Characterization of in vitro models for evaluation of
molecular mechanisms involved in the action of
Acetyl-L-Carnitine
Dissertação para obtenção do grau de Mestre
Mestrado em Biotecnologia para as Ciências da Saúde
JOANA FILIPA RAMOS LAPA
Trabalho de tese realizado sob a orientação da
Professora Doutora Maria Teresa Burnay Summavielle
Characterization of in vitro models for evaluation of
molecular mechanisms involved in the action of
Acetyl-L-Carnitine
Dissertação para obtenção do grau de Mestre
Mestrado em Biotecnologia para as Ciências da Saúde
JOANA FILIPA RAMOS LAPA
Trabalho de tese realizado sob a orientação da
Professora Doutora Maria Teresa Burnay Summavielle
Composição do Júri:
___________________________________________
___________________________________________
___________________________________________
Este trabalho foi realizado no IBMC sob a supervisão da Professora Doutora Maria Teresa Burnay Summavielle.
V
ACKNOWLEDGEMENTS
Chegado ao fim desta caminhada, gostaria de expressar o meu reconhecimento pelo apoio e disponibilidade concedidas por diversas pessoas que contribuíram para a realização deste trabalho. A elas, os meus mais sinceros agradecimentos:
À Professora Doutora Teresa Summavielle quero agradecer desde já ter aceite ser minha orientadora e por toda a ajuda prestada durante a escrita desta dissertação. Um obrigado por tudo aquilo que me ensinou, pois irá ajudar-me durante todo o meu percurso de vida.
À Sílvia Fernandes, por ter sido uma ajuda imprescindível durante o meu percurso no laboratório, por toda a paciência e conselhos, tanto durante o trabalho prático como durante a escrita da dissertação. Pela sua boa disposição, doces e travessuras!
À Joana Bravo pelos conselhos, pelos desabafos e pela amizade. À Andrea Lobo pela ajuda prestada, por todas as dúvidas esclarecidas.
Ao Manuel João pelos conselhos, pelas tardes na biblioteca e por todos os momentos de alegria! Por me ter feito conhecer a Maria!
À Maria por toda a dedicação e ajuda prestada. Por me ter motