UN I VERSI DADE FEDERAL DA PARAÍ BA CEN TRO DE CI ÊN CI AS EXATAS E D A N ATUREZA
DEPARTAM EN TO DE QUÍ M I CA
PROGRAM A DE PÓS- GRADUAÇÃO EM QUÍ M I CA
LI GAÇÕES DE HI DROGÊN I O I N TRAMOLECULARES: UM
ESTUDO TEÓRI CO DE COM POSTOS DI - CARBON Í LI COS
DI SSERTAÇÃO DE MESTRADO
ALI N E FON SECA BEZERRA
UN I VERSI DADE FEDERAL DA PARAÍ BA CEN TRO DE CI ÊN CI AS EXATAS E D A N ATUREZA
DEPARTAM EN TO DE QUÍ M I CA
PROGRAM A DE PÓS- GRADUAÇÃO EM QUÍ M I CA
DI SSERTAÇÃO DE MESTRADO
LI GAÇÕES DE HI DROGÊN I O I N TRAMOLECULARES: UM
ESTUDO TEÓRI CO DE COM POSTOS DI - CARBON Í LI COS
ALI N E FON SECA BEZERRA*
Dissert ação apresent ada com o requisit o para obt enção do t ít ulo de M est re em Quím ica pela Universidade Federal da Paraíba.
Orient ador( a) : Profa. Dr. Regiane de Cássia M . U. de Araúj o 2 º Orient ador: Prof. Dr. Boaz Galdino de Oliveira
* Bolsist a CAPES
B574l Bezerra, Aline Fonseca.
Ligações de hidrogênio intram oleculares: um estudo teórico de com postos di- carbonílicos / Aline Fonseca Bezerra.- João Pessoa, 2009.
129f.
Orientadores: Regiane de Cássia M. U. de Araúj o, Boaz Galdino de Oliveira
Dissert ação ( Mestrado) – UFPB/ CCEN
“H á um t em po em que é preciso abandonar as roupas
usadas, que j á t êm a form a do nosso corpo, e esquecer os
nossos cam inhos que nos levam sem pre aos m esm os
lugares. É o t em po da t ravessia e, se não ousarm os fazê- la,
t erem os ficado, pra sem pre, à m argem de nós m esm os.”
AGRADECI MEN TOS
A Deus, por estar sem pre do m eu lado em todos os m om entos
dessa longa cam inhada, erguendo m eu corpo sem pre que ele insistia em cair.
A m inha Mãe, Rosanira, por ser m eu porto seguro, m eu
exem plo de vida, m eu am or m ais sincero. A m inha fam ília pelo zelo e pela força.
Ao Edson, por ser m eu com panheiro de todos os m om entos,
sem pre carinhoso e paciente com igo. A toda fam ília de Edson por todo carinho e apoio.
A Profa. Dra. Regiane de Cássia M. U. de Araúj o, pela
orientação e, em especial, pelo carinho, am izade, com preensão e alegria em todos os m om entos.
Ao Prof. Dr. Boaz G. Oliveira por sua disponibilidade em sem pre
esclarecer m inhas dúvidas e por sua am izade.
Aos professores Dr. Antônio Bezerra, Dr. Mário Vasconcelos, Dr.
Sidney, Dr. Otávio, Dr. Gerd e todos que fazem parte do LQQC pelas aj udas valiosas ao longo desse trabalho.
Aos m eus am igos Arm strong, Lenildo, Tião, Bilu, Juscélia,
Andréa, Luci, Veruschka e Andreza por serem com panheiros de longa data na grande cam inhada da vida.
Aos am igos do LQQC em especial Kelson e Arquim edes por toda
aj uda e am izade em todos os m om entos.
A todos que estiveram com igo, m esm o que em pensam ento,
torcendo para que este obj etivo fosse alcançado.
RESUMO
I Tít ulo: Ligações de H idrogênio I nt ram oleculares: Um Est udo Teórico de Com post os Di- carbonílicos.
Aut ora: Aline Fonseca Bezerra.
1 ° Orient adora: Profa. Dra. Regiane de Cássia M.U. de Araúj o.
2 ° Orient ador: Prof. Dr. Boaz Galdino de Oliveira.
A ligação de hidrogênio intram olecular ocorre quando um a m esm a m olécula apresenta, sim ultaneam ente, um grupo doador e outro receptor de próton, em configuração espacial favorável à form ação dessa int eração. É im portante salient ar as m udanças nas propriedades estruturais, eletrônicas e vibracionais que ocorrem devido à form ação dessa interação. Na form ação da ligação de hidrogênio ocorre um fenôm eno im portante denom inado de “ transferência de carga” , onde parte da densidade eletrônica da espécie receptora de próton, Y, é transferida para a espécie doadora de próton, HX. Com respeito aos espectros vibracionais, são observadas m odificações nos m odos de estiram ento das espécies doadora e receptora de próton. Com postos di-carbonílicos (C3H2O2R2)
com suas substituições (R= CH3, CN, H, NH2, OH e SH) foram
estudados enfocando as análises energética, estrutural, vibracional e de densidade eletrônica. I nicialm ente foram realizadas as análises energéticas e estruturais a partir da geom etria otim izada das m oléculas. Foram avaliados a força da ligação de hidrogênio e do com prim ento da ligação intram olecular. O estudo usando a QTAI M foi realizado para adquirir os valores de densidade eletrônica e do Laplaciano da densidade eletrônica e verificar a existência do ponto crítico de ligação na ligação de hidrogênio intram olecular. A partir dos espectros vibracionais harm ônicos foi possível ident ificar as variações no infraverm elho, referentes à form ação da interação intram olecular.
ABSTRACT
II I nt ram olecular H ydrogen Bonds in Di- carbonyl Com pounds: A Theoret ical St udy
The intram olecular hydrogen bond occurs when the sam e m olecules has bot h proton donor and proton acceptor groups in satisfactory configuration space for the form ation of this interaction. I t is im portant to note the changes in the struct ural, electronic and vibrat ional properties that occur due to the form ation of this interaction. I n t he hydrogen bonding form ation is an im portant phenom enon called “ charge transfer” , where part of the electronic density of the proton acceptor species, Y, is transferred o the proton donor specie, HX. With respect to the vibrational spectrum are observed changes in the way of straightening of donor and acceptor proton species. Di-carbonyl com pounds (C3H2O2R2) with their
subst ituent groups (R= CH3, CN, H, NH2, OH and SH) were studied
focusing on the energetic, structural, vibrational and electron density analysis. I nitially the energy and struct ural analysis were carried out starting from the m olecules opt im ized geom etry. We also evaluated of the strength’s hydrogen bonding and t he length’s intram olecular bond. The QTAI M study was perform ed to obtain t he electron density’s values and the electron densit y’s Laplacian values and verify the existence of the bond crit ical point in the intram olecular hydrogen bond. From the harm onic vibrat ional spectra was possible to identify changes in the vibrat ional m odes, related the intram olecular interaction’s form ation.
