnicas de caracterização descritas neste capítulo.
I.2.1. Adsorção de gases
O método da adsorção de gases permite a caracterização da micro e mesoporosidade dos materiais, revelando-se por isso de extrema utilidade para a caracterização dos mesmos.
A adsorção é considerada como sendo um processo espontâneo que ocorre sempre que a superfície de um sólido é exposta a um gás ou a um fluído. Ao fazer contactar o gás com a superfície do sólido, dá-se o aumento da concentração do gás na vizinhança do sólido e cria-se uma interface gás-sólido.
A terminologia utilizada no processo de adsorção refere-se ao termo adsorvente para designar o material sólido com capacidade de adsorção, adsorvível ao gás ou fluído que irá ser adsorvido na superfície sólida e adsorvato quando esse mesmo gás ou fluído já foi adsorvido na superfície do sólido.
13
e o adsorvato. Os dois fenómenos de adsorção podem ser distinguidos de acordo com os seguintes critérios:
- A adsorção física apresenta um baixo grau de especificidade, enquanto que a adsorção química depende da reactividade existente entre o adsorvente e o adsorvível, conferindo-lhe maior grau de especificidade.
- Ao ocorrer a adsorção química de moléculas, estas encontram-se ligadas a centros activos existentes na superfície, pelo que o processo de adsorção ocorre em monocamada. Para pressões relativas elevadas, a adsorção física ocorre, normalmente, em multicamada.
- As moléculas adsorvidas fisicamente mantêm a sua identidade após ocorrer a dessorção, retornando à sua forma original. No entanto se uma molécula for adsorvida quimicamente, é sujeita a uma reacção ou dissociação, perdendo a sua identidade original.
- O processo de adsorção química está associado a uma energia da mesma ordem de grandeza que uma reacção química, enquanto que, apesar de a adsorção física ser sempre um processo exotérmico, a energia envolvida é da mesma ordem de grandeza da energia de condensação do adsorvível. No entanto, esta energia poderá ser mais baixa se a adsorção ocorrer ao nível de poros muito estreitos.
- Na adsorção física o equilíbrio termodinâmico é rapidamente atingido, uma vez que a energia de activação é dispensável para a ocorrência do processo, ao contrário do que acontece com a adsorção química, em que, a baixas temperaturas, não é garantido que esse equilíbrio seja atingido.
- Como já foi referido acima, a adsorção química implica o estabelecimento de ligações químicas, logo o calor de adsorção é da mesma ordem de grandeza que o calor de reacção.
-196 °C, uma vez que a sua obtenção com um elevado nível de pureza é relativamente
acessível, sendo igualmente fácil de atingir e manter a temperatura de -196 °C, em
laboratório.
I.2.1.1. Isotérmicas de adsorção
O processo de adsorção de gás pode ser estudado e aprofundado através da realização de isotérmicas de adsorção, obtidas a partir da representação gráfica da extensão da
adsorção, isto é nads (usualmente expressa em mmol/g) em função da pressão relativa
(p/p0), onde p0 é a pressão de saturação do adsorvato, à temperatura a que decorre o
ensaio.
Pela observação das isotérmicas experimentais é possível tirar algumas conclusões sobre a textura porosa do material sendo, por isso, uma ferramenta importante no estudo da caracterização de um carvão activado.
15
Figura I. 8 – Classificação da IUPAC para as isotérmicas de adsorção de gases (adaptada de [30]).
■ Isotérmicas tipo I: caracterizam-se pela existência de um patamar que começa a ser
definido a partir de pressões relativas baixas. Estão associadas a materiais exclusivamente microporosos, como alguns carvões activados e zeólitos. O patamar corresponde ao preenchimento dos microporos e sua altura está relacionada com o volume deste tipo de porosidade. Quanto mais rectangular for a curva, mais estreita será a distribuição de tamanho de poros. Este tipo de isotérmicas está também associada ao fenómeno de adsorção química, onde o valor limite corresponderá à adsorção de uma monocamada nos centros activos.
■ Isotérmicas tipo II: traduzem uma adsorção em multicamada e estão associadas à
adsorção física que pode ocorrer em sólidos não porosos. São caracterizadas por possuir duas inflexões, uma para valores de pressão relativa menores que 0,1 e outra para valores superiores a 0,9. A primeira inflexão, assinalada na figura como o ponto B, é considerada como indicando o preenchimento da primeira camada adsorvida.
■ Isotérmicas tipo III: são pouco frequentes e traduzem-se numa isotérmica convexa
energética, comparativamente com a que ocorre no caso das isotérmicas tipo II. Correspondem a um mecanismo de adsorção em que as interacções gás-sólido são particularmente fracas.
■ Isotérmicas tipo IV: apresentam um patamar a pressões relativas altas, e podem
também apresentar um ciclo de histerese, que ocorre quando o mecanismo de preenchimento de mesoporos por condensação capilar é diferente do mecanismo de dessorção dos mesmos.
■ Isotérmicas tipo V: são características de materiais com micro e mesoporos, em que a
adsorção é baseada numa fraca interacção gás-sólido. Devido a este facto, são de difícil interpretação e pouco comuns.
■ Isotérmicas tipo VI: estão associadas a superfícies uniformes não porosas e traduzem
um mecanismo de adsorção em multicamada. Cada patamar, ou degrau, representa a formação de uma camada, a pressões relativas diferentes. Nestes casos ocorre o fenómeno de adsorção cooperativa em que as camadas que vão sendo adsorvidas vão facilitar a adsorção da camada seguinte, ou seja, a interacção entre as camadas é superior à afinidade entre a superfície e o adsorvato.
I.2.1.2. Histerese e condensação capilar
Quando é feita a adsorção de gases em sólidos porosos, pode ocorrer que a curva de adsorção não coincida com a curva de dessorção, formando-se um ciclo de histerese. Isto deve-se ao facto de, no interior dos poros, a condensação e a evaporação do adsorvato ocorrerem a valores de pressão diferentes.
17
Figura I. 9 – Classificação dos ciclos de histerese (processos de adsorção-dessorção irreversíveis), de acordo com a IUPAC (adaptado de [30]).
Histerese tipo H1: caracteriza-se por apresentar dois ramos da isotérmica praticamente paralelos, que serão tanto mais verticais quanto mais estreita for a distribuição de tamanho de mesoporos [27]. Este tipo de histerese está associada a materiais porosos constituídos por aglomerados rígidos de partículas esféricas de tamanho uniforme e ordenadas de forma regular.
Histerese tipo H2: apenas o ramo correspondente à dessorção é praticamente vertical. Está associada aos diferentes tipos de mecanismo de condensação e evaporação em poros em forma de garrafa.
Histerese tipo H3: ambos os ramos constituintes da isotérmica, adsorção e
dessorção, são assimptóticos relaticamente à recta p/p0=1. Ocorre normalmente
para agregados não rígidos de partículas em forma de placa, que dão origem a poros em fenda.
I.2.1.3. Métodos de análise das isotérmicas de adsorção
Existem vários métodos para analisar as isotérmicas de adsorção, de modo a obter os diversos parâmetros estruturais (área específica, volume poroso, distribuição de tamanho de poros).
De seguida apresentam-se os métodos usados no decorrer deste trabalho.
I.2.1.3.1. Modelo e equação de Brunauer-Emmet-Teller (BET)
O modelo de Brunauer, Emmet e Teller (BET), aplicado à análise das isotérmicas de adsorção, constitui uma poderosa ferramenta para a determinação da área superficial dos carvões activados.
Este modelo foi proposto em 1938 [31], para interpretar a adsorção em multicamada em sólidos não porosos com base na adsorção física devido a forças de van der Waals. Foi pois desenvolvido para analisar isotérmicas de adsorção do tipo II.
O modelo de BET admite o estabelecimento de um equilíbrio dinâmico entre a adsorção e a dessorção, considerando a possibilidade de ocorrer adsorção em multicamadas. Estas permissas são passíveis de explicar a forma geral da isotérmica tipo II e são compatíveis com o fenómeno de adsorção física, mas excluem o fenómeno de adsorção química.
A equação que define o modelo BET pode ser deduzida tendo em conta que:
›
A velocidade de adsorção é igual à velocidade de dessorção, para cada camadaadsorvida.
›
A partir da adsorção da segunda camada, o calor de adsorção mantém-se constante eigual ao calor de condensação do gás.
›
Quando se verifica a igualdade p=p0, o adsorvato condensa como um líquido ordinário19
Sendo assim, obtém-se a equação BET, que normalmente é utilizada na forma linear:
(
0)
00 1 1 1 p p c n c c n p p n p p a m a m − + = −
ads Equação I. 1
Onde nads corresponde à quantidade adsorvida à pressão p e à temperatura T; p0 é a
pressão de saturação do adsorvato à temperatura T; a
m
n a quantidade adsorvida necessária
para preencher uma monocamada e c é a constante de BET, dada por c = exp (E1 – EL/RT),
em que E1 e EL correspondem, respectivamente, ao calor de adsorção relativo à primeira
camada adsorvida e ao calor de condensação do adsorvato.
Esta equação foi aplicada aos dados experimentais obtidos através dos ensaio de adsorção de
azoto a -196 °C, representando-os graficamente na forma
(
0)
01 p p n
p p
−
ads vs
0
p p . O
declive (m) e a ordenada na origem (b) da recta ajustada, para uma gama restrita de pressões
relativas, permitem calcular a capacidade da monocamda a m
n (mol/g), pela aplicação da
equação:
b m
a
m = +
1
n Equação I. 2
Após determinar o número de moles adsorvidas na monocamada é então possível calcular a área específica, relacionando-as, pela fórmula:
ABET = nma NA am Equação I. 3
Onde NA corresponde ao número de Avogadro (6,022×1023 mol-1) e am é a área média
ocupada por uma molécula de adsorvato na monocamada. Para a adsorção de azoto a
-196 °C, am = 0,162 nm2 [32].
Como se referiu anteriormente a equação de BET foi originalmente deduzida e proposta para ser aplicada a isotérmicas tipo II, apresenta geralmente uma validade máxima de
desvios a partir de p/p° próximos de 0,1. No caso de materiais microporosos, a área superficial determinada pelo método BET pode não ter um verdadeiro significado físico, uma vez que a adsorção que ocorre neste tipo de materiais não é feita por sobreposição de camadas mas pelo preenchimento da microporosidade e, sendo assim, a área superficial determinada pelo método BET (ABET) deverá ser encarada com uma área superficial aparente.
I.2.1.3.2. Método αs
Este método tem como base a comparação da isotérmica de adsorção experimental com a isotérmica padrão do mesmo adsorvato num material não poroso com características de química superficial semelhantes ao material em estudo [27, 32].
Com o objectivo de determinar o volume microporoso, Sing [32], substituiu a variável
nads para ns, que traduz a quantidade adsorvida a um determinado valor de pressão
relativa (p/p0)s. Em termos práticos (p/p0)s=0,4 de modo a garantir o preenchimento
total dos microporos. Sendo assim é traçada uma isotérmica padrão, que divide os
valores de adsorção padrão, n, por n0,4, a quantidade adsorvida pelo material de
referência a (p/p0)s=0,4. O quociente n/n0,4 é assim designado por αs e a isotérmica para
o material de referência é obtida representando graficamente αs em função de p/p0.
Conjugando a quantidade adsorvida obtida a partir da isotérmica de adsorção
experimental em função dos valores de αs obtidos para a isotérmica de referência,
obtêm-se uma representação gráfica que, no caso dos carvões activados, apresenta duas secções lineares (Figura I.10). A primeira secção observa-se normalmente para valores de αs<1 (p/p0<0,4), e a intersecção com o eixo das ordenadas indica o volume dos
microporos mais estreitos. A segunda zona linear, encontra-se normalmente para
valores de αs mais elevados (1 < αs <2) e a intersecção com o eixo das ordenadas fornece
21
Figura I. 10 – Representação αs hipotética para adsorventes microporosos.
A Figura I.11(a) representa a recta 1, característica de sólidos não porosos e a curva 2, o resultado da análise de isotérmicas de materiais mesoporosos. Se a amostra em estudo diferir da amostra de referência apenas na área superficial e não na porosidade, então a
representação αs irá ser uma recta que passa pela origem. Se, pelo contrário, a amostra
em estudo apresentar meso ou microporosidade irão observar-se desvios à linearidade. Na Figura I.11.(b) a recta 3 corresponde à analise referente a um material microporoso e a recta 4, a um material micro e mesoporoso. Os desvios negativos à linearidade indicam a presença de microporosidade. Como foi dito anteriormente, a extrapolação da parte linear até ao eixo das ordenadas permite estimar o volume microporoso, convertendo a quantidade adsorvida em volume líquido através da densidade do adsorvato no estado liquido.
αs αs
Figura I. 11 – Esquema da representação αs.
O método αs foi aplicado para os ensaios de adsorção de azoto a -196 °C realizados no
I.2.1.3.3. Equação de Dubinin-Radushkevich (DR)
A equação de Dubinin-Radushkevich (equação DR) admite que o fenómeno de adsorção envolve o preenchimento do volume microporoso e não a formação de várias camadas nas paredes dos poros.
A equação DR é usada na sua forma linearizada:
(
0)
p p T
B w
wads log log /
log 2 2 0 − =
β onde
(
)
2 0 / 304 ,
5 R E
B= Equação I. 4
onde wads traduz o volume ocupado pela fase adsorvida, w0 o volume microporoso, que
se irá designar por VDR, B uma constante independente da temperatura e característica
da estrutura porosa do adsorvente, E0 a energia característica, T a temperatura a que o
ensaio de adsorção ocorreu, β é o coeficiente de afinidade (apresenta o valor de 0,33
para quando o adsorvato é o azoto, e 0,35 para o dióxido de carbono), p pressão de
equilíbrio e, por fim, p0 é a pressão de saturação.
A representação gráfica de log wads vs log2(p/p0) traduz a equação DR numa recta cuja
ordenada na origem é dada por log w0 e declive B(T/β)2. Esta representação gráfica
apenas é linear para valores de pressão relativa baixos, verificando-se desvios à linearidade à medida que se aumenta o valor da pressão relativa, para os sólidos que não apresentam exclusivamente microporosidade.
Na aplicação da equação DR aos ensaios de adsorção de dois adsorvatos, N2 e CO2, com o objectivo de determinar o volume microporoso de um material, pode observar-se as seguintes situações:
VDR(N2)<VDR(CO2), situação que se verifica quando o material possui microporos
muito estreitos, o que dificulta o estabelecimento do equilíbrio de adsorção das moléculas de azoto à temperatura a que o ensaio é realizado.
VDR(CO2)≈VDR(N2), ocorre quando o material apresenta uma estrutura
microporosa suficientemente larga que evita a ocorrência de limitações
23
VDR(N2)>VDR(CO2), ocorre em carvões de apresentam elevados graus de
activação. Quando não é possível obter resultados para a adsorção de azoto a
-196 °C a baixas pressões relativas (p/p0=0,01), a extrapolação da curva
característica para o azoto a -196 °C e para o CO2 a 0 °C não é coincidente.
Sendo assim a gama de pressões relativas a que se aplica a equação DR não é a mesma o que faz com que o volume microporoso obtido a partir da adsorção de azoto seja superior ao obtido pela adsorção de dióxido de carbono.
Na caracterização das amostras preparadas no decorrer deste trabalho, usou-se este
método aplicado à adsorção de azoto a -196 °C e de dióxido de carbono a 0 °C.
I.2.2. pH no ponto de carga zero (pH
pzc)
A caracterização dos centros ácidos na superfície é fundamental para se compreender a actividade e selectividade do catalisador uma vez que fornece informação relativamente aos grupos funcionais presentes na superfície do carvão [3].
A medição do valor do pHPZC (PZC advém do acrónimo inglês para Point of Zero Charge) é
uma das ferramentas de caracterização da química superficial mais usada.
Seguindo o modelo apresentado por Noh e Schwarz [33] considera-se que quando o valor do pHPZC é 7 (valor do pH da água) então o valor de pHPZC para a mistura carvão-água em estudo é independente da massa de amostra existente nessa mistura.
Neste trabalho, a determinação do valor de pHPZC das amostras foi feita através de titulação mássica, com base no procedimento proposto por Noh e Schwarz [33].
