Universidade de Lisboa
Faculdade de Farmácia
Molecular insights into the role of alpha-synuclein
phosphorylation in synucleinophathies
Inês Filipa Caldeira Brás
Dissertação orientada por:
Prof. Doutor Tiago Fleming Outeiro
Prof. Doutora Cecília Rodrigues
Mestrado em Ciências Biofarmacêuticas
Universidade de Lisboa
Faculdade de Farmácia
Molecular insights into the role of alpha-synuclein
phosphorylation in synucleinophathies
Inês Filipa Caldeira Brás
Dissertação orientada por:
Prof. Doutor Tiago Fleming Outeiro
Prof. Doutora Cecília Rodrigues
Mestrado em Ciências Biofarmacêuticas
The studies presented in this dissertation were performed within the Cell and Molecular Neuroscience research group, at the Chronic Diseases Research Center (CEDOC), Nova Medical School, Universidade Nova de Lisboa, under the supervision of Tiago F. Outeiro, Ph.D. and Cecília M. P. Rodrigues, Ph.D.
Publications
Abstract
Inês Brás*, Carolina Madaleno*, Sandra Tenreiro, Tiago F. Outeiro. Molecular insights into the role of alpha-synuclein phosphorylation in synucleinopathies. 1st CEDOC Symposium on Chronic Diseases. 30 June – 1 July 2016, Lisbon, Portugal.
I
Resumo
As doenças neurodegenerativas, tal como a Doença de Parkinson (DP) e a Doença de Alzheimer resultam da perda progressiva de populações neuronais específicas levando ao aparecimento de sintomas clínicos característicos de cada doença. Estas pertencem a uma vasta superfamília de patologias conhecidas como sendo doenças onde existe a perda da conformação correta de proteínas, resultante da aquisição de uma conformação aberrante por parte de proteínas específicas, o que as torna mais propensas a agregar e formar estruturas em folha-beta como a estrutura amiloide. A composição dos agregados, assim como a sua localização em células, tecidos e órgãos, é específica de cada doença.
A DP é a segunda doença neuronal degenerativa mais comum. A perda progressiva de neurónios dopaminérgicos na substantia nigra pars compacta (SNpc) e a acumulação de Corpos de Lewy (CL) são os marcadores patológicos nesta doença. A alfa-sinucleína (aSyn) é o maior componente dos CLs e é uma proteína chave em outras doenças neurodegenerativas comumente conhecidas como sinucleinopatias, tal como a demência com Corpos de Lewy e a Atrofia de Múltiplos Sistemas (AMS). Embora multiplicações e mutações no gene que codifica a aSyn terem sido associadas a formas familiares da DP, a maioria dos casos são esporádicos. O mecanismo preciso envolvido na perda de conformação e agregação da aSyn permanece por elucidar. Por exemplo, é também incerto se as inclusões proteicas atuam como espécies protetoras ou tóxicas na patogénese da DP. Portanto, investigar as vias moleculares envolvidas na agregação proteica é essencial para a compreensão da progressão da doença e pesquisa de novos alvos terapêuticos para a DP e para as outras doenças relacionadas.
A fosforilação é uma modificação pós-tradução (MPT) importante para a sinalização celular, e está envolvida na agregação e toxicidade de proteínas em várias doenças neurodegenerativas. O estudo da fosforilação na aSyn é um tópico importante de investigação, dado que aproximadamente 90% da aSyn depositada em CL é fosforilada na serina 129 (S129), enquanto que menos de 4% da aSyn é fosforilada na proteína solúvel. No entanto, ainda não está elucidado se esta ou outras MPT na aSyn poderão ter um papel na doença, ou como poderão influenciar a agregação e a toxicidade da proteína. A fosforilação da aSyn tem sido abordada em diferentes aspetos, como a pesquisa de cinases e fosfatases capazes de modular esta MPT. Dadas as associações entre fosforilação e agregação, a modulação desta MPT elevou-se como uma estratégia terapêutica atrativa. A contribuição da fosforilação na aSyn neste e em outros resíduos na agregação e toxicidade continua elusiva apesar do grande esforço para desvendar o
II
seu envolvimento no processo de doença. Avanços nesta área poderão fornecer novos alvos terapêuticos para o tratamento de sinucleinopatias.
Esta dissertação focou-se na identificação de novas cinases e fosfatases envolvidas na patobiologia da aSyn usando um modelo bem estabelecido para a DP: a levedura Saccharomyces cerevisiae (S. cerevisiae). S. cerevisiae é um organismo eucariota simples e muito bem caracterizado, tanto a nível celular como ao nível genético. Além disso, este modelo tem sido capaz de fornecer conhecimentos sobre os mecanismos moleculares fundamentais da aSyn e de recapitular várias características importantes, como a formação de acumulações citoplasmáticas similares às inclusões da aSyn humana e alteração no tráfego vesicular. Apesar de a levedura carecer de ortólogos óbvios para alguns dos genes associados a DP, como a aSyn, tem sido utilizada extensivamente como modelo para o estudo da aSyn. Além disso, a descoberta de que a aSyn faz parte de uma diversa rede de interações incluindo cinases e fosfatases foi identificada em grandes estudos genéticos feitos em levedura e que foram posteriormente confirmados em células de mamífero. Em levedura, a aSyn é submetida a várias MPT observadas em células de mamífero e em cérebros de pacientes com DP, nomeadamente a fosforilação na S129. Apesar da relevância da fosforilação da aSyn na S129, tem sido exposta a importância de explorar as interações entre as diferentes MPT. Portanto, neste trabalho investigou-se o papel de um grupo de cinases e fosfatases na fosforilação da aSyn, formação de inclusões e toxicidade através da sua co-expressão com a aSyn humana. Demonstrou-se que não existe uma correlação estrita entre a percentagem de células que formam inclusões de aSyn e a toxicidade. Cdc28, uma cinase envolvida no ciclo celular, foi capaz de reduzir a fosforilação da aSyn na S129 de uma forma dependente do tempo. Os resultados revelaram ainda que os papéis desempenhados pelas cinases e fosfatases na toxicidade da aSyn estarão relacionados, pelo menos em parte, com outras funções desempenhadas por estas proteínas na célula, e não com uma alteração direta dos níveis de fosforilação na aSyn.
A função da aSyn ainda não está totalmente clarificada devido à sua aparente participação em diversos processos celulares. No entanto, observações concordantes sugerem um papel como regulador da reciclagem vesicular na sinapse. Em células de levedura, o tráfego vesicular entre o retículo endoplasmático (RE) e o Complexo de Golgi (Golgi) foi observado como um defeito inicial após a expressão da aSyn. Um potencial avanço na compreensão da toxicidade da aSyn na PD resultou da realização de grandes estudos genéticos através da sobre-expressão de genes em busca de supressores da toxicidade da aSyn em levedura, proporcionando uma oportunidade para a procura de genes cuja sobre-expressão em conjunto com a aSyn
III
restabeleceria a viabilidade celular. Estudos posteriores demonstraram que a toxicidade originada pela expressão da aSyn poderia ser suprimida pela sobre-expressão de proteínas envolvidas na maquinaria de transporte entre o RE e o Golgi. Desde a validação da levedura como modelo celular para o estudo da patobiologia da aSyn, os seus recursos genéticos permitiram a realização de grandes estudos, nos quais foram identificados modificadores da toxicidade gerada pela expressão da aSyn. Estes conhecimentos impulsionaram o estudo do papel da Sly41 na formação de inclusões e toxicidade da aSyn, e uma possível interação entre as proteínas. Estudos anteriores demonstraram que a expressão em multicópia do gene SLY41 suprimiu a perda do gene YPTI, que codifica uma Rab GTPase requerida para o transporte entre o RE e o Golgi. A sobre-expressão da Sly41 na via secretória eleva os níveis de cálcio no citosol para suprimir os defeitos na formação de vesículas. Diversas evidências indicam que o cálcio desempenha um papel na regulação do tráfego membranar através da via secretória. Além disso, a Sly41 foi identificada como sendo eficientemente adicionada a vesículas COPII para o seu tráfego entre o RE e o Golgi, e a sua correta localização nas membranas do RE e Golgi é requerida para a sua função como supressor. Observou-se neste trabalho que a sobre-expressão da aSyn com a Sly41 em células de levedura levou a um aumento da percentagem de células com inclusões de aSyn, com a particularidade de estas apresentarem uma única inclusão, um fenótipo que é diferente da expressão isolada da aSyn. Curiosamente, a sobre-expressão da SLY41 per si é muito tóxica e a sua deleção aparenta ser protetora com a expressão da aSyn, sugerindo um potencial efeito tóxico da sobre-expressão da Sly41 relacionado com a sua função na regulação dos níveis intracelulares de cálcio. A construção do gene SLY41 com um marcador fluorescente permitiu o estudo da co-localização entre a Sly41 e a aSyn, sugerindo que a forma citotóxica da aSyn em levedura pode associar-se com vesículas de transporte tal como a aSyn normalmente se associa com as vesículas sinápticas, e que foi anteriormente descrito para a Ypt1. No geral, a regulação da homeostasia intracelular do cálcio é complexa e permanece pouco compreendida, e a análise do seu possível papel na agregação da aSyn deve fornecer conhecimentos relevantes para a patobiologia da DP.
Claramente, os desafios para o futuro irão continuar na pesquisa do efeito da toxicidade originada pela aSyn e o entendimento de como esta proteína patológica se torna fosforilada e a medida a que as MPT têm impacto na agregação da proteína. Estes esclarecimentos poderão fornecer conhecimentos chave para a etilogia da DP. Notavelmente, o nosso estudo identificou modificadores adicionais da agregação e toxicidade da aSyn envolvidos numa variedade de
IV
funções celulares, sugerindo uma variedade de novas proteínas capazes de modular a patobiologia da aSyn.
V
Abstract
Neurodegenerative disorders, such as Parkinson’s disease (PD) and Alzheimer’s disease result from the progressive loss of specific neuronal populations, leading to the appearance of the clinical symptoms that are distinctive of each disorder. They belong to a wide superfamily of pathologies known as protein misfolding disorders, resultant from the aberrant folding of particular proteins, which makes these proteins more prone to aggregate and form amyloid-like beta-sheet structures. The composition of the aggregates, as well as their localization in cells, tissues and organs, is specific of each disease.
PD is the second most common neuronal degenerative disorder. The progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the accumulation of Lewy bodies (LBs) are the pathological hallmarks in this disorder. aSyn is the main component of the LBs and one key protein in other neurodegenerative diseases commonly known as synucleinophathies, such as dementia with LBs and Multiple Systems Atrophy (MSA). Although multiplications and missense mutations in the gene encoding for aSyn have been associated with familial forms of PD, the majority of cases are sporadic. The precise mechanisms involved in aSyn misfolding and aggregation remain unclear. For example, it is also uncertain whether LBs are protective or toxic species in the PD pathogenesis. Therefore, defining the molecular pathways involved in protein aggregation is essential for understanding the disease progression and for defining new targets for intervention in PD and related diseases.
Phosphorylation is a key posttranslational modification (PTM) for cellular signaling, and is involved in protein aggregation and toxicity in several neurodegenerative disorders. The study of aSyn phosphorylation is an important topic of research, given that approximately 90% of aSyn in LBs is estimated to be phosphorylated at serine 129 (S129), whereas less than 4% of aSyn is phosphorylated in the soluble protein. However, it is still not clear whether this and other PTMs in aSyn play a role in the disease, or how they can influence the aggregation and toxicity of the protein. The phosphorylation of aSyn has been addressed from different angles, such as the search of kinases and phosphatases able to modulate this PTM. Given the associations between phosphorylation and aggregation, modulation of this PTM increased as an attractive therapeutic strategy. The contribution of aSyn phosphorylation on this and in other residues in aggregation and toxicity remains elusive despite the great effort to discover its involvement towards the disease process. Advances in this area may provide new therapeutic targets to treat synucleinopathies.
VI
This dissertation focused on the identification of novel kinases and phosphatases involved in the pathobiology of aSyn using one well-established model for PD: the yeast Saccharomyces cerevisiae (S. cerevisiae). S. cerevisiae is a simple and very well characterized eukaryotic organism, both at genetic and cellular levels. Moreover, this model has been able to provide insights into the fundamental molecular mechanisms of aSyn and recapitulate several important features, such as the formation of cytoplasmic accumulations similar to human aSyn inclusions and alterations of vesicular trafficking. Even though yeast lacks obvious orthologs for some PD-associated genes, as aSyn itself, it has been extensively used as a model to study aSyn. Furthermore, the finding that aSyn is part of a diverse interaction network including kinases and phosphatases was identified in genetic screens performed in yeast and was further validated in mammalian cells. In yeast, aSyn is subjected to several PTM observed in mammalian cells and in PD patient’s brains, namely phosphorylation at S129. Despite the relevance of aSyn phosphorylation on S129, the importance to explore the cross talk between different PTMs has been presented. Therefore, we investigated the role of a set of kinases and phosphatases in aSyn phosphorylation, inclusion formation and toxicity through its co-expression with human aSyn. We showed that there was no strict correlation between the percentage of cells that formed aSyn inclusions and toxicity. Cdc28, a kinase involved in cell cycle, was able to reduce aSyn phosphorylation on S129 in a time-dependent manner. Our results further revealed that the roles of kinases and phosphatases on aSyn toxicity seem to be related, at least in part, to other functions that these proteins play in the cell, and not with a direct alteration of aSyn phosphorylation levels.
The function of aSyn is far from clear due to its apparent involvement in many cellular processes. However, converging observations suggest a role as regulator of synaptic vesicular recycling. In yeast cells, vesicular trafficking between the endoplasmic reticulum (ER) and Golgi apparatus (Golgi) was observed as an early defect following aSyn expression. A potential breakthrough in understanding aSyn toxicity in PD resulted from high-copy suppressor screens conducted in yeast, providing the opportunity to search for genes whose co-overexpression restored viability. Subsequent studies showed that aSyn toxicity could be suppressed by overexpression of proteins involved in ER-to-Golgi transport machinery. Since the validation of yeast as a cell model to study aSyn pathobiology, its genetic resources have enabled several screens, in which modifiers of aSyn toxicity were identified. This prompted us to investigate the role of Sly41 in aSyn inclusion formation and toxicity, and the possible interaction between the proteins. Multicopy expression of SLY41 was shown to suppress the loss of YPT1, an essential Rab GTPase required for ER- Golgi transport. The overexpression of Sly41 to the early secretory
VII
pathway elevates cytosolic calcium levels to suppress vesicle-tethering mutants. Several lines of evidence indicate that calcium plays a role in regulation of membrane trafficking through the early secretory pathway. In addition, Sly41 was identified as being efficiently packaged into coat protein complex II (COPII) vesicles for trafficking between the ER and Golgi compartments, and its proper localization into ER and Golgi membranes is required for its suppressive activity. We found that yeast cells overexpressing aSyn with Sly41 showed an increase in the percentage of cells with aSyn inclusions, in particular in cells displaying a single inclusion, a phenotype that is different from the expression of aSyn alone. Interestingly, overexpression of SLY41 per si is very toxic and its deletion seems protective under aSyn expression, suggesting a potential toxic effect of Sly41 overexpression related with its function in regulating intracellular calcium levels. Sly41 tagging with a fluorescent protein allowed us to study the co-localization between Sly41 and aSyn, suggesting that the cytotoxic form of aSyn in yeast may associate with transport vesicles as aSyn normally does with synaptic vesicles and that was previously described for Ypt1. Overall, the regulation of intracellular calcium homeostasis is complex and remains poorly understood, and the dissection of its possible role in aSyn aggregation may provide valuable insights into PD pathobiology.
Clearly, the challenges for the future will continue in exploring the effect of the aSyn proteotoxicity and understanding how this major pathological protein becomes phosphorylated and the extent to which PTMs impact upon the aggregation of the protein. This may provide key insights into PD etiology. Notably, our study identified additional modifiers of aSyn aggregation and toxicity involved in a variety of cellular functions, suggesting a variety of novel proteins able to modulate aSyn pathobiology.
IX
Acknowledgements
“With work and determination, you can accomplish all that you want, the important is to have health to fight for it.”
Graça Brás
First and foremost, I would like to express my sincere gratitude to my supervisor Prof. Tiago Fleming Outeiro for his guidance and support during my master degree and the opportunity given to be part of his group in CEDOC. It was a pleasure and a rewarding journey for my desire to become an excellent scientist. Also, to Prof. Cecília Rodrigues for the assistance and time dedicated to this dissertation.