Í NDI CE
I I I
SUM ÁRI O
Lista de Figuras
V
Lista de Tabelas I X
Lista de Nom enclat uras, Abreviaturas, Siglas e Sím bolos XI
1. I ntrodução 1
1.1. A Ligação de Hidrogênio 1
1.2. Com postos Di-carbonílicos 9
2. Obj etivos 13
2.1. Obj etivo Geral 13
2.2. Obj etivos Específicos 14
3. Fundam entação Teórica 15
3.1. I ntrodução 15
3.1.1. Operador Ham iltoniano 16
3.1.2. O Método Hartree-Fock-Roothaan 18
Í NDI CE
I V
3.3. Teoria do Funcional da Densidade 23
3.4. Funções de Base 26
3.5. Análise Populacional 29
3.5.1. Análise Populacional de Mulliken 31
3.5.2. Cargas Atôm icas Derivadas do Potencial Eletrostático
34
3.5.3. Cargas de Bader 36
3.6. Teoria Quântica de Átom os em Moléculas 38
4. Metodologia 43
5. Resultados e Discussões 45
5.1. Caracterização da Ligação de Hidrogênio 45
5.1.1. Propriedades Energéticas e Estruturais 45
5.1.2. Espectros Vibracionais 55
5.2. Análise Populacional 65
5.3. Propriedades Topológicas da Densidade Eletrônica 71
5.4. Efeito dos Grupos Substituintes na Form ação da Ligação de Hidrogênio I ntram olecular nos com postos Di-carbonílicos
Í NDI CE
V
6. Conclusões 85
7. Perspectivas 88
Í NDI CE
VI
LI STA DA FI GURAS
Figura 1 .1 . Moléculas de água unidas por ligações de hidrogênio 2
Figura 1 .1 .2 . Ligação de Hidrogênio I nterm olecular entre dois fenóis
3
Figura 1 .1 .3 . Ligação de Hidrogênio I ntram olecular na m olécula de 2- am ino fenol
3
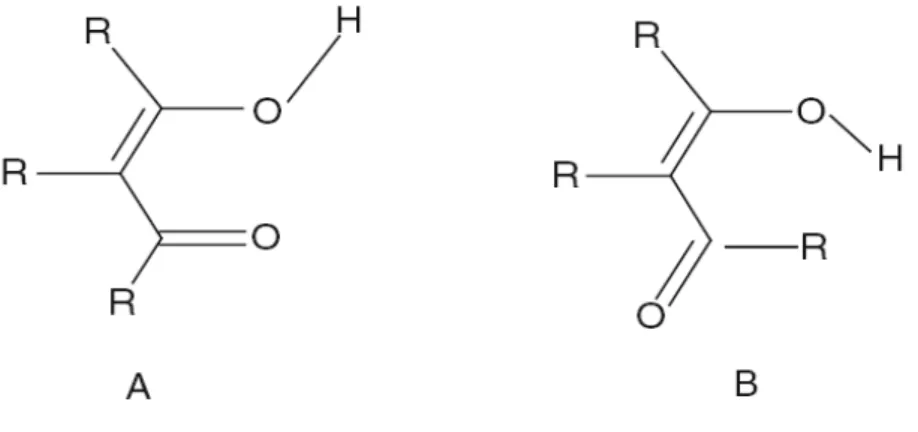
Figura 1 .1 .4. Conform ações abertas em A e B para a m olécula de m alonaldeido
7
Figura 1 .2 .1 . Condensação de Claisen form ando β-ceto- éster 9
Figura 1 .2 .2 . Condensação de Dieckm ann 10
Figura 1 .2 .3 . Grupo central di- carbonílico 10
Figura 1 .2 .4 . Com posto di- carbonílico e as substituições propostas para estudo neste trabalho, através de m ét odos teóricos
11
Figura 3 .1 . I lustração da densidade eletrônica na form a de linhas de contorno para a m olécula de etileno
Í NDI CE
VI I
Figura 3 .2 . Vetor cam po gradiente, ∇�(�⃗), para a m olécula de
etileno
39
Figura 3 .3 . I lustração dos BCP’s na m olécula de antraceno. Os pontos em cor verm elha presentes nas ligações entre átom os
consecutivos são os BCP’s 40
Figura 5 .1 . Estruturas de m ínim o com as respectivas geom etrias otim izadas dos com postos di- carbonílicos com seus respectivos
substituintes na conform ação fechada 45
Figura 5 .2 . Estruturas de m ínim o com as respectivas geom etrias otim izadas dos com postos di- carbonílicos com seus respectivos
substituintes na conform ação aberta 46
Figura 5 .3 . Gráfico dos valores absolutos de energia da ligação de hidrogênio intram olecular obt idos através da diferença entre a conform ação fechada e a conform ação aberta dos com postos di-carbonílicos calculados com os m étodos DFT/ B3LYP e MP2. Valores em kJ.m ol-1
50
Figura 5 .4 . Valores DFT/ B3LYP e MP2 para o com prim ento da ligação de hidrogênio int ram olecular, RH-- - O, versus os substituintes
dos com postos di- carbonílicos 54
Figura 5 .5 . Valores DFT/ B3LYP e MP2 para os increm entos nos com prim entos da ligação O- H devido a form ação da ligação de hidrogênio intram olecular, δrO-H versus os substituintes dos
Í NDI CE
VI I I com postos di- carbonílicos
Figura 5 .6 . Valores DFT/ B3LYP de energia da ligação de hidrogênio intram olecular, ∆E, versus os valores dos deslocam entos na freqüência de est iram ento OH, δνOH 60
Figura 5 .7 . Valores MP2 de energia da ligação de hidrogênio intram olecular, ∆E, versus os valores dos deslocam entos na freqüência de est iram ento OH, δνOH 60
Figura 5 .8 . Espectro vibracional obt ido teoricam ente através do m étodo DFT/ B3LYP para o com posto di- carbonílico subst ituído com CN nas conform ações aberta e fechada 62
Figura 5 .9 . Espectro vibracional obt ido teoricam ente através do m étodo DFT/ B3LYP para o com posto di- carbonílico substituído com H
nas conform ações aberta e fechada 63
Figura 5 .1 0 . Espectro vibracional obtido teoricam ente através do m étodo DFT/ B3LYP para o com posto di- carbonílico subst ituído com SH nas conform ações aberta e fechada 63
Figura 5 .1 1 . Espectro vibracional obtido teoricam ente através do m étodo DFT/ B3LYP para o com posto di- carbonílico subst ituído com CH3 nas conform ações aberta e fechada 64
Í NDI CE
I X m étodo DFT/ B3LYP para o com posto di- carbonílico subst ituído com
OH nas conform ações aberta e fechada
Figura 5 .1 3 . Espectro vibracional obtido teoricam ente através do m étodo DFT/ B3LYP para o com posto di- carbonílico subst ituído com NH2 nas conform ações aberta e fechada 65
Figura 5 .1 4 . I lustração dos BCP’s em todos os com postos di-carbonílicos substituídos estudados, presentes entre a ligação RH---O
caracterizando a ligação de hidrogênio intram olecular na
conform ação fechada 73
Figura 5 .1 5 . Gráfico dos valores de densidade eletrônica no BCP,
�(�⃗), versus os valores do com prim ento da ligação de hidrogênio
intram olecular, RH---O, m ostrando que há um a relação linear entre
esses dois parâm etros 74
Figura 5 .1 6 . Gráfico da relação entre a densidade eletrônica no BCP e a variação da freqüência vibracional do grupo O- H para os com postos di- carbonílicos substituídos 75
Figura 5 .1 7 . Os BCP’s nos com postos di- carbonílicos substit uídos estudados, entre a ligação O- - - O, caracterizando a interação intram olecular na conform ação aberta 78
Figura 5 .1 8 . Estrutura de ressonância m ostrando o favorecim ento da form ação da ligação de hidrogênio intram olecular no m alonaldeido
Í NDI CE
X
Figura 7 .1 . Com postos propostos para estudo: ( 1) ácido salicílico, ( 2) ácido 5- fluorosalicílico, ( 3) ácido salicilsalicílico, ( 4) 2- m
etil-acetoacetanilida 89
Figura 7 .2 . Com postos trifluorm et il β-dicatonatos com suas substituições: ( 1) 1- fenil- 4,4,4- trifluor- 2- m etil- 1,3- butanodiona, ( 2) 5- ( 2- m etoxifenil) - trifluor- 2,4- pentanodiona, ( 3) 6- fenil-
1,1,1-trifluor- 2,4- hexanodiona 89
Í NDI CE
XI
LI STA DE TABELAS
Tabela 1 .1 .1 . Característ icas gerais dos três principais t ipos de ligação
de hidrogênio 6
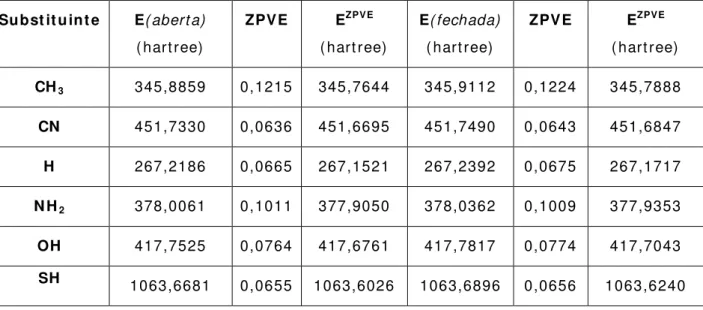
Tabela 5 .1 . Valores de energia, em m ódulo, e suas respect ivas correções ZPVE, Ecorr , para os com post os di-carbonílicos subst ituídos nas conform ações abert a e fechada em nível DFT-B3LYP/ 6- 311+ + G* * 47
Tabela 5 .2 . Valores de energia, em m ódulo, e suas respect ivas correções ZPVE, Ecorr , para os com post os di-carbonílicos subst ituídos nas conform ações abert a e fechada em nível MP2/ 6- 311+ + G* * 48
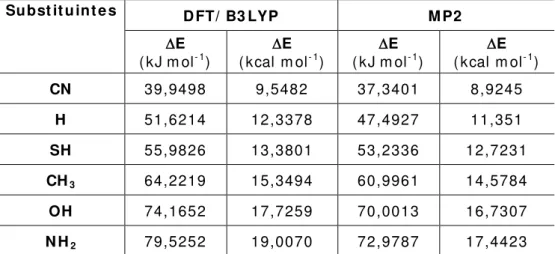
Tabela 5 .3 . Valores DFT/ B3LYP e MP2 com o conj unt o de base 6-311+ + G* * da energia da ligação de hidrogênio intram olecular para os com post os di- carbonílicos, com suas respectivas subst ituições. Unidades
em kJ m ol-1 e kcal m ol-1 49
Tabela 5 .4 . Valores dos com prim ent os de ligação rO-H nas conform ações abert a e fechada e as correspondent es diferenças entre esses valores, com provando o aum ent o do com prim ent o da ligação O- H devido a form ação da ligação de hidrogênio intram olecular, δrO-H. Unidades em Å
51
Tabela 5 .5 . Valores do com prim ent o DFT/ B3LYP e MP2 da ligação de hidrogênio intram olecular, RH---O present es nas conform ações fechadas.