A medição deste parâmetro permite fazer uma caracterização química da superfície dos carvões, e pode ser determinado para várias soluções da mesma amostra, fazendo variar a percentagem mássica da mesma para cada uma dessas soluções, ou então, partindo de uma única solução e fazendo diluições e medições de pH sucessivas.
Independentemente do método escolhido, a amostra de carvão tem que ser seca, antes dos ensaios, sendo representativa de uma amostra não contaminada [33].
É necessário ter em atenção que a razão sólido/água não deve ultrapassar os 10%, uma vez que, para valores superiores a solução estará demasiado concentrada para que a medição do valor de pH seja correcta.
I.2.3. Dessorção a temperatura programada
Num ensaio de dessorção a temperatura programada (TPD) o sólido é aquecido sob um fluxo de gás inerte. O aumento progressivo da temperatura vai provocar a dessorção das espécies presentes na superfície do carvão [3], as quais são analisadas por diferentes técnicas onde se incluem a espectrometria de massa, a cromatografia gasosa ou a análise gravimétrica.
Os perfis de dessorção térmica obtidos nestes ensaios apresentam vários picos sendo necessário fazer uma desconvulsão dos mesmos, recorrendo a uma função Gaussiana múltipla [34], de modo a ser possível calcular a quantidade de grupos superficiais.
25
temperatura à qual se decompõe um determinado grupo funcional, é possível ter uma estimativa desse valor com base nos resultados já apresentados na literatura. Deste modo, os grupos carboxílicos decompõem-se em CO2 e água, sofrendo uma reacção de descarboxilação, enquanto que os grupos do tipo quinona se decompõem em CO e os fenólicos em CO e água, sofrendo uma reacção de descarbonilação.
Analisando os gases libertados e a temperatura a que ocorre a decomposição é possível identificar a natureza dos grupos funcionais presentes na superfície do carvão (Figura I.12) e quantificá-los. Os valores dos intervalos de temperatura associados aos grupos funcionais que se decompõem encontram-se na Tabela I.1.
Tabela I. 1 – Grupos funcionais e respectiva temperatura de decomposição por dessorção a temperatura programada e gás libertado [34].
Grupo funcional Gás libertado Temperatura (°°°°C)
Ácido carboxílico CO2 100-400
Lactona CO2 350-750
Fenol CO 600-700
Carbonilo CO 700-1000
Anidrido carboxílico CO+ CO2 300-400/600-650
Éter CO 600
Figura I. 12 - Grupos funcionais que podem ser encontrados na superfície dos carvões activados e libertados por dessorção, a temperatura programada, na forma de CO ou CO2.
A análise de TPD permite caracterizar os grupos funcionais presentes na superfície do carvão e fornecer informação quanto ao carácter ácido do material em estudo.
I.2.4. Análise elementar
Os carvões activados são constituídos maioritariamente por átomos de carbono, podendo no entanto apresentar na sua estrutura heteroátomos ligados quimicamente à superfície dos carvões. Estes heteroátomos, apesar de constituírem uma pequena percentagem comparativamente à percentagem de carbono, influenciam as características químicas do material e o seu comportamento como adsorventes, assumindo assim um papel de extrema relevância. A quantidade de heteroátomos presente na superfície do material pode resultar da matéria-prima utilizada, ou pode resultar da activação ou de tratamentos posteriores.
Os heteroátomos que se quantificam por análise elementar são o oxigénio, o hidrogénio, Ácido Carboxílico
Lactona
Fenol
Carbonilo
Anidrido carboxílico
Éter
27
percentagem destes átomos é possível ter uma estimativa dos possíveis grupos funcionais existentes na superfície da amostra.
Para que seja feita a determinação da percentagem de cada um dos elementos as
amostras são queimadas a elevadas temperaturas (∼1200 °C) sob fluxo de oxigénio.
Numa atmosfera com excesso de oxigénio e após uma combustão completa o carbono passa a CO2, o hidrogénio a vapor de água, o enxofre a óxido de enxofre e os óxidos de azoto são reduzidos a N2 por limalhas de cobre [3].