To the Cell and Molecular Neuroscience Group, Dra. Sandra Tenreiro, Carolina Madaleno and Dr. Hugo Miranda, I want to thank for their insightful comments and encouragement during the development of my research. A very special thanks goes to Dra. Sandra Tenreiro, without whose motivation, immense knowledge and precious support it would not be possible to conduct this research. It was through her persistence and kindness that she taught me to not limit the goals for the future, despite where we come from. After all, it is the spark of science that make us build our own path in the scientific world.
To CEDOC, as an institution and community, I want to express my gratefulness for receiving so well the new students and sympathy demonstrated when I needed assistance. Furthermore, a special thanks goes to the flow cytometry facility for the assistance in the experiments.
I would also like to thank my friends and colleagues for the encouragement and help when I almost need it.
Last but not least, I am grateful to my parents, Graça and Celestino Brás, and to my brother Nelson Brás for the unconditional support in my life. Without them, I would not have been able to reach so far. Also, a dearest thanks to Rui Duarte that gave me strength through these years.
XI
Table of contents
I. Introduction 1
1.1. Protein misfolding diseases: synucleinopathies 3
1.2. Parkinson’s disease 4
1.3. Alpha-synuclein: shift from normal to toxic function 6
1.4. Posttranslational modifications in alpha-synuclein 10
1.4.1. Regulation of alpha-synuclein phosphorylation: kinases and phosphatases 11 1.5. Saccharomyces cerevisiae as a model system for studying the molecular basis of Parkinson’s disease 14
1.6. A yeast-based screen to identify genes involved in alpha-synuclein pathobiology 17
1.7. SLY41, a gene that restores defects in vesicular trafficking 21
II. Objectives 23
III. Materials and methods 27
Cultivation conditions and media 29
Cultivation of Escherichia coli 29
Cultivation of Saccharomyces cerevisiae 29
Isolation of plasmid DNA from Escherichia coli 29
Transformation of Escherichia coli 30
Transformation of Saccharomyces cerevisiae 30
DNA restriction assays 32
DNA ligation – Gateway reaction 32
Electrophoresis 32
DNA sequencing 33
XII
Electrophoretic separation of proteins 33
Western blot analyses 34
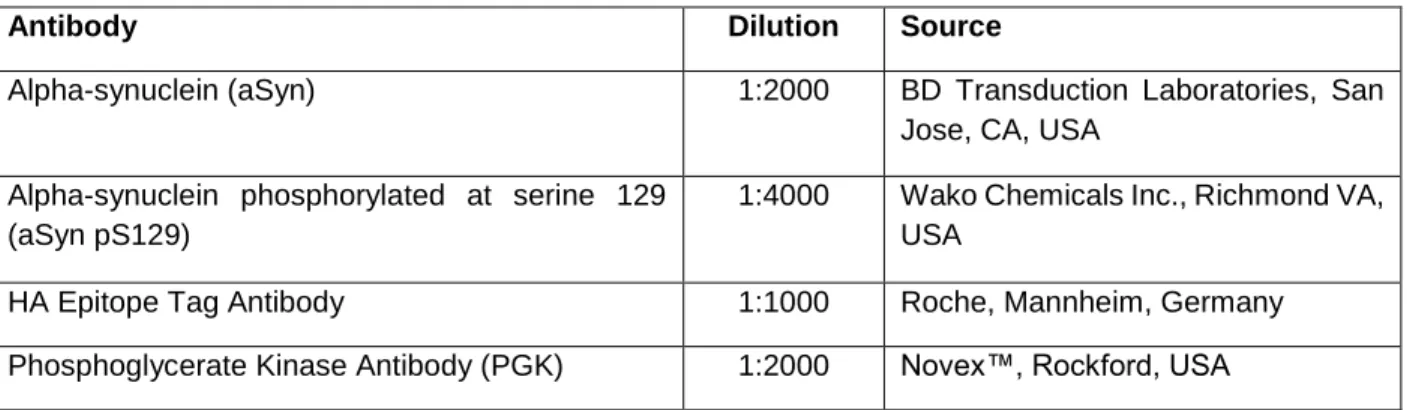
Fluorescence microscopy 35 Confocal microscopy 35 Flow cytometry 36 Toxicity assay 36 Statistics 36 IV. Results 37
4.1. Screening of selected kinases and phosphatases on aSyn phosphorylation, inclusion formation and toxicity 39
4.2. Sly41 expression enhance alpha-synuclein inclusion formation and toxicity with formation of single inclusions 55
4.2.1. Phenotypic analyses of ∆SLY41 strain 59
4.2.2. Subcellular imaging of alpha-synuclein single inclusions and Sly41 localization 64
V. Discussion 71
VI. General conclusions and future perspectives 79
XIII
Abbreviations
ALP – Autophagy-lysosomal pathway aSyn – Alpha-synuclein
ATP13A2 – ATPase 13A2 CK – Casein kinase
COPII – Coat protein complex II DJ-1 – Protein deglycase 1
DLB – Dementia with Lewy Bodies DNA – Desoxyribonucleic acid DsRed – Red fluorescent protein ER – Endoplasmic reticulum
ERAD – ER-associated degradation GFP – Green fluorescent protein Golgi – Golgi apparatus
GRK – G protein-coupled receptor kinase H. Sapiens – Homo sapiens
kDa – Kilo Dalton LBs – Lewy bodies LN – Lewy neurites
LRRK2 – Leucine- rich repeat kinase 2 mM – Milimolar
MSA – Multiple System atrophy
NBIA – Neurodegeneration with brain iron accumulation type I ORF – Open reading frame
PD – Parkinson’s disease PGK – Phosphoglycerate kinase
PINK1 – PTEN-induced putative kinase 1 PLKs – Polo-like kinases
XIV PP2A – Protein phosphatase 2A
pS129 aSyn – aSyn phosphorylated at S129 PTMs – Posttranslational modifications RFP – Red fluorescent protein
SNARE – Soluble NSF Attachment Protein Receptor
SNpc – Substantia nigra pars compacta S. cerevisiae – Saccharomyces cerevisiae
S87 – Serine 87 S129 – Serine 129
TH – Tyrosine hydroxylase
UPS – Ubiqiuitin–proteasome system
UCHL1 – Ubiquitin carboxy-terminal hydrolase L1 WT – Wild-type
Y39 – Tyrosine 39 µl – Microliter µg – Microgram 2µ – 2-micron plasmid
XV
Index of figures
Figure 1. Representative image of aSyn in LBs. 3 Figure 2. Schematic representation of human aSyn. 6 Figure 3. Relation between aSyn structure, function and toxicity. 8 Figure 4. Schematic representation illustrating the residues in aSyn that can be phosphorylated, the familial mutations, and the kinases or phosphatases able to phosphorylate or dephosphorylate aSyn. 11
Figure 5. Yeast model for aSyn-triggered cytotoxicity. 15 Figure 6. Schematic representation of genes coding the proteins involved in the screening in
Saccharomyces cerevisiae and homologs genes in Homo sapiens. 17
Figure 7. Effect of kinases and phosphatases expression on aSyn inclusions formation. 43 Figure 8. Effect of Plk2 in the formation of aSyn inclusions. 44 Figure 9. Immunoblot analysis of the levels of pS129 aSyn in yeast cells co-expressing aSyn with kinases or phosphatases. 45
Figure 10. Immunoblot analysis of the levels of pS129 aSyn in yeast cells co-expressing aSyn with Plk2. 47
Figure 11. Toxic effect of kinases and phosphatases expression in yeast cells. 49 Figure 12. Toxic effect of Plk2 expression in yeast cells. 50 Figure 13. Effect of kinases and phosphatases expression in cell viability. 52 Figure 14. Immunoblot analysis of the pS129 aSyn in yeast cells co-expressing aSyn with kinases or phosphatases. 54
Figure 15. Effect of Sly41 expression on aSyn inclusions formation. 55 Figure 16. Immunoblot analysis of the levels of aSyn in yeast cells co-expressing aSyn with Sly41. 56
XVI
Figure 17. Toxic effect of Sly41 expression in yeast cells. 57 Figure 18. Effect of Sly41 expression in cell viability. 57 Figure 19. Immunoblot analysis of the levels of pS129 aSyn in yeast cells co-expressing aSyn with Sly41. 59
Figure 20. Toxic effect of Sly41 expression in ∆SLY41 strain. 60
Figure 21. Toxic effect of aSyn expression in ∆SLY41 strain. 60
Figure 22. Effect of Sly41 expression in cell viability in ∆SLY41 strain. 62
Figure 23. Immunoblot analysis of the levels of aSyn in cells co-expressing aSyn with Sly41, in ∆SLY41 strain. 64
Figure 24. Analyses of the subcellular localization of aSyn-GFP inclusions in yeast cells co-expressing aSyn and Sly41. 65
Figure 25. Investigation of the subcellular localization of aSyn-GFP inclusions in yeast cells co-expressing aSyn and Sly41 using yeast strains with a genome marker of the spindle pole, nucleolus and actin cytoskeleton. 66
Figure 26. Schematic map showing the steps for the construction of pAG415GAL_ccdb_DsRed_SLY41 vector. 68
Figure 27. Imaging of the subcellular localization of aSyn-GFP inclusions and SLY41-DsRed. 69 Figure 28. Confocal analysis of the co-localization between Sly41-DsRed with aSyn-GFP. 70
XVII
Index of tables
Table 1. Summary of the Saccharomyces cerevisiae strains used in this study. 30 Table 2. Summary of the plasmids used in the yeast transformations. 31 Table 3. List of primary antibodies used for the immunoblotting analyses. 34 Table 4. Nomenclature of Saccharomyces cerevisiae genes, and respective homologs in Homo sapiens, of the proteins comprised in this screening. 39
1
3
1.1. Protein misfolding diseases: synucleinopathies
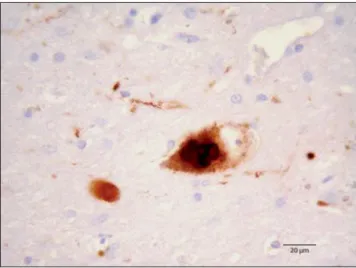