Unidades em Å 53
Tabela 5 .6 . Valores de int ensidades dos m odos de est iram ent o para a espécie O-H dos com post os di- carbonílicos subst it uídos nas conform ações fechadas e abert as obtidas at ravés dos cálculos DFT- B3LYP/ 6-311+ + G* *
Í NDI CE
XI I
e MP2/ 6- 311+ + G* * . Unidades em km m ol-1
Tabela 5 .7 . Valores da freqüência de est iram ent o para a espécie O- H dos com post os di- carbonílicos subst ituídos nas conform ações fechada e abert a obt idas através dos cálculos DFT- B3LYP/ 311+ + G* * e MP2/
6-311+ + G* * . Unidades em cm-1 58
Tabelas 5 .8 . Valores DFT/ B3LYP/ 6- 311+ + G* * das cargas de Mülliken e as correspondent es diferenças ent re as cargas de Mülliken nas conform ações aberta e fechada, δq(fechada-abert a). Unidade eletrônica de
carga, e 67
Tabelas 5 .9 . Valores DFT/ B3LYP/ 6- 311+ + G* * das cargas atôm icas derivadas do pot encial elet rostát ico e as correspondent es diferenças entre as cargas nas conform ações aberta e fechada, δq(fechada-abert a).
Unidade eletrônica de carga, e 68
Tabelas 5 .1 0 . Valores DFT/ B3LYP/ 6-311+ + G* * das cargas de Bader e as correspondent es diferenças entre as cargas de Bader nas conform ações aberta e fechada, δq(fechada-abert a). Unidade eletrônica de
carga, e 69
Í NDI CE
XI I I Tabela 5 .1 2 . Valores DFT/ B3LYP/ 6- 311+ + G* * da densidade, ρ(�⃗), e do
vet or Laplaciano, ∇2ρ(�⃗
) para as ligações O- H (hidroxila, doadora de
prót on) e RH---O ( ligação de hidrogênio int ram olecular) nos com postos di-carbonílicos subst ituídos de conform ação fechada. Unidade de ρ(�⃗) em
e/ a03 e de ∇2ρ(�⃗) em e/ a05
72
Tabela 5 .1 3 . Valores DFT/ B3LYP/ 6- 311+ + G* * da densidade ρ(��⃗), vet or
Laplaciano ∇2ρ(
�⃗) para o com prim ent o da ligação O-- - O e para as
ligações O- H, doadoras de prót on, nos com post os di- carbonílicos subst ituídos de conform ação aberta. Unidade de ρ(�⃗) em e/ a03 e de
∇2ρ(�⃗
) em e/ a05
Í NDI CE
XI V
LI STAS DE N OMEN CLATURAS, ABREVI ATURAS,
SI GLAS E SÍ MBOLOS
Ab init io – Prim eiros Princípios.
BCP – Bond Crit ical Point (Ponto Crítico de Ligação).
DFT – “ Density Funct ional Theory” (Teoria do Funcional da Densidade) .
H F – Hartree-Fock.
LCAO – “Linear Com bination of Atom ic Orbitals” (Com binação Linear de Orbitais Atôm icos).
LYP – Lee-Yang-Parr.
M Pn – Teoria da Perturbação de MØller e Plesset de ordem n.
QTAI M – Quantum Theory Atom s in Molecules.
RH F - Restricted Hartree-Fock.
SCF – “ Self Consist ent Field” (Cam po Auto-Consistente).
STO – “ Slater Type Orbitals” (Orbitais do tipo Slater).
B – Funcional de Becke .
H – Operador Ham iltoniano.
Ô – Operador Herm itiano.
T – Energia cinética da part ícula.
Í NDI CE
XV Γ – Operador Densidade.
Ψ – Função de Onda.
ρ(��⃗) – Densidade Eletrônica.
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 1
-1 . I N TRODUÇÃO
Quando átom os, m oléculas ou íons se aproxim am uns dos outros podem ocorrer dois fenôm enos dist intos, eles podem reagir ou eles podem interagir. A reação quím ica é um processo de m udança quím ica, isto é, a conversão de um a ou m ais substâncias em outras substâncias havendo, geralm ente, um a variação de energia nesse processo. Um a interação quím ica, por sua vez, im plica em atração ou repulsão das m oléculas entre si, sem que ocorra quebra ou form ação de novas ligações quím icas. No segundo caso, têm -se as interações ou forças interm oleculares e as energias envolvidas nesse processo são, em geral, bem m enores que aquelas envolvidas em um a reação quím ica [ 1].
As forças interm oleculares estão intim am ente relacionadas à diversas propriedades de sólidos, líquidos e gases, sendo de extrem a im portância para o entendim ento de sistem as quím icos em nível m olecular. Dentre as interações interm oleculares podem ser evidenciadas as interações de van der Waals e as ligação de hidrogênio. Enfat izando as ligações de hidrogênio, que consistem do fenôm eno investigado nesse trabalho, um a busca na literatura especializada evidencia que este fenôm eno vem sendo estudado há décadas, devido, principalm ente, aos diversos com portam entos da m atéria observados nas áreas da Quím ica, Física e Biologia[ 2-4].
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 2
-outros exem plos relevante[ 5-7]. Na Figura 1.1, a ilustração serve para
visualizar as ligações de hidrogênio presentes na água [ 8].
Figura 1 .1 . Moléculas de água unidas por ligações de hidrogênio.
1 .1 . A LI GAÇÃO DE H I DROGÊN I O
A ligação de hidrogênio é um a interação inter ou intram olecular, que ocorre entre um hidrogênio deficiente de elétrons e um a região de alt a densidade eletrônica[ 9].
Y + HX →← YHX (1.1.1)
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 3
-Na Figura 1.1.2 podem os visualizar a form ação da ligação de hidrogênio interm olecular existente ent re duas m oléculas de fenol.
Figura 1 .1 .2 . Ligação de Hidrogênio I nterm olecular entre dois fenóis.
Por outro lado, a ligação de hidrogênio intram olecular ocorre quando um a m esm a m olécula apresenta, sim ultaneam ente, um grupo doador e outro receptor de próton, em configuração espacial favorável à form ação dessa interação, com o podem os ver na Figura 1.1.3.
Figura 1 .1 .3 . Ligação de Hidrogênio I ntram olecular na m olécula de 2- am ino fenol.
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 4
-sabe-se que ocorre um aum ento pronunciado no com prim ento de ligação da espécie doadora de próton, HX, ocasionando na m esm a m olécula um efeito red shift no espectro infraverm elho, sendo esse efeito observado para a m aioria dos com prim entos de ligação, sej a da espécie doadora, quanto receptora de próton, porém esse aum ento ocorre em m enor escala.
O efeito red shift ocorre quando parte da densidade eletrônica é transferida do receptor de prótons, Y, para o orbital sigm a antiligante (σ* ) do doador de próton, HX, causando um enfraquecim ento da ligação H-X e, consequentem ente, um decréscim o no valor de sua frequência de estiram ento[ 10]. Pode ocorrer ainda o efeito contrário no que diz respeito ao estiram ento vibracional de HX, ou sej a, quando o m odo vibracional de estiram ento HX é deslocado para um a freqüência m aior no com plexo de hidrogênio do que na espécie livre e, cham am os esse com portam ento de efeito blue shift.
A prim eira evidência experim ental do efeito blue shift na form ação de um com plexo de hidrogênio foi observada por Hobza e colaboradores[ 11]. Outra evidência experim ental desse efeito foi notada por Arnold e colaboradores para o efeito blue shift relativo ao m odo de estiram ento C–H em com plexos de triform ilm etano-clorofórm io (Cl3C-H)[ 12].
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 5
-duas espécies, doadora e receptora de prótons. Outra m aneira de verificar a redistribuição de cargas nos com plexos de hidrogênio é analisando a direção e o m ódulo do m om ento de dipolo elétrico do agregado m olecular[ 13].
Resultados experim entais e teóricos m ostram que ligações de hidrogênio não-usuais podem ocorrer entre elétrons π, por exem plo, presentes em hidrocarbonetos insaturados (que at uam com o receptores de próton), e o hidrogênio deficiente em elétrons de espécies HX[ 14-16]. Foi ainda const atado que ligações pseudo-π
presentes, por exem plo, no ciclopropano, são eficientes receptores de próton na form ação de ligações de hidrogênio. Um outro aspecto a destacar são as forças das ligações de hidrogênio envolvendo essas espécies não usuais com o receptoras de próton, sendo essas forças consideravelm ente m ais elevadas do que o esperado.
Com respeito aos fatores energéticos, a energia da ligação de hidrogênio estende-se de 15 a 40 kcal m ol-1 [ 17,18]. A energia em
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 6
-Tabela 1 .1 .1 . Caract eríst icas gerais dos três principais t ipos de ligação de hidrogênio[ 21].
Parâm et ros da ligação de
hidrogênio Fort e Moderado Fraco
Tipo de int eração Fort em ent e
covalent e
Geralm ent e
elet rost át ica Elet rost át ica
Com prim ent o de ligação
( H⋅⋅⋅Y [ em Å] ) 1,2 – 1,5 1,5 – 2,2 > 2,2
Alongam ent o do
com prim ent o X–H ( em Å ) 0,08 – 0,25 0,02 – 0,08 < 0,02
H- X versus H⋅⋅⋅Y X–H ≈ H⋅⋅⋅Y X–H < H⋅⋅⋅Y X–H < < H⋅⋅⋅Y
Com prim ent o da ligação de
hidrogênio ( X- H⋅⋅⋅Y) ( em Å) 2,2 – 2,5 2,5 – 3,2 > 3,2
Direcionam ent o Fort e Moderado Fraco
Ângulo da ligação de
hidrogênio ( em graus) 170 - 180 > 130 > 90
Energia da ligação de
hidrogênio ( Kcal m ol- 1) 15 - 40 4 - 15 < 4
Mudanças relat ivas no
infraverm elho ( cm- 1) 25% 10 – 25% < 10%
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 7
-∆
E = E
Com plexo- H-
Σ
E
Monôm eros ( 1.1.2)onde, ∆E corresponde a energia da ligação de hidrogênio; ECom plexo-H é a energia do com plexo de hidrogênio, Y⋅⋅⋅HX;
EMonôm eros é a energia das espécies receptora e doadora de próton, Y e
HX, respectivam ent e.
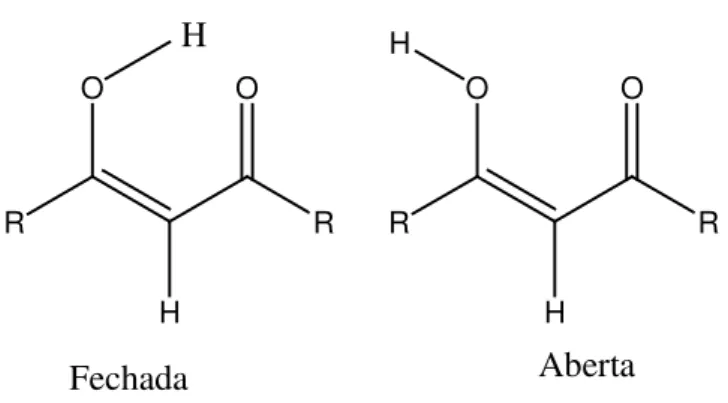
Por sua vez, a energia da ligação de hidrogênio intram olecular, geralm ente, é definida pela diferença de estabilidade entre as conform ações fechada e aberta da m olécula onde, na conform ação aberta se assum e que não há a form ação da ligação de hidrogênio e não há outro tipo de atração nem repulsiva nem atrativa com outros grupos. O caso inverso diz respeito à conform ação fechada, onde ocorre a form ação de um a ligação de hidrogênio[ 25].
Pode-se obter dois tipos de conform ações abertas para o m alonaldeido, dependendo de sua referência, com o m ostra a Figura 1.1.4.
Figura 1 .1 .4. Conform ações abertas em A e B para a m olécula de m alonaldeido.
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 8
-am bos os casos. I sto é um a definição im portante, pois a conform ação aberta é assum ida com o a origem na escala da energia de ligação de hidrogênio e os resultados de energia podem ser diferentes, dependendo da form a com o a conform ação de referência é adotada[ 25].
Algum as considerações são feitas para um bom entendim ento e segurança na interpretação num érica dos resultados:
A diferença entre as energias das conform ações fechada e aberta inclui term os que são determ inados por parâm etros geom étricos diferentes nos dois casos (por exem plo, ângulos de ligação).
As m oléculas que possuem ligação de hidrogênio intram olecular são, geralm ente, planares.
A conform ação aberta em O-H é com um ente m ais utilizada com o referência para calcular valores de energia de ligação de hidrogênio. Essa conform ação é preferencial porque preserva a configuração cis e apresenta duas ligações C= C na estrutura fechada.
A existência da ligação de hidrogênio intram olecular é afetada não apenas pela eletronegatividade dos heteroátom os envolvidos na m olécula, m as tam bém pelo tam anho da estrutura fechada[ 25].
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 9
-1 .-1 . COM POSTOS DI - CARBON Í LI COS
Muitas das m et odologias desenvolvidas pelos quím icos orgânicos de síntese, na verdade se baseiam em processos usados pela nat ureza para a construção de ligações carbono–carbono em biom oléculas. A condensação aldólica, um m étodo im portante de conversão de aldeídos e cetonas em com postos β-hidróxi– carbonilados, é um exem plo im portante. Por exem plo, na condensação de Claisen o ataque de um enolato ao grupo carbonila gera um a ligação carbono–carbono e, seus produtos são com postos 1,3-dicarbonilados, m ais conhecidos com o com postos β -dicarbonilados, im portantes por sua versatilidade em sínteses orgânicas[ 26]. Na Figura 1.2.1. é ilust rada um a reação de síntese através da condensação de Claisen.
Figura 1 .2 .1 . Condensação de Claisen form ando β-ceto- éster
Os hidrogênios ligados ao átom o de carbono localizado ent re as duas carbonilas no com posto β-dicarbonilado são ácidos, devido ao efeito indutivo das carbonilas, que retira elétrons e, tam bém , porque o ânion resultante da desprotonação é estabilizado por ressonância.
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 10
-Figura 1 .2 .2 . Condensação de Dieckm ann
Os com plexos m etálicos form ados por β-dicetonas têm sido em pregados com o adit ivos para com bustíveis, fluidos supercríticos, em inúm eras reações de condensação, principalm ente em reações orgânicas [ 27], na produção de film es supercondutores e de catalisadores hom ogêneos e heterogêneos[ 28]. Os β-dicetonatos de lantanídeos possuem am plas aplicações na separação de m ist uras de lantanídeos com outros m etais por sublim ação fracionada, em crom atografia gasosa, em crom atografia liquida de adsorção, em análises espectrofot om étricas, com o oxidantes e com o reagentes de deslocam ento em espectroscopia de RMN[ 29,30].
O tautôm ero m ais estável do com posto β-dicarbonílico, na form a cetoenólica, é o 3-hidroxipropenal, caracterizado por um sistem a t ipo anel fechado assim étrico estabilizado por um a ligação de hidrogênio intram olecular O-H⋅⋅⋅O m uito intensa[ 31], conform e
m ostrado na Figura 1.2.3.
O H O
R1 R2
H
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 11
-A intensidade da ligação de hidrogênio intram olecular nesses com postos pode ser afetada pela presença de substituintes que, dependendo de sua natureza, dim ensão e posição podem induzir diferentes efeitos na deslocalização dos elétrons π[ 32,33].
Devido à im portância de com postos dessa natureza em diversas áreas científicas e tam bém pela riqueza de interações que sua estrutura possui, esse trabalho se propõe a analisar a form ação da ligação de hidrogênio intram olecular nesses com postos utilizando alguns subst ituintes que possuem nat urezas distintas (subst ituintes doadores ou receptores de próton) com o m ostrado na Figura 1.2.4. Outra variante nesse estudo diz respeit o à conform ação do com posto com o pode ser observado:
O
O O O
H
R
H
R R
H
R
Fechada Aberta
H
onde R = CH3, CN, H, NH2, OH, SH
Figura 1 .2 .4 . Com posto di-carbonílico e as subst ituições propostas para estudo neste trabalho, através de m étodos t eóricos.
Esse tipo de com posto tem sido bastante invest igado em estudos que enfocam o fenôm eno da ligação de hidrogênio intram olecular. Efeitos com o deslocalização de elétrons π[ 34], ligação
di-hidrogênio intram olecular[ 35], efeitos de substituintes [ 36], entre
outros, podem ser com preendidos através de com postos dessa natureza.
CAPÍTULO 1 - INTRODUÇÃO
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 12
-linear entre: (i) os valores de deslocam ento do m odo de estiram ento O-H, δνO-H, versus os valores de increm ento no com prim ento da
ligação O-H, δrO-H, e, (ii) ainda, neste contexto, se pretende verificar
se essa relação linear pode ser estendida para os valores do m odo de estiram ento da ligação de hidrogênio intram olecular, O-H⋅⋅⋅O, versus os valores de increm ento no com prim ento da ligação O-H, δrO-H,
am bos os itens (i) e (ii) devidos à form ação da ligação de hidrogênio intram olecular.
Ainda, neste trabalho, pretende-se caracterizar a form ação da ligação de hidrogênio intram olecular nos com postos di- carbonílicos com suas respectivas substit uições, usando a Teoria Quântica de Átom os em Moléculas, QTAI M[ 37] e, na m edida do possível, fazer um estudo com parat ivo da força da ligação de hidrogênio intram olecular ao longo da série de subst ituintes propostos, H, CH3, CN, NH2, OH e
CAPÍTULO 2 – OBJETIVOS
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 13
-2 . OBJETI VOS
2 .1 . OBJETI VO GERAL
O desej o desse trabalho é caracterizar teoricam ente a ligação de hidrogênio intram olecular a partir de resultados de cálculos teóricos analisando propriedades eletrônicas, geom étricas e vibracionais. Para isso serão utilizados com postos di-carbonílicos (C3H2O2R2) com substituintes de natureza distintas (R= CN, H, SH,
CH3, OH, NH2) analisados através de m étodos quânt icos
com putacionais selecionados. Espera-se avaliar a natureza dessa interação nesses com postos bem com o o efeito que os substituintes podem exercer sobre a form ação da ligação de hidrogênio intram olecular.
O
O O O
H
R
H
R R
H
R
Fechada Aberta
H
CAPÍTULO 2 – OBJETIVOS
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 14
-2 .-2 OBJETI VOS ESPECÍ FI COS
Otim izar a geom etria dos com postos di-carbonílicos C3H2O2R2 com R= H, CH3, CN, NH2, OH e SH nas conform ações aberta
e fechada, encontrando o m ínim o global das barreiras rotacionais e obter as respectivas freqüências vibracionais harm ônicas;
I nvestigar as propriedades estruturais enfat izando os com prim entos de ligação O-H, rO-H, da espécie doadora de próton e
da ligação-H intram olecular, rO⋅⋅⋅H;
I nvestigar os principais m odos vibracionais e suas m odificações devido a form ação da ligação-H intram olecular, enfocando o deslocam ento do m odo de estiram ento da espécie doadora de próton, νO-H;
Caracterizar os novos m odos vibracionais que surgem devido a form ação da ligação-H intram olecular, enfocando a ligação de hidrogênio, νO-H⋅⋅⋅O;
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 15
-3 .1 . I N TRODUÇÃO
Os m odelos que descrevem os sistem as m acroscópicos e m icroscópicos são diferentes. Enquant o os sistem as m acroscópicos são bem descritos por m odelos físicos clássicos, algum as evidências experim entais m ostram que, para se descrever sistem as m icroscópicos há necessidade do uso de m odelos físicos diferentes denom inados de física quântica. Um a das diferenças fundam entais entre os m odelos clássicos e os m odelos quânt icos é que estes últ im os estão em basados em princípios probabilíst icos. Por exem plo, dada um a função de onda, é possível determ inar a densidade de probabilidade de se encontrar um a partícula em certa região do espaço e não a posição exata desta partícula [ 38].
Os postulados da m ecânica quânt ica consideram que qualquer sistem a pode ser com pletam ente descrito por um a função de onda (Ψ) . O quadrado desta função de onda, (Ψ2) (ou |Ψ|2, se Ψ for
com plexa), define a densidade de probabilidade das partículas serem encontradas em det erm inada região do espaço.
Um operador herm it iano, atuando nesta função de onda, fornece um a observável do sistem a. Em notação m atem ática, tem os a equação do autovalor, eq. (3.1) :
Ô
Ψ
=
o
Ψ
(3.1)
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 16
-��
Ψ
= E
Ψ
(3.2)A eq. (3.2) é a equação fundam ental da m ecânica quântica, conhecida com o Equação de Schrödinger. As várias soluções possíveis para a equação de Schrödinger correspondem aos diferent es estados estacionários da partícula ou da m olécula. A solução que tem a m enor energia é cham ada de estado fundam ental [ 39].
3 .1 .1 . OPERADOR H AM I LTON I AN O
O operador Ham ilt oniano para um dado sistem a eletrônico é descrito com o a som a das contribuições cinética e potencial para este sistem a, eq. (3.3) :
��
=
��
+
��
(3.3)
O operador Ham iltoniano que descreve um sistem a m olecular com n elétrons e M núcleos engloba cinco contribuições para a energia deste sistem a: os term os cinéticos referentes aos elétrons e aos núcleos, os term os de atração e repulsão eletrônica e o term o de repulsão nuclear, eq. (3.4) :
��=− � ħ
2
2�� ∇�
2− � ħ 2
2�� �
�
∇�2− � ��
2�
�
��� + � �
�2
��� + � �
�2�
���
��� �
�>�
�
� �
�>�
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 17
-onde, os índices i e j referem -se aos elétrons, A e B aos núcleos, ħ é a constante de Planck dividida por 2π, me é a m assa do elétron, mA é
a m assa do A-ésim o núcleo, ∇2 é o operador laplaciano, e é a carga
do elétron, Z é o núm ero atôm ico,riA é a distância elétron- núcleo, rij é
a dist ância intereletrônica e RAB é a separação internuclear [ 40].
A resolução da equação de Schrödinger, ut ilizando o operador Ham iltoniano da eq. (3.4), é difícil para a m aioria dos sistem as de interesse quím ico.