As técnicas de detecção variam consoante o aparelho comercial escolhido, mas pretende-se sempre que o seu grau de precisão seja o mais elevado possível, apresentando erros absolutos na ordem dos 0,3% (m/m) e limites de detecção entre 0,01 e 0,1%. Nestas técnicas incluem-se a reacção com diferentes reagentes (como exsicantes para remover a água, NaOH para remover o dióxido de carbono), quimiluminescência, coulometria ou sensores químicos de base electroquímica [3].
I.2.5. Difracção de raios X
Na difracção de raios X (método dos pós) usa-se uma radiação monocromática de comprimento de onda λ e aplica-se a amostras constituídas por um elevado número de cristalites dispostas aleatoriamente. Considera-se que um certo número dessas cristalites estão em posição de Bragg para uma dada família de planos hkl, ou seja, que obedecem à Lei de Bragg dada por nλ = 2 d(hkl) sen(θ), onde n corresponde a um número inteiro de comprimentos de onda, θ ao ângulo de Bragg, d à distância entre os planos inter-reticulares e hkl os índices de Miller (números inteiros que correspondem à razão entre as dimensões da malha cristalina e as distâncias de intersecção dos respectivos planos nos eixos cristalográficos). A fórmula de Bragg define assim as possíveis direcções dos raios difractados[27].
e característico podendo assim fazer-se a identificação das várias fases cristalinas presentes numa amostra comparando o difractograma obtido com os difractogramas de referência disponíveis da literatura.
I.2.6. Espectroscopia de infravermelho por reflectância difusa
No espectro electromagnético a região do visível divide-se em três zonas: longínquo
(400-10 cm-1); intermédio (4000-400 cm-1) e próximo (4000-14000 cm-1). A
espectroscopia de infravermelho (IV) por reflectância difusa aplica-se à região intermédia do espectro e é usada na caracterização de amostras sólidas.
Ao irradiar uma superfície, a luz é absorvida selectivamente consoante a frequência específica de vibração (níveis vibracionais) das moléculas constituintes. Sendo assim, os grupos funcionais idênticos irão apresentar bandas de adsorção características no espectro de IV. No caso da análise de carvões a identificação das bandas obtidas e, consequentemente, dos grupos funcionais presentes na superfície do carvão, é feita fazendo a comparação com os espectros obtidos para materiais orgânicos que na sua constituição apresentam grupos funcionais semelhantes [3].
O resultado da análise por espectroscopia de infravermelho depende das condições experimentais, nomeadamente do ângulo de incidência e de reflexão e da espessura e estado da superfície da amostra.
As amostras devem ser previamente reduzidas a pó, num almofariz, pois a razão entre a intensidade das bandas não é constante relativamente à alteração de tamanho das partículas [35].
I.2.7. Microscopia electrónica de transmissão
A microscopia electrónica de transmissão, de abreviatura TEM (advém do acrónimo
inglês para Transmission Electron Microscopy), contribui para a caracterização da
29
Para a realização desta análise usa-se um microscópio electrónico de transmissão (TEM) onde uma amostra muito fina (5 nm a 0,5 μm de espessura máxima) é irradiada com um feixe de electrões monocromático, que vai interagir com o material através de colisões elásticas e inelásticas.
O feixe de electrões ao atravessar a amostra cristalina sofre uma difracção que respeita a lei de Bragg. Os feixes resultantes convergem pela lente da objectiva de forma a criar um padrão de difracção.
II. Preparação e técnicas de caracterização de
carvões activados a partir de biomassa
II. 1. Preparação das amostras
Na primeira parte do estudo que se apresenta nesta tese foram preparados carvões activados usando como material de partida borras de café. As amostras foram obtidas por activação química com K2CO3, seguindo o procedimento descrito na referência [11], dado ser uma metodologia que tem sido utilizada com sucesso para a activação de outros precursores.
O precursor foi cedido pelo grupo Engenharia das Reacções (Departamento de Química, REQUIMTE) da Faculdade de Ciências e Tecnologia da Universidade Nova de Lisboa (FCT-UNL). Uma parte das borras de café sofreu um tratamento prévio, realizado na FCT-UNL, o qual consistiu numa lavagem em Soxhlet com água desionizada, durante cerca de três a quatro dias. A água foi trocada sempre que se apresentava acastanhada e o processo foi dado como terminado quando a água ficou límpida. Outra parte das borras de café foi usada sem qualquer processo de lavagem prévio.