The prevalence of chronic neurodegenerative diseases is increasing due to the extension of life expectancy and the aging of the general population. A diverse group of neurodegenerative proteinopathies comprising idiopathic Parkinson’s disease (PD), dementia with Lewy bodies (DLB), multiple systems atrophy (MSA), and other rare clinical disorders as neurodegeneration with brain iron accumulation type I disease (NBIA or Hallervorden–Spatz disease), are associated with a common enigmatic protein: alpha-synuclein (aSyn) (Wakabayashi et al., 1998a; Goedert, 2001). In all these disorders aSyn is found to aggregate in the nervous system, but each disorder is characterized by different clinical phenotypes and differential morphological and spatiotemporal distributions of aSyn aggregates. Together, these disorders are known as “synucleinopathies” (Spillantini and Goedert, 2000; reviewed in Peelaerts and Baekelandt, 2016). aSyn was identified as the major component of neuronal deposits known as Lewy bodies (LBs) (Figure 1) (Spillantini et al. 1997). In PD and DLB patients, LBs were found in neurons throughout the brain (Spillantini et al. 1998), in contrast with MSA patients were aSyn inclusions were predominantly found in oligodendroglia and Schwann cells (Wakabayashi et al.1998b; Nakamura et al., 2015).
Despite the continuous effort, the main molecular and cellular mechanisms regulating aSyn aggregation and toxicity remain to be elucidated.
Figure 1. Light microscopy of a neuron full of aSyn in LBs. The neuron is located in the substantia nigra of
4
1.2.
Parkinson’s disease
PD is the most prevalent age-related movement disorder, and is characterized by a number of motor and non-motor features that can impact on multiple functions. The cardinal manifestations of PD include tremor at rest, bradykinesia (slowed movements), rigidity (increased muscular tone), postural instability, and gait impairment. Many patients also suffer from non-motor symptoms, including disturbances of autonomic functions and deterioration of cognition (Lees et al., 2009). Currently, only symptomatic therapies that ameliorate the motor symptoms are available, but the benefits are only temporary and disappear when the treatment is stopped, since they do not interfere with the progression of the disease.
A region-specific selective loss of dopaminergic, neuromelanin-containing, neurons from the pars compacta of the substantia nigra is the pathological hallmark of PD (Damier et al., 1999; Dickson et al., 2009). Furthermore, the consequent striatal dopamine deficiency is seen in association with the development of neuronal intracytoplasmic inclusions that are termed LBs, when they are present in cell bodies, and Lewy neurites (LNs) when they are present in neuronal processes (Spillantini et al., 1998). Because specific populations of catecholaminergic neurons in the substantia nigra and midbrain contain neuromelanin, cell loss within these regions is accompanied by depigmentation, readily visible in post-mortem analysis of human brain tissue. aSyn misfolded, posttranslationally modified and ubiquitylated is a major component of LBs found in affected neurons in PD (Spillantini et al., 1997; Uversky et al., 2007) and it is usually accepted that these cytoplasmic inclusions are central to the pathological process in the disease. LBs also contains numerous others proteins, including components of the ubiquitin–proteasome system (UPS) and molecular chaperones, as synphilin, parkin and ubiquitin (Halliday et al., 2005; Uryu et al., 2006; Uversky et al., 2007).
According to Braak’s model, PD is as a progressive disease that appeared initially in the olfactory bulb and the dorsal motor nucleus of the vagus nerve. From these areas the lesions ascend through the brain, impacting in different neural networks and leading to non-motor symptoms such as depression and cognitive decline (Braak et al., 2002, 2003a, b). This distribution of pathology could have suggested that the pathology was transmitted between neurons via the spreading of the misfolded aSyn, rising the “prion-like” hypothesis (Kordower et al., 2008; Li et al., 2008; Mendez et al., 2008). This hypothesis suggests that aSyn may self-propagate and, once transferred into a new cell, can act as a seed that recruits endogenous aSyn, leading to the formation of larger aggregates and contributing to the progression of the disease.
5
Most of PD cases have been considered sporadic, with the minority being linked to genetic factor cases. Mutations in the SNCA gene encoding for the protein aSyn - A53T (Polymeropoulos et al., 1997), A30P (Kruger et al., 1998), E46K (Zarranz et al., 2004), H50Q (Appel-Cresswell et al., 2013), G51D (Lesage et al., 2013), A53E (Pasanen et al., 2014) - result in the expression of mutant aSyn variants. In addition, duplications and triplications of SNCA, leading to elevated levels of aSyn, cause PD in some familiar forms of the disorder (Singleton et al., 2003; Chartier-Harlin et al., 2004). Apart from the genes causing the six monogenic forms of PD, changes in a large number of additional genes were considered PD-causative. These genes were identified by linkage analysis or a candidate gene approach, such as parkin, ubiquitin carboxy-terminal hydrolase L1 (UCHL1), protein deglycase 1 (DJ-1 or PARK7), PTEN-induced putative kinase 1 (PINK1), leucine-rich repeat kinase 2 (LRRK2) and ATPase 13A2 (ATP13A2) (Gasser, 2001; reviewed in Klein and Westenberger, 2012), which play important roles in mitochondrial function, UPS, autophagy-lysosomal pathway (ALP) and membrane trafficking (Hatano et al., 2009; Wales et al., 2013). Other studies also showed that polymorphisms in the SNCA gene lead to increased risk for developing PD (Satake et al., 2009; Simon-Sanchez et al., 2009; Edwards et al., 2010; Nalls et al., 2014). In addition to genetic factors, aging is the major risk factor for sporadic PD, as well as environmental triggers.
6
1.3. Alpha-synuclein: shift from normal to toxic function
aSyn is a 14.5 kDa protein of 140 amino acids abundantly expressed in the brain, primarily found in pre-synaptic terminals and in the cell nucleus (Maroteaux et al., 1988). It has also been reported to be expressed in other organs including kidney, liver and heart (Baltic et al., 2004), as well as being present in blood cells (Nakai et al., 2007). It is part of the synuclein family, which has two other members, beta-synuclein and gamma-synuclein (Lavedan et al., 1998).
Structurally, aSyn can be divided in three regions (Figure 2). The N-terminal is highly-conserved and an amphipathic region (residues 1-60), containing a series of imperfect KTKEGV repeats, and with propensity to form alpha-helical structures upon binding (Eliezer et al., 2001; Chandra et al., 2003). This region confers membrane binding ability to aSyn (Vamvaca et al., 2009; Bartels et al., 2010) and remarkably all six PD-related mutations described thus far are located in this domain (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Lesage et al., 2013; Pasanen et al., 2014) (Figure 2). The second is a central hydrophobic region (NAC region), between 61-95 residues, which stands for a non-amyloid component and this region is indispensable for the aggregation of aSyn (Giasson et al., 2001). Lastly, the third region is the C-terminal part, an acidic and proline-rich area negatively charged and highly unstructured, where several proteins have been reported to bind (Dev et al., 2003; Burré et al., 2012). In addition, this region has been suggested to play a role on the protein’s chaperone-like activity (Souza et al., 2000).
Figure 2. Schematic representation of aSyn featuring the three main regions and the six
7
aSyn is considered a natively unfolded protein, but it can misfold and aggregate, forming beta-sheet structures (Weinreb et al., 1996; Fauvet et al., 2012). However, it has also been reported that aSyn may occur as a natively folded helical tetramer (Bartels et al., 2011; Wang et al., 2011; Dettmer et al., 2013). Large, insoluble protein inclusions, inside or outside cells, where initially thought to be toxic, and some authors still claim that the final mature aggregates are the most dangerous for cell homeostasis (El-Agnaf et al., 1998; Tanik et al., 2013). However, current evidence indicates that they are rather cytoprotective (Bodner et al., 2006), and that the smaller, more soluble protein dimers and oligomers are the ones that exert toxicity (Outeiro et al., 2008; Karpinar et al., 2009; Winner et al., 2011). The toxicity induced through the aSyn pathological species comprise several mechanisms such as the normal function disruption of aSyn in neurotransmission release (possibly acting as a negative regulator of dopamine release) (Abeliovich et al., 2000; Cabin et al., 2002; Chen et al., 2013; DeWitt and Rhoades, 2013), mitochondrial structure and complex I activity impairment, as well as mitochondrial dynamics and mitophagy deficiency (Devi et al., 2008; Liu et al., 2009; Chinta et al., 2010; Nakamura et al., 2011), endoplasmic reticulum (ER) - Golgi apparatus (Golgi) vesicular transport disruption culminating in toxic ER stress (Cooper et al., 2006; Gitler et al., 2008; Thayanidhi et al., 2010) and protein- degradation mechanisms impairment (Martinez- Vicente and Vila, 2013), interfering with the normal physiology of the cell.
Although the physiological function(s) of aSyn remains unknown, substantial evidence suggests it plays a role in the vesicle trafficking during the neurotransmission release, synaptic function and plasticity (reviewed in Lashuel et al., 2013). This hypothesis has been supported through aSyn localization at presynaptic terminals (Jakes et al., 1994; Iwai et al., 1995; Withers et al., 1997), its association with the distal reserve pool of synaptic vesicles (Kahle et al., 2000; Lee et al., 2008; Zhang et al., 2008) and the deficiencies in synaptic transmissions observed in response to knockdown (Abeliovich et al., 2000; Cabin et al., 2002) or overexpression of aSyn (Scott et al., 2010; Nemani et al., 2010). Moreover, its possible role in regulating synaptic homeostasis comprises not only to direct interaction with synaptic vesicles, but also aSyn can interact with synaptic proteins controlling vesicle exocytosis, such as phospholipase D2 (Payton et al., 2004) and the family of RAB small GTPases (Dalfo et al., 2005; Chen et al., 2013). aSyn has been described as molecular chaperone in the SNARE formation complex, being involved in the release of neurotransmitters, including dopamine (Burré et al., 2010). Triple knockout mice exhibit a mild phenotype with alterations in synaptic structure and transmission, age-dependent neuronal dysfunction, as well as reduced survival (Greten-Harrison et al., 2010). Moreover, individual synucleins knockouts are viable, suggesting that these proteins, although not essential
8
components of the neurotransmitter release machinery, may contribute to the long- term regulation and maintenance of nerve terminal function (Chandra et al., 2004).
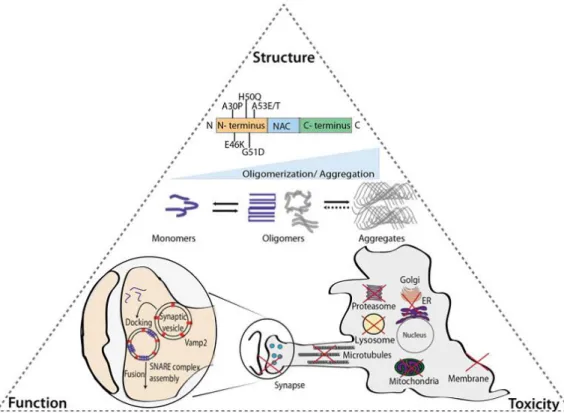
Significant findings have been disclosed in the field to elucidate the molecular bases of synucleinopathies, specifically PD. Still, understanding the complex triangle formed between the structure, function and toxicity of aSyn remains crucial for the development of novel therapeutic strategies (Figure 3) (Villar-Piqué et al., 2015).
Figure 3. Relation between aSyn structure, function and toxicity. As shown, the six pathological mutations
of aSyn identified so far are located at the N-terminal. aSyn is characterized by a conformational plasticity, being classified as an “intrinsically disordered protein”. However, the progression of protein oligomerization occurs under pathological conditions where the final outcome is the formation of aSyn aggregates with beta-sheet structures. The aSyn structure determines it fate as a functional (blue) or dysfunctional (grey) protein. aSyn has been implicated in essential cellular processes such as the vesicular trafficking in the synapse. The molecular mechanism of aSyn transformation and the relation between different structural species and their functional and pathogenic roles remain unknown. Still, dysfunction in proteasome, lysosome, synapse, microtubules, ER-to-Golgi trafficking, mitochondria and membranes were described as expected from aSyn toxic species, between other damaging consequences on several organelles and pathways (from Villar-Piqué et al., 2015).
9
A crucial process in cell homeostasis is the degradation of the differential aSyn structures (reviewed in Lopes da Fonseca et al., 2015). Monomeric aSyn can be degraded by both UPS and the ALP (Xilouri et al., 2013; Vilchez et al., 2014). A number of studies thereafter support that soluble aSyn can be a target for the 26S proteasome (Bennett et al., 1999; Tofaris et al., 2001). In contrast, aSyn oligomeric forms and aggregates cannot be subjected to proteasomal degradation and are targeted to the ALP (Lee et al., 2004; Ebrahimi-Fakhari et al., 2011).
In secretory function, aSyn is able to inhibit the ER- Golgi transport of coat protein complex II (COPII) vesicles, thereby disrupting normal transit and processing of proteins within the secretory pathway (Cooper et al., 2006; Thayanidhi et al., 2010). Several studies have found that aSyn inhibited SNARE and RAB function leading to declines in ER- Golgi transit of COPII vesicle while overexpression of several secretory accessory proteins reduced aSyn dependent toxicities in cellular PD models (Cooper et al., 2006; Gitler et al., 2008; Thayanidhi et al., 2010; Sancenon et al., 2012). A hypothetical mechanism was proposed where a primary defect in ER- to- Golgi transport could selectively cause dopaminergic neuronal death (Cooper et al., 2006; Gitler et al., 2008). The inadequate delivery of proteins such as the vesicular monoamine transporter to synaptic sites could allow dopamine and its by-products to accumulate in the synaptic cytoplasm, causing oxidative stress and thus, the inability to properly compartmentalize dopamine metabolism could selectively kill dopaminergic neurons (Thayanidhi et al., 2010).
10
1.4. Posttranslational modifications in alpha-synuclein
Several posttranslational modifications (PTMs) have been described to exist in aSyn. These include ubiquitination (Shimura et al., 2001), phosphorylation at serine 129 (S129) (Fujiwara et al., 2002), C-terminal truncation (Li et al., 2005), N–terminal acetylation (Bartels et al., 2014), nitration on tyrosine residues (Giasson et al., 2000), glycosylation and SUMO modification (Dorval and Fraser, 2006).
Homeostatic control of phosphorylation is critical to maintain physiological functions in cells, with phosphorylation networks being necessary for a wide range of cellular signaling processes (Olsen et al., 2006). Alterations in the equilibrium of phosphorylation can lead to significant cellular dysfunction and ultimately cell death (Braithwaite et al., 2012). In the particular case of neurodegenerative and protein misfolding diseases, phosphorylation has been shown to be involved in both protein aggregation and toxicity. Despite the extensive research, there is still no consensus on the beneficial or harmful effect of phosphorylation on aSyn-mediated toxicity and aggregation because the results in different model systems are controversial (reviewed in Tenreiro et al., 2014; Oueslati et al., 2016). More recently, Buck and colleagues proposed that phosphorylation of aSyn at the S129 residue per si is not sufficient to trigger neurodegeneration (Buck et al., 2015).
aSyn can be putatively phosphorylated in three other serine, four tyrosine and ten threonine residues (reviewed in Tenreiro et al., 2014). In synucleinophathies, increased levels of phosphorylated serine 87 (S87) were also reported (Paleologou et al., 2010). Moreover, phosphorylation on tyrosine 39 (Y39) and 125 (Y125) was described as occurred in human brains, but no correlation was established between the levels of phosphorylation in these residues and the pathological condition (Chen et al., 2009; Mahul-Mellier et al., 2014). The majority of focus has been on the role of phosphorylation at S129. While only 4% of the soluble, monomeric aSyn appears phosphorylated under physiological conditions in vivo, approximately 90% is phosphorylated in LB lesions of patients with PD (Fujiwara et al., 2002; Anderson et al., 2006), enhances aSyn aggregation in vitro (Fujiwara et al., 2002), and leads to neurodegeneration (Chen and Feany, 2005). However, other studies did not observe effects of aSyn phosphorylation at S129 on aSyn-mediated toxicity and aggregation (Azeredo da Silveira et al., 2009; McFarland et al., 2009). Studies in Drosophila melanogaster (Chen and Feany, 2005) and transgenic mouse models (Freichel et al., 2007) revealed pathogenic roles of aSyn phosphorylation on S129. Studies in rats (Gorbatyuk et al., 2008) and Caenorhabditis elegans (Kuwahara et al., 2012) showed an opposite protective effect of S129 phosphorylation against neuronal dysfunction.
11
Addressing this impaired homeostatic control of phosphorylation in PD has been the focus of great discovery efforts, primarily on kinases, while the relevant phosphatases are only recently being explored (Braithwaite et al., 2012).
1.4.1. Regulation of alpha-synuclein phosphorylation: kinases and
phosphatases
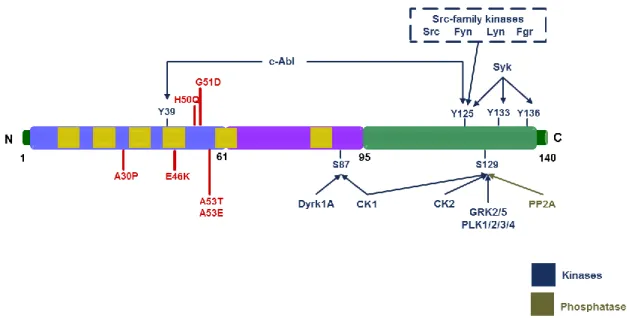
The relationship between aSyn phosphorylation and the PD progression has been further elucidated with the discovery of kinases and phosphatases that phosphorylate or dephosphorylate aSyn, respectively (reviewed in Tenreiro et al., 2014) (Figure 4).
Figure 4. Illustration of the various residues that can be phosphorylated in aSyn. The three regions in which
the protein is typically divided are reported. In blue, the amphipathic N-terminal of the protein with the six mutations associated with familial PD (in red); the hydrophobic central region that contains the non-amyloid-beta component (NAC) domain is represented in purple and, in last, the acidic C-terminal is represented in green. The protein contains imperfect KTKEGV repeats, which are represented in the figure as yellow rectangles. The kinases or phosphatases described as being able to phosphorylate or dephosphorylate, respectively, each of the indicated residues are also indicated (S, serine; Y, tyrosine) (adapted from Tenreiro
12
Several tyrosine kinases phosphorylate aSyn, including c-Abl (Mahul-Mellier et al., 2014), Syk (Negro et al., 2002) and the Src-family kinases - Src (Ellis et al., 2001), Fyn (Nakamura et al., 2001), Lyn (Negro et al., 2002) and c-Frg (Negro et al., 2002). The Dyrk1A and CK1 were found to phosphorylate aSyn at S87 (Okochi et al., 2000; Waxman and Giasson, 2008). S129 can be phosphorylated by casein kinases (CK1, CK2) (Waxman and Giasson, 2008; Zabrocki et al., 2008), members of the G protein-coupled receptor kinases (GRK1, GRK2, GRK3, GRK4) (Pronin et al., 2000; Arawaka et al., 2006; Sakamoto et al., 2009), LRRK2 (Qing et al., 2009) and the Polo-like kinases (PLKs) (Inglis et al., 2009; Mbefo et al., 2010; Oueslati et al., 2013) (Figure 4).
Phosphorylation of aSyn by GRK5 plays a crucial role in the pathogenesis of PD (Arawaka et al., 2006). In particular, PLK2 and PLK3 were found to be the most active kinases within the PLK family in vitro by phosphorylating aSyn selectively at S129, being PLK2 the most efficient PLK (Inglis et al., 2009; Mbefo et al., 2010). Overexpression of the PLK2 kinase increases aSyn phosphorylation and mediates its selective clearance through autophagic degradation (Oueslati et al., 2013). Moreover, Bergeron and colleagues found a 25–50% reduction of pS129 levels in various forebrain and midbrain regions of PLK2 knockout mice, indicating that PLK2 was one of the main endogenous kinases phosphorylating aSyn in vivo (Bergeron et al. 2014).
Several mammalian kinases possess yeast orthologues that also phosphorylate aSyn on S129 site. Therefore, the molecular impact of phosphorylation in this residue was further elaborated by heterologous expression of human kinases and co-expression with human aSyn in yeast cells. The overexpression of yeast PLK2 ortholog, Cdc5, rescues aSyn toxicity through the pS129 aSyn (Gitler et al., 2009; Wang et al., 2012). It was shown that yeast CK1 and CK2 casein kinases increases pS129 aSyn and overexpression of yeast Yck3, a vacuolar localized CK1 kinase, reduces the aSyn toxicity (Zabrocki et al., 2008). It was also revealed that the genetic background of yeast cells influence the phosphorylation effect observed in cells (Sancenon et al., 2012). Recent studies in yeast revealed that expression of S129A or S129G variants, which are phosphorylation deficient mutants, promoted the toxicity and aSyn inclusion formation (Tenreiro et al., 2014a).
Protein phosphatase 2A (PP2A) is the major serine/threonine phosphatase in the brain and can act as a master regulator of phosphorylation status for a number of important neuronal proteins such as tau (Liu et al., 2005). Also, it is a major regulator of cellular growth, death and survival (Kong et al., 2004; Strack et al., 2004). In PD, there have been reports of PP2A activation
13
being both beneficial (Lee et al., 2011) and detrimental (Lou et al., 2010; Peng et al., 2005) (Figure 4). These discrepant reports may be due to the different compositions of the PP2A holoenzyme studied (see description in section 1.6). Beyond the effect on neurodegeneration, aSyn phosphorylation can cause functional consequences in dopamine neurons. In particular, aSyn has previously been suggested to play a role in the regulation of dopamine synthesis by inhibiting tyrosine hydroxylase (TH) enzyme resulting in a decrease of dopamine levels (Perez et al., 2002). Overexpression of aSyn was shown to reduce serine 40 phosphorylation of TH, which is caused by enhanced activity of PP2A, and in turn leads to reduced TH enzyme activity, both in vitro and in vivo (Peng et al., 2005; Lou et al., 2010).
Downstream of nigral dopaminergic neuron degeneration, a range of striatal proteins involved in synaptic plasticity are hyperphosphorylated (Brown et al., 2005), appearing to be associated with decreased activity of protein phosphatase 1 (PP1) (Brown et al., 2008). Therefore, enhancing PP1 activity may be beneficial in improving synaptic function and motor symptoms in PD (Braithwaite et al., 2012).
Between the kinases and phosphatases proteins, the advantages of manipulating serine/threonine phosphatases in PD include that there is no significant redundancy as there are fewer phosphatases when compared to kinases, and also the fact that phosphatases can be pharmacologically activated, unlike kinases that can only be inhibited (Braithwaite et al., 2012).
14
1.5. Saccharomyces cerevisiae as a model system for studying the
molecular basis of Parkinson’s disease
The unicellular eukaryote S. cerevisiae has become an established model system to analyze the biology of human proteins, including aSyn (Tenreiro and Outeiro, 2010; Tenreiro et al., 2013). Several molecular aspects of PD have been exhibited in yeast, even though yeast lacks orthologs for some PD-associated genes. A yeast homolog of the SNCA gene encoding human aSyn is not present in the yeast genome. However, several relevant aspects of the PD phenotype are recapitulated when human aSyn is heterologous expressed in yeast cells (Outeiro and Lindquist, 2003). These findings confirm that yeast models expressing neurotoxic proteins are very helpful in elucidating novel paradigms of pathobiology in neurodegenerative disorders.
Among the different pathways involved in aSyn toxicity in yeast, induction of apoptosis, lipid droplet accumulation (Outeiro and Lindquist 2003), mitochondrial dysfunction (Büttner et al., 2008; Su et al., 2010), proteasome impairment (Outeiro and Lindquist, 2003; Chen et al., 2005; Sharma et al. 2006), oxidative stress (Sharma et al., 2006; Witt and Flower, 2006), autophagy/ mitophagy dysfunction (Petroi et al., 2012; Sampaio-Marques et al., 2012), vesicle trafficking defects (Outeiro and Lindquist 2003; Soper et al., 2008), and ER- to- Golgi trafficking impairment (Cooper et al. 2006), seem to be the most remarkable (Figure 5).
When expressed in yeast, aSyn binds to vesicles of the secretory pathway, leading to its localization in the plasma membrane (Outeiro and Lindquist, 2003; Sharma et al., 2006; Zabrocki et al., 2008). This depends on aSyn phosphorylation (Basso et al., 2013; Tenreiro et al., 2014a). Upon high expression levels, aSyn forms cellular inclusions in a nucleation-dependent manner, which starts at the plasma membrane and eventually leads to cytoplasmic inclusions (Outeiro and Lindquist, 2003). These inclusions co-localize with markers of different vesicles, including Ypt1 (ER-to-Golgi), Ypt31 (late Golgi), Sec4 (secretory vesicles to plasma membrane), Ypt6 (Golgi), Vps21 and Ypt52 (early-to-late endosome) and Ypt7 (late endosome-to-vacuole) (Gitler et al., 2008). aSyn expression also affects vesicular trafficking, including ER-to-Golgi transport, endocytosis, vesicular recycling back to the plasma membrane, and vacuolar fusion (Cooper et al., 2006; Gitler et al., 2008; Zabrocki et al., 2008; Basso et al., 2013).
The relevance of the different pathways in modulating aSyn-triggered cytotoxicity might depend on aSyn itself, e.g., the expression levels, the PTMs, the cellular localizations, folding or distinct aggregation conditions (Braun et al., 2015).
15
Overexpression screens were also performed to identify modifiers of aSyn toxicity (both enhancers and suppressors). It is the case of Ypt1, a protein involved in vesicular trafficking, identified as suppressors of aSyn toxicity and, ultimately, resulted in the finding that the mammalian Ypt1 protein homolog, Rab1, was also a suppressor of dopaminergic cell loss in animal models of PD (Cooper et al. 2006).
Figure 5. Model for aSyn-triggered cytotoxicity in S. cerevisiae. aSyn is a protein that can be bound to the
plasma or vesicle membranes and upon high expression levels or upon mutation forms smaller and larger inclusions, which can be ubiquitylated (1). aSyn can be degraded through the UPS (2), autophagy (3), and possibly via the multivesicular bodies pathway (4). The aSyn inclusions causes reactive oxygen species, mitochondrial dysfunction and mitochondria-dependent cell death (5), as well as ER stress and the UPR (6). These cytotoxic effects can be in part clarified by inhibition of the proteasome (7), the ER-associated degradation (ERAD) pathway (8), or vesicular trafficking (9) through aSyn inclusion formation. The impairment in vesicular trafficking includes (but is not limited to) ER-to-Golgi transport, which can be restored by YPTI gene expression (10) (from Braun et al., 2015).
16
Furthermore, it was recently observed that aSyn is part of a diverse and highly conserved interaction network including proteins with very diverse functions (namely kinases, phosphatases, deubiquitinating enzymes and metal transporters). This network was identified in genetic screens in yeast and was further validated in rat primary neuronal cultures and in nematode models (Gitler et al., 2009; Yeger- Lotem et al., 2009). This conserved network is evident in the observation that in yeast, aSyn is subjected to several posttranslational modifications observed in mammalian cells and in PD patient’s brains, namely phosphorylation at S129 and acetylation of the N-terminal (Anderson et al., 2006; Zabrocki et al., 2008; Chen et al., 2009).
17
1.6. A yeast-based screen to identify genes involved in alpha-synuclein
pathobiology
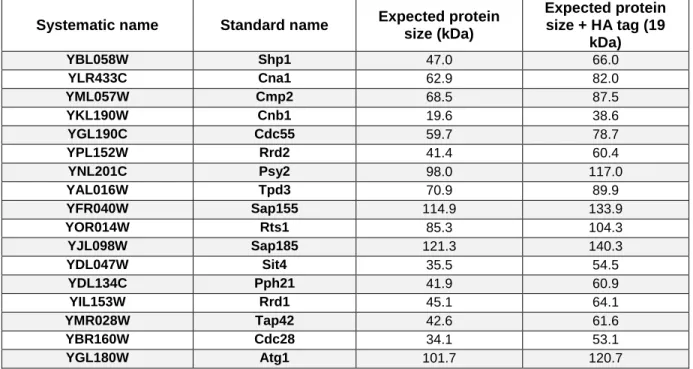
In this study, genetic approaches were employed to identify relevant kinases and phosphatases in PD using the yeast model. Namely, 17 proteins were selected according to their functional conservation with human orthologues and expression in the nervous system (Figure 6). An overview of the kinases and phosphatases included in this functional screening is following described.
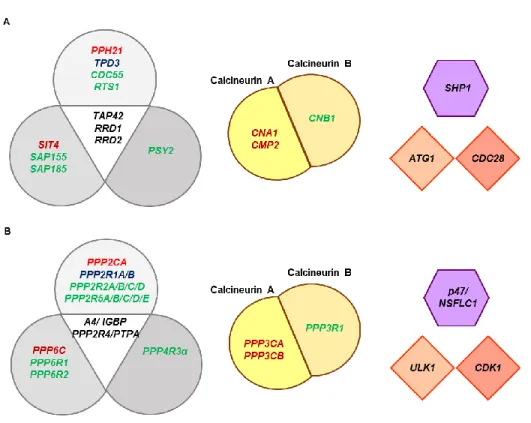
Figure 6. Schematic representation of genes encoding the proteins involved in the screening (A) in S.
cerevisiae, with homologs genes in (B) Homo sapiens (H. sapiens). Boxes with similar shapes and colours
intent to facilitate the schematic representation of the homologs between species. (Left) The three closely related groups of serine/threonine protein phosphatases (PP). PP2A, PP4 and PP6 are conserved throughout eukaryotes. Genes coding catalytic subunits are shown in red, canonical scaffolding subunits in blue and regulatory subunits are in green; putative common interactors for the subunits are shown in black in the central triangle. (Middle) Calcineurin consists in a catalytic subunit calcineurin A and a tightly associated calcium-binding regulatory subunit, calcineurin B. (Right upper) Shp1, p47/NSFLC1 H. sapiens homolog, promotes cell cycle progression by positive regulation of PP1 (Lower right) Atg1 and Cdc28 are conserved proteins that functions as serine/threonine kinases.
18
Autophagy is an intracellular degradation system conserved among eukaryotes. Autophagosome formation, a landmark event in autophagy, is accomplished by the concerted actions of autophagy-related (Atg) proteins, including Atg1 kinase in yeast and its counterpart in higher eukaryotes, serine/threonine-protein kinase (ULK1) (Mizushima, 2010; Noda and Fujioka, 2015). In S. cerevisiae, the cyclin-dependent kinase (CDK) Cdc28 (also known as Cdk1) is necessary and sufficient for cell cycle regulation by phosphorylating a large number of substrates to coordinate cell cycle events (Enserink et al., 2010). It was recently shown that Cdc28 also boosts the expression of a specific subset of housekeeping genes that encode proteins involved in energy supply, translation, cell wall integrity and chromatin architecture (Chymkowitch et al., 2012).
Protein phosphatases fall into major families established on different structures, specificity and catalytic mechanisms. Eukaryotic protein phosphatases can be usually divided into four distinct gene families each with different active site signatures: (1) PPP (serine/ threonine- specific phosphoprotein phosphatases); (2) PPM/ PP2C (magnesium- dependent protein phosphatases); (3) aspartate- based protein phosphatases; and (4) phosphotyrosine phosphatases (PTP). The PPP family are amongst the most highly conserved proteins across eukaryotic species, and can be further divided into subgroups: PP1, PP2/ PP2A, PP3/ PP2B (reviewed in Lillo et al., 2014).