Para sim plificar esse problem a, algum as aproxim ações são em pregadas com o, por exem plo, a aproxim ação de Born-Oppenheim er (BO). Esta sim plifica o problem a m olecular através da separação dos m ovim entos dos núcleos e dos elétrons. Esta aproxim ação é razoável, se considerarm os a escala tem poral do m ovim ento relativo dos elétrons e núcleos [ 40], onde os prim eiros se aj ustam instantaneam ente as m udanças nas posições nucleares. Logo, a distribuição eletrônica dentro de um sistem a m olecular depende das posições dos núcleos, e não de suas velocidades. Desse m odo o operador Ham iltoniano pode ser separado em um a parte eletrônica (��el) e outra nuclear (��N), eq. (3.5):
��
=
��
el+
��
N (3.5)
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 18
-ℎ
�
�=
−
1
2
∇
�2
−
�
�
��
�� ��=1
(3.6)
3 .1 .2 . O M ÉTODO H ARTREE– FOCK
O term o de repulsão intereletrônica da eq. 3.4 é a principal dificuldade para se resolver a equação de Schrödinger no caso de sistem as m ult ieletrônicos. Esse term o pode ser dividido, considerando apenas as com ponentes m onoeletrônicas.
Assim a equação de Schrödinger para um sistem a contendo n elétrons é dada por um conj unto de n equações do tipo:
����+ � ���� �
�=1
+ ���� �� = ����,� = 1, … ,� (3.7)
onde ��� é o operador de energia cinética do i- ésim o elétron,
��
�� o term o de atração de todos os N núcleos pelo i- ésim o elétron e��
� o operador de repulsão eletrônica efetiva do i- ésim o elétron. As funções�
�, denom inadas orbitais, correspondem a funções m onoeletrônicas
que descrevem o estado do i- ésim o elét ron com energia ��.
Douglas Hartree trabalhou com produtos das funções �� da seguinte form a[ 41]:
Ψ (1, 2,..., n) = ϕ1 ( 1) ϕ2 (2) ... ϕn (n) (3.8)
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 19
-a indistinguibilid-ade dos elétrons e -a -anti-sim etri-a d-as funções de onda que os representa.
Vladim ir Fock ent ão utilizou um a função com a seguinte form a[ 42]:
Ψ = 1
√�! �
Ψ1(1) Ψ2(1) ⋯ Ψ�(1) Ψ1(2) Ψ2(2) ⋯ Ψ�(2)
⋮ ⋮ ⋱ ⋮
Ψ1(�) Ψ2(�) … Ψ�(�)
� (3.9)
sendo, N o núm ero total de elétrons, 1
√�! é o fator de norm alização das funções de onda.
A ant i-sim etria é autom aticam ente sat isfeita pela representação da função de onda na form a de determ inantes, um a vez que, a troca de duas coordenadas dos spin-orbitais, que equivale a t rocar duas linhas do determ inante, faz com que a função de onda m ude de sinal devido aos elétrons serem partículas de spin fracionário, sendo por isso denom inados de férm ions. Esta form a de representação das funções de onda foi proposta por Slat er[ 43], sendo denom inados os determ inantes de “ determ inantes de Slater” e as funções de “ funções spin-orbitais” .
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 20
-constit ui a prim eira correção dos efeitos de correlação eletrônica no m odelo de Hartree.
Entretanto, a aplicação inicial do m étodo se restringia apenas aos átom os. Apenas em nível atôm ico as equações de Hartree-Fock podiam ser resolvidas num ericam ente. Modificações no procedim ento de Hartree-Fock para descrever as m oléculas foi proposto por Roothaan em 1951[ 44].
A proposta de Roothaan consist ia em com binar orbitais atôm icos que, para o caso de sistem as com m uitos elétrons, são funções aproxim adas, para form ar os orbitais m oleculares. Este procedim ento é conhecido com o “ Com binação Linear de Orbitais Atôm icos” (Linear Com bination of Atom ic Orbitals - LCAO). A sugestão de Roothaan não foi a criação das com binações lineares dos orbitais atôm icos, m as a sua ut ilização nas equações de Hartree-Fock.
O bom desem penho de um cálculo em nível HF depende da escolha apropriada dos conj untos de base. A energia eletrônica obt ida é variacional e o aum ento da qualidade das funções de base torna os resultados cada vez m ais precisos, até que se atinj a o lim it e Hartree-Fock. Pelo princípio variacional, para um dado sistem a, a energia no lim ite Hartree-Fock (EHF) é sem pre m aior que a energia exata (E),
obtida pela resolução da equação de Schrödinger não relativíst ica, dentro da aproxim ação de Born-Oppenheim er. Esta diferença de energia é conhecida com o energia de correlação eletrônica (Ecorr).
E
corr= E – E
HF ( 3.10)CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 21
-fundam ento para posteriores aproxim ações, que incluem efeitos de correlação eletrônica. Os m étodos que incluem a energia de correlação eletrônica são cham ados de m étodos pós-HF e serão descritos adiantes.
3 .2 . TEORI A DE PERTURBAÇÃO D E M∅LLER- PLESSET
Existem diversas teorias de perturbação de m uitos corpos, desenvolvidas para incluir a correlação eletrônica ao m étodo Hartree-Fock.
Em 1934, C. Möller e M. S. Plesset propuseram um tratam ento perturbativo, onde o Ham iltoniano total de um dado sistem a é dividido em duas partes. A prim eira corresponde ao Ham ilt oniano não perturbado (��° ), que possui autofunções e autovalores conhecidos e
a segunda representa um a pequena perturbação no �′�° . Est a teoria é conhecida com o Teoria de Perturbação de Muitos Corpos de Møller-Plesset (Many Body Perturbation Theory - MBPT)[ 40], eq. (3.11).
��
=
��
° +
λ
��′
(3.11)Considerando a teoria de perturbação MBPT, para um sist em a no estado fundam ental de cam ada fechada, a Equação de Schrödinger pode ser escrita com o, eq. (3.12):
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 22
-Expandindo Ψn e En em séries de Taylor em relação a λ
(parâm etro relacionado à perturbação) , obtém -se os valores Ψ�(�) e
��(�), que são as respectivas correções de ordem k na função de onda e na energia, eqs. ( 3.13) e (3.14):
Ψ
�(�)=
�
λ
�
�
!
∞
�=0
∂
�Ψ
�
∂λ
� (3.13)
�
�(�)=
�
λ
�
�
!
∞
�=0
∂
��
�∂λ
� (3.14)
Percebe-se que a correção de prim eira ordem para a energia do estado fundam ental [ E0(1)] é a própria energia HF. Sendo assim , a
energia HF será substituída por um a versão m elhorada a partir da correção de segunda ordem , denom inado m étodo MP2, ou ordens m aiores, n ( Møller-Plessetn).
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 23
-3 .-3 . TEORI A DO FUN CI ON AL DA DEN SI DADE
A função de onda eletrônica de um a m olécula de n elétrons depende de 3n coordenadas espaciais e n coordenadas de spins. O operador Ham iltoniano contém som ente os term os espaciais dos elétrons, a energia m olecular pode ser escrita em term os de integrais que fornecem apenas seis coordenadas espaciais. Assim , a função de onda de um a m olécula polieletrônica contém m ais inform ações que o necessário e falta um significado físico direto. Este fato deu inicio a busca por funções que fornecessem m enos variáveis que as funções de onda e que pudessem ser utilizadas para calcular a energia e outras propriedades[ 40].
Na década de 1920 alguns cient istas com partilhavam a idéia de que a energia de um sistem a pode ser expressa com o um a função de sua densidade eletrônica, ρ(r), ou sej a, E = E[ρ(r) ] . A densidade eletrônica é um a função das coordenadas espaciais, r. Por esta razão diz-se que a energia é um “ funcional” da densidade eletrônica, pois o papel de variável é desem penhado por um a outra função, ρ(r).
Pode-se dizer que a estrutura m oderna da Teoria do Funcional da Densidade (Density Functional Theory, DFT) deve-se aos trabalhos de Hohenberg e Kohn, em 1964 [ 46], e Kohn e Sham , em 1965 [ 47].
Na abordagem DFT, os elétrons interagem uns com os out ros e com um potencial externo. O prim eiro teorem a de Hohenberg-Kohn estabelece que este potencial externo sej a um potencial único de ρ(r) , além de um a constante adit iva. Dessa form a, um sistem a m ult ieletrônico pode ser descrito pelo seguinte Ham iltoniano, eq. (3.15):
��
=
��
+
����
+
��
(3.15)CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 24
-sendo �� o operador energia cinética, ���� o operador de repulsão eletrônica e Û o potencial externo, descrito por ∑υ(�). Considerando que a energia de um sistem a é dada por �Ψ����Ψ�, tem -se que essa energia é um funcional da sua densidade eletrônica, eq. (3.16):
�
[
�
] =
�
Ψ
���
+
����
+
���
Ψ
�
�
[
�
] =
�
Ψ
���
+
�����
Ψ
�
+
�
Ψ
����
Ψ
�
�
[
�
] =
�
[
�
] +
�
υ
(
�
)
ρ
(
�
)
��
(3.16)
sendo F o funcional universal de ρ, que independe do potencial externo υ (r) [ 48].
Baseado no teorem a variacional, o segundo teorem a de Hohenberg e Kohn estabelece que o m ínim o do funcional de energia se obtém quando ρ é a densidade do estado fundam ental, associada ao potencial externo υ (r) , ou sej a, eq. (3.17):
�
[
�
] +
�
υ
(
�
)
ρ
(
�
)
��
≥
�
[
ρ
]
(3.17)A determ inação da energia do est ado fundam ental de um sistem a m ult ieletrônico, sob um potencial externo é feita através do m étodo de Kohn- Sham , que utiliza o conceito de um sistem a de referência de partículas independentes, eq. (3.18).
� [�] = ��[�] + �υ(�)ρ(�)��+1
2 �
�(�)�(�′) |� − �′| ����
′ + �
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 25
-sendo ��[�] o funcional de energia cinét ica de um sistem a de elétrons que não interagem , tendo a m esm a densidade eletrônica de um sistem a que interage e ���[�] o term o de t roca e correlação, que é desconhecido [ 49].
Na m aioria das vezes o funcional de troca e correlação é separado: ���[�] = ��[�] + ��[�]. É interessante ressaltar que Ec não
corresponde a m esm a definição de correlação dos m étodos ab initio, enquanto que Ex possui a m esm a definição para o term o de troca
dada pelo m étodo HF.
Logo após, Becke[ 50] percebeu que haveria vantagens em m esclar os m étodos de Hartree-Fock e DFT, o que deu origem aos m étodos híbridos. O m ais popular deles nos dias de hoj e é conhecido com o B3LYP, sigla que identifica o uso do funcional de troca-correlação de Becke no qual está incluído o funcional de troca-correlação desenvolvido por Lee, Yang e Parr[ 51]. O núm ero três vem do uso de três parâm etros em píricos ut ilizados para com por o funcional Ex. Por
utilizar esses parâm etros em píricos, é com um não classificar o m étodo B3LYP com o ab init io. Convém m encionar que nem todos os m étodos DFT são híbridos ou ut ilizam parâm etros e, assim , poderiam ser cham ados ab initio. Todavia, os parâm etros em píricos estão presentes nos m ais ut ilizados e é com um considerar a DFT propriam ente com o um a classe à parte para diferenciá-la de m étodos com o HF e MP2. Um a vantagem im portante da DFT sobre os dem ais m étodos de correlação eletrônica é sua rapidez, o que fez com que essa teoria se tornasse popular nos últim os anos entre a com unidade quím ica.
Diante dessa vantagem e tendo em vista o sucesso dessa teoria para descrever a ligação de hidrogênio[ 52,53] nosso estudo foi
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 26
-3 .4 . FUN ÇÕES DE BASE
É a part ir das funções atôm icas, que são por sua vez descritas pelas funções de base, que são construídos os orbitais m oleculares e, sendo assim , a escolha correta do conj unto de funções de base é essencial para um bom resultado, sej a em term os de exatidão ou de rapidez.
A escolha m ais óbvia nos prim eiros tem pos da quím ica com putacional foi à adoção do cham ado conj unto de base m ínim o, com posto apenas um a ou algum as poucas funções de base para representar os orbitais atôm icos do átom o livre. Assim , o conj unto m ínim o para o hidrogênio é um único orbital 1s, ao passo que para o carbono tem os 1s, 2s, 2px, 2py e 2pz.
Os orbitais para o átom o de hidrogênio, soluções exatas da equação de Schrödinger, são “ orbit ais do tipo hidrogenóides e possuem um a dependência radial exp (-ξr) . O problem a com essas funções é a sua form a m atem ática, que acaba por dificultar os cálculos.
Por outro lado, funções Gaussianas, exp (-ξr2) , possuem
propriedades que facilitam sua m anipulação m atem ática e se m ostram atrat ivas para a subst ituição dos STO (orbital do tipo Slater)
[ 54]. Porém , um a única Gaussiana não é capaz de reproduzir um STO,
principalm ente próxim o à origem . Faz- se necessário então com binar várias Gaussianas para aproxim ar o com portam ento de um STO, m as ainda assim o uso das Gaussianas traz sim plificações.
Um dos prim eiros conj untos construídos com este intuito foi o STO-3G [ 55,56], no qual três funções gaussianas são com binadas para
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 27
-A base STO-3G, apesar de ter sido am plam ente ut ilizada nos prim eiros tem pos da quím ica com putacional, possui pouca flexibilidade do pont o de vista quím ico. Por exem plo, um orbital 2s de um carbono próxim o a um átom o m ais eletronegativo deve ser m ais com pacto do que próxim o a um átom o de m aior eletropositividade. Mas no STO-3G am bos os am bientes são representados pela m esm a base 2s.
Visando resolver este problem a, foram desenvolvidos os conj unto de bases com “ valência dividida” . Por exem plo, ao invés de com binar três Gaussianas para descrever um orbital de valência num a única contração, pode-se dividir essas três funções em dois grupos, um com duas e outro com um a única Gaussiana. Esses grupos podem ent ão ser otim izados de form a separada e produzir orbitais que se adaptam m elhor às particularidades do am biente quím ico.
A base 3-21G representa cada orbital interno por um a com binação de três Gaussianas prim itivas, e cada orbital de valência é representado por dois grupos de funções, um form ado por duas Gaussianas prim itivas e o outro por apenas um a.
É possível ainda m elhorar a base aum entando-se o núm ero de Gaussianas. Esse é o caso da base 6-31G, que utiliza 6 Gaussianas prim itivas para representar cada orbit al interno e, à sem elhança da 3-21G, divide cada orbital de valência em dois grupos, sendo agora de 3 e de 1 Gaussianas prim itivas. Os resultados das bases com valência dividida são bastante superiores aos dos conj untos m ínim os, m as em com pensação o tem po de cálculo aum enta substancialm ente.
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 28
-efeito pode ser tratado acrescentando- se “ funções de polarização” ao conj unto de bases.
As funções de polarização são fundam entais para a correta descrição dos ângulos de ligações. Segundo Jasien et al.[ 57], essas funções são tam bém necessárias para um a descrição adequada das barreiras rotacionais em diversos com postos. Denotam -se as funções de polarização por um a letra entre parênteses, ou por “ asteriscos” : 6-31G(d,p) ou 6-31 G* * indica o uso da base 6-31G, com o a definim os acim a, com a inclusão de funções do tipo p no hidrogênio e funções do tipo d nos átom os pesados.
Há sistem as quím icos cuj a descrição em regiões distantes do núcleo m erece ser considerada com m ais cautela, com o é o caso dos ânions ou dos átom os com pares isolados. Para esses sistem as, adicionam -se m ais um a Gaussiana ao conj unto de bases, possuindo esta Gaussiana um coeficiente (ξ) pequeno. I sto faz com que a função adicionada decaia “ lentam ente” com o raio atôm ico e possua valores significativos nas regiões afastadas do núcleo. Por essa razão, são cham adas “ funções difusas” e são identificadas pelo sím bolo “ + ” . Assim , a base 6-31+ G(d,p) possui todos os atributos anteriores e ainda um a função difusa em cada átom o pesado (heteroátom o). Pode-se ainda adicionar funções difusas ao hidrogênio e aí teríam os 6-31+ + G(d,p) .
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 29
-3 .5 . AN ÁLI SE POPULACI ON AL
A função de onda obtida a partir de cálculos de orbitais m oleculares não pode ser interpretada fisicam ente. Ao contrário, a densidade de carga ou distribuição de densidade eletrônica em um a m olécula, representada por �(�⃗), possui um a interpretação segundo a física clássica. Essa é um a propriedade classificada com o “ local” , isto é, definida para cada ponto do espaço especificado pelo vetor posição
�⃗, podendo ser calculada, segundo a t eoria dos orbitais m oleculares
[ 40], eq. (3.19):
�
(
�⃗
) = 2
�
|
Ψ
�(
�⃗
)|
2�/2
�=1 (3.19)
em que Ψi representa o i- ésim o orbital m olecular duplam ente
ocupado em um sist em a contendo n elétrons.
A densidade eletrônica é um fator im portante que influencia as propriedades físicas e quím icas de um a m olécula, sendo aplicadas em estudos de reatividade quím ica, interações interm oleculares, sim ilaridade m olecular, etc. A im portância da densidade de carga na área da Quím ica foi consolidada pela form ulação da Teoria do Funcional da Densidade proposta por Hohenberg e Kohn na década de 1960.
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 30
-expansão m ultipolar da densidade eletrônica de carga m olecular
[ 62,63].
Consequentem ente, as cargas atôm icas fornecem a representação m ais sim ples possível da distribuição de carga total na m olécula. Ao contrário da densidade eletrônica, as cargas atôm icas não podem ser calculadas teoricam ente sem arbitrariedades, pois estas não são valores esperados da função de onda, isto é, não são observáveis físicas. Deste m odo, todos os m étodos para cálculo teórico de cargas atôm icas são invariavelm ente arbitrários, resultando, em geral, em um a grande variação nos valores absolutos das cargas atôm icas.
Apesar de sua natureza arbitrária, as cargas atôm icas fazem parte do pensam ento quím ico, encontrando aplicações em vários estudos, com o por exem plo: estudo do efeito de substituintes; avaliação de interações intra e interm oleculares em Métodos de Mecânica Molecular, Dinâm ica Molecular, entre outros; análises conform acionais; estudos de correlações entre a estrutura m olecular e a atividade biológica; etc.
Existem diversos m étodos em pregados atualm ente e outros continuam sendo desenvolvidos, na tentat iva de se buscar um a análise m ais precisa e com putacionalm ente viável das características físicas e quím icas de um a m olécula através do estudo das cargas atôm icas.
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 31
-3 .5 .1 . AN ÁLI SE POPULACI ON AL DE M ÜLLI KEN
Este m odelo consiste no m ais tradicional e de uso m ais difundido entre os quím icos. Mülliken propôs, baseado na teoria de orbitais m oleculares, que um conj unto de orbitais m oleculares, (Ψk),
sej a definido por um a com binação linear de k orbitais atôm icos duplam ente ocupados (Xj) [ 64], eq. (3.20).
Ψ
�=
�
(
�
���
�)
�
�=1
(3.20)
Os coeficientes Cj k, são determ inados pelo m étodo
Hartree-Fock.
A densidade eletrônica associada ao k- ésim o orbital m olecular é expressa em term os de orbitais atôm icos com o vem os na eq. 3.21:
�
�(
�⃗
) =
� � �
���
��
� ��=1
�
�=1
(3.21)
��� = 2∑ ��� ����� são elem entos da m atriz densidade. I ntegrando a
eq. 3.21 tem os a eq. (3.22):
�
ρ
(
�⃗
)
��⃗
=
� � �
���
��=
�
�
�=1
�
�=1
(3.22)
onde, ��� = ∫ ������⃗ é cham ada de “ integral de recobrim ento”
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 32
-cada orbital atôm ico Xi, denom inadas de “ população orbital” e
representada por
η
i, eq. (3.23):η
�=
�
��= 2
���
���
2�
�=1
(3.23)
No local da região de recobrim ento, a população,
Λ
ij, éexpressa pela eq. ( 3.24), onde i é diferente de j :
Λ
��= 2
�
���
��= 4
�
(
�
���
��)
�
�=1
�
��(3.24)
Pij são elem entos da m atriz densidade que com põem a população.
A população eletrônica atribuída às regiões de ligação (eq. 3.24) pode ser dividida entre átom os individuais. A população bruta, pi, no orbital atôm ico i é definida pela eq. (3.25):
�
�=
η
�+
1
2
�
Λ
���≠�
(3.25)
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 33
-�
�=
� �
� ��∈�
(3.26)
�
�=
�
�−
�
� (3.27)
onde, PA representa a população brut a para o átom o A, ZA é a carga
nuclear do átom o A.
Devido à facilidade de cálculo, a análise de Mülliken é bastante popular. Todas as variáveis necessárias para essa análise são obtidas diretam ente, sem nenhum custo com putacional adicional. Porém , este m étodo tem lim itações im portant es [ 65,66].
A principal fonte de problem as é a divisão das populações nas regiões de recobrim ento em contribuições atôm icas. O fator arbitrário “ ½ ” na equação 3.25 divide a população eletrônica de ligação igualm ente entre cada um dos dois át om os que com põem a ligação.
Esta part ição equitativa não pode ser considerada num a ligação quím ica onde existem dois átom os com eletronegatividades diferentes.
Outro problem a dessa análise populacional é a alta dependência do conj unto de base em pregado no cálculo. Em bora em alguns casos a análise populacional de Mülliken forneça resultados satisfatórios, na m aioria dos casos ela caracteriza m ais os detalhes do conj unto de base, do que a estrutura eletrônica da m olécula.
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 34
-3 .5 .2 . CARGAS ATÔM I CAS DERI VADAS DO POTEN CI AL ELETROSTÁTI CO
Classicam ente, um conj unto de N cargas elétricas puntiform es, num determ inado instante, gera um cam po elétrico E no espaço, o qual aplica um a força F= q0 E, em um a carga de prova q0. Associado a
esse cam po, pode-se definir um a grandeza escalar denom inada potencial eletrostático V, que possui arbitrariam ente valor nulo a um a distância infinita do conj unto de cargas. Em um ponto do espaço, este potencial eletrostático, tam bém denom inado potencial Coulôm bico, é dado pela equação 3.28:
�
=
1
4
��
�� �
�
��
��
��=1
(3.28)
onde,
ε
o é a perm issividade no vácuo, qi é a m agnitude da i-ésim acarga elétrica, ri é a distância da i-ésim a carga até o ponto onde V é
calculado.
O potencial eletrostático m olecular quântico para um ponto p na posição ����⃗�, dado por ���(�⃗) em torno de um a m olécula com posta de n elétrons e N átom os, é definido com o a força eletrostática que age sobre um a carga positiva unitária neste ponto, segundo a equação 3.29:
�
��(
�⃗
) =
�
�
��
�⃗
�− ��⃗
��
��=1
−
�
�
(
�⃗
)
��⃗
�−
�⃗�
��⃗
CAPÍTULO 3 – FUNDAMENTAÇÃO TEÓRICA
DISSERTAÇÃO DE MESTRADO – ALINE FONSECA BEZERRA - 35
-em que, ZA é a carga do núcleo A localizado na posição �����⃗� e ρ(�⃗) é a
densidade eletrônica total.
O prim eiro term o da equação 3.29 corresponde à contribuição dos núcleos para o potencial eletrostático total, em que a lei de Coulom b é ut ilizada para calcular o potencial de repulsão entre as cargas pontuais ZA e a carga unitária em ����⃗�. O segundo term o dessa
equação corresponde ao potencial de atração eletrostática envolvendo a distribuição de cargas eletrônicas em todo o espaço e a carga unitária positiva em ����⃗�. Um a vez que a função de onda do
sistem a é calculada através de m étodos ab initio ou sem i- em píricos, o potencial eletrost ático pode ser avaliado através da equação 3.29.
É necessário estabelecer a localização e o núm ero de pontos que serão utilizados no cálculo do pot encial eletrostát ico e posterior aj uste ao m odelo de cargas pontuais. Usualm ente, selecionam -se pontos situados em um conj unto de cam adas além da superfície de Van der Waals na m olécula. Foram efetuados estudos com o obj etivo de determ inar o m enor núm ero de pontos necessários para proporcionar um a qualidade de aj uste aceitável, os quais resultaram em valores ótim os para o núm ero de cam adas, para a distância da cam ada m ais interna e superfície m olecular.
O m étodo CHELP (Charges from Eletrostat ic Potencial), proposto por Chirlian e Francl [ 67], tornou-se popular. Com a utilização de um a m alha regular de pontos, norm alm ente, é observada um a variação das cargas atôm icas com a rotação da m olécula, a qual é logicam ente indesej ada e representa um problem a sério em análises conform acionais. Neste contexto, o algorítm o geralm ente utilizado para efetuar o aj uste das cargas é o CHELPG (Charges from Eletrostatic Potencial Grid Based) [ 68].