II.1.1. Impregnação e activação
Antes da impregnação secaram-se as borras de café a 100 °C durante uma noite. De
seguida, pesou-se a massa de precursor e de carbonato de potássio necessárias para se obterem as proporções K2CO3:biomassa (em massa) de 0,5:1, 1:1 e 2:1.
31
Figura II. 1 – Representação esquemática da montagem para carbonizações sob fluxo de azoto.
As amostras foram activadas a 600 e a 700 °C, de acordo com o perfil térmicos indicado
na Figura II.2. O fluxo de N2 foi ajustado com uma válvula, medindo–se o caudal
(2 cm3/s), através de um fluxímetro de bolha.
No final do processo de activação e antes da remoção da barquinha do interior do forno,
aguardou-se que este alcançasse uma temperatura inferior a 80 °C, mantendo-se o fluxo
de azoto.
Figura II. 2 – Perfil térmico das activações.
II.1.2. Lavagem
Após a etapa de activação, retirou-se a amostra de carvão da barquinha e colocou-se no interior de uma manga de diálise, juntamente com água destilada. Fechou-se a manga, em ambas as extremidades, e colocou-se dentro de um copo de vidro com água destilada e sob agitação.
Tambiente
T = 600 ºC ou 700 °C t = 1h
10 ºC / min
Fluxímetro de bolha
Forno tubular horizontal, Thermolyne
Entrada de fluxo de N2
Torneira de vidro
Trocou-se a água do copo com regularidade, até obter pH neutro. Após este período de lavagem a amostra foi retirada da manga e colocada num copo de vidro, ao qual se
adicionou água destilada. Após ser mantida em aquecimento a, aproximadamente, 90 °C,
durante 20 minutos, deixou-se repousar a mistura até à deposição do carvão, sendo então decantada a fase líquida e adicionado novo volume de água destilada. Repetiu-se o processo de aquecimento sob agitação.
Foram necessários, pelo menos, cerca de quatro dias para a lavagem de cada amostra.
No final das lavagens as amostras foram secas na estufa, à temperatura de 100 °C
durante uma noite, após o que se procedeu à sua pesagem para se determinar o rendimento da preparação. Os carvões foram guardados em frascos fechados até à sua utilização.
Na tabela II.1 encontram-se os nomes e condições de preparação das várias amostras. Os carvões preparados a partir de borras de café lavadas são identificados com a letra “L” e, as amostras obtidas a partir de borras de café não lavadas com a designação “NL”.
Amostra
Temperatura de Carbonização
(°°°°C)
Precursor
NL/0,5:1/700 700
NL/1:1/600 600
NL/1:1/700 700
NL/2:1/700 700
borras de café não lavadas
L/1:1/700 700
L/2:1/700 700
borras de café lavadas
33
II.2. Técnicas de caracterização
II.2.1. Adsorção de gases
II.2.1.1. Adsorção de N
2a -196
°°°°
C
Nos ensaios de adsorção de azoto foram utilizados cerca de 50 mg de amostra e os mesmos foram realizados num equipamento automático ASAP 2010 Micromeritics (Figura II.3).
Figura II. 3 – Equipamento ASAP 2010.
Antes da adsorção de azoto a -196 °C as amostras foram desgaseificadas a 120 °C
durante 7 horas, sob vácuo melhor que 10-2 Pa. As quantidades adsorvidas foram

![Figura I. 8 – Classificação da IUPAC para as isotérmicas de adsorção de gases (adaptada de [30])](https://thumb-eu.123doks.com/thumbv2/123dok_br/16494275.733497/33.892.310.627.89.561/figura-classificação-iupac-para-isotérmicas-adsorção-gases-adaptada.webp)
![Tabela I. 1 – Grupos funcionais e respectiva temperatura de decomposição por dessorção a temperatura programada e gás libertado [34]](https://thumb-eu.123doks.com/thumbv2/123dok_br/16494275.733497/43.892.134.811.430.698/funcionais-respectiva-temperatura-decomposição-dessorção-temperatura-programada-libertado.webp)