PP2A refers to a ubiquitous, highly conserved family of serine/threonine phosphatases that play a role in the regulation of various cellular functions (reviewed in Virshup and Shenolikar, 2009). This protein is a heterotrimeric complex including a scaffolding A subunit (PPP2R1A or PPP2R1B isoforms), the catalytic C subunit (PPP2CA or PPP2CB isoforms) and a regulatory B subunit, which belongs to one of four distinct families: B (PPP2R2), B’(PPP2R5), B” (PPP2R3), or B”’/striatins (PPP2R4). PP2A regulatory subunits are expressed in a cell and tissue-specific manner, which is responsible for the isoform-specific distribution of PP2A in the brain. In S. cerevisiae, PP2A is represented by a single A subunit, Tpd3, two B regulatory subunits, Cdc55 and Rts1, and two catalytic subunits, Pph21 and Pph22. The assembly and activity of PP2A is regulated by multiple interacting proteins designated as PP2A intracellular inhibitors and PP2A activators (PTPA). PTPA activates PP2A (and probably PP4 and PP6) by acting as a chaperone and allowing the activation of the phosphoserine and threonine phosphatase activity (Longin et al., 2004). Two PTPA orthologues, Rrd1 and Rrd2, exist in S. cerevisiae.
S. cerevisiae has one PP4 catalytic subunit named Pph3, and is found complexed with the specific regulators Ybl1046w and Psy2. Functional homologs (PP4R2 and PP4R3) of these two regulators are present in mammals. Also, in mammals PP4 was shown to be essential for
19
development in a specific group of white blood cells, and depletion of PP4 was lethal emphasizing that PP4 could not be replaced by other protein phosphatases (Shui et al., 2007). The complex, PP4-PP4R2-PP4R3 (Pph3-YBL1046W-Psy2), is conserved between humans and S. cerevisiae, and are involved in DNA damage responses in both organisms (Wu et al., 2004; Gingras et al., 2005; Chowdhury et al., 2008; Lee et al., 2012).
In humans and S. cerevisiae only one gene is encoding a PP6 catalytic subunit. The S. cerevisiae PP6, called Sit4, is identified as part of a dimer with regulatory SIT4-associated proteins (SAPs) (Stefansson and Brautigan, 2006). The human PP6 is also part of a dimer, and the regulatory subunits PP6R1, PP6R2 or PP6R3 are orthologues of yeast Sap155, Sap185, Sap190, respectively. Both yeast and human PP6 are necessary for progression in the cell cycle and were found to be essential in initiating repair of double strand DNA breaks (Douglas et al., 1997; Hosing et al., 2012). Also, PP6 is important for ER-to-Golgi traffic through regulation of COPII dephosphorylation (Bhandari et al., 2013).
Human immunoglobulin-α-binding protein 1 (α4/ IGBP) and S. cerevisiae Tap42 form stable dimers with PP2A, PP4 and PP6 catalytic subunits (Zabrocki et al., 2002; reviewed in Sents et al., 2013). Both IGBP and Tap42 are essential proteins involved in the TOR signaling pathway (Di Como and Arndt, 1996).
PP1 functions in several aspects of eukaryotic cell physiology and consists in the catalytic subunit and various regulatory subunits that confer substrate specificity, regulation of activity and subcellular localization (Ceulemans and Bollen, 2004). In the budding yeast, the essential gene GLC7 encodes the catalytic subunit of PP1, which, in turn, regulates glycogen metabolism, actin cytoskeleton, gene expression, translation, mitosis and meiosis (Cannon, 2010). Ubx1 (also known as Shp1) was originally isolated as a mutant protein that suppresses the toxic effect of Glc7 overexpression (Zhang et al., 1995). Subsequent studies further support the hypothesis that Shp1 positively regulates Glc7 to counteract Ipl1/Aurora B activity for mitotic progression (Böhm and Buchberger, 2013). Cdc28- Shp1 acts by, at least in part, promoting nuclear localization of PP1 (Cheng and Chen, 2010; Cheng et al., 2015). In addition, Shp1 serves as a cofactor for Cdc48 in ubiquitin- dependent protein degradation (Hartmann- Petersen et al., 2004), and it interacts with the ubiquitin- fold autophagy protein Atg8 during autophagosome biogenesis (Krick et al., 2010). The mammalian homolog p47 is involved in post- mitotic Golgi membrane reassembly (Rabouille et al., 1998).
20
Calcineurin is a eukaryotic calcium and calmodulin-dependent serine/threonine protein phosphatase. It is a heterodimeric protein consisting of a catalytic subunit calcineurin A, and a tightly associated, calcium-binding subunit, calcineurin B. Calcineurin is widely distributed in mammalian tissues, with the highest levels found in the brain. As a serine/threonine protein phosphatase, calcineurin participates in a number of cellular processes and calcium-dependent signal transduction pathways, including cell cycle, brain ischemia and programmed cell death (Morioka et al., 1999; Rusnak et al., 2000). There are two genes for the catalytic subunit of calcineurin in S. cerevisiae (CNA1 and CMP2) and only one gene for the B subunit (CNB1). Between the physiological roles of calcineurin in S. cerevisiae are cation resistance, calcium homeostasis and calcium-mediated cell cycle arrest (Rusnak et al., 2000).
21
1.7. SLY41, a gene that restores defects in vesicular trafficking
The potential of specific suppressors and enhancer genes to rescue or exacerbate trafficking defects, as well as to rescue or exacerbate aSyn toxicity, support that ER-to-Golgi vesicular transport is susceptible to aSyn accumulation. Indeed, it has been reported that expression of aSyn causes deficits in ER- Golgi trafficking (Cooper et al., 2006) and that yeast cells expressing aSyn are sensitive to disruption of vesicular trafficking (Outeiro and Lindquist, 2003; Zabrocki et al., 2008). Additionally, unfolded protein response to accumulation of misfolded proteins within the ER is affected by aSyn expression. aSyn is able to inhibit the processing of ATF6 directly through physical interactions and indirectly through restricted incorporation into COPII vesicles. Impaired ATF6 signaling was accompanied by decreased ERAD function and increased pro-apoptotic signaling (Credle et al., 2015).
Several studies have found that overexpression of several secretory accessory proteins, such as Ypt1, reduced aSyn dependent toxicities in cellular PD models (Cooper et al., 2006; Gitler et al., 2008; Thayanidhi et al., 2010; Sancenon et al., 2012), providing an impulse for the discovery of novel genes able to modify aSyn-induced toxicity.
SLY41 gene encodes one of the proteins discovered as suppressors of YPT1 deletion and capable to restore ER-to-Golgi transport defects in YPT1- depleted cells (Ossig et al., 1991). This is not an essential gene and yeast cells overexpressing SLY41 where viable even after disruption of the YPT1 gene (Dascher et al., 1991). Sly41 is a membrane protein that shares sequence homology to the SLC35 family of solute carriers (Dascher et al., 1991), but the Sly41 subgroup (SLC35E) comprises orphan transporters with no known physiological substrates (Hadley et al., 2014). Sly41 bears 24% identity to its human homolog, SLC35E1, which is also of unknown function (He et al., 2009).
COPII vesicle assembled at the ER transports nascent secretory proteins forward to the Golgi complex. A recent report identified Sly41 as a COPII vesicle protein and its overexpression to the early secretory pathway elevated cytosolic calcium levels to suppress vesicle-tethering YPT1 mutant cells. Also, indication that calcium positively regulates the SNARE-dependent fusion stage of ER–Golgi transport was shown (Margulis et al., 2016). For many years, it has been known that an increase in cytosolic calcium triggers the fusion of secretory granules and synaptic vesicles with the plasma membrane (Hay et al., 2007).
23
25
To modulate the pathogenic activities of aSyn one possibility is to identify potential kinases and phosphatases involved in regulation of aSyn phosphorylation at S129, clarify its effect in cell toxicity and determine if it influences inclusion formation. With this information it would then be possible to evaluate the potential of targeting kinases/ phosphatases as therapeutic strategies to treat PD and related synucleinopathies. Another possibility is to identify additional genetic modifiers of aSyn toxicity, particularly those that could restore aSyn-induced transport block.
In this context, the aims of this study were:
1) To identify novel kinases/ phosphatases of aSyn and clarify their role in aSyn phosphorylation, inclusion formation and toxicity.
2) To elucidate the role of Sly41 in aSyn inclusion formation and toxicity and the possible interaction between the proteins.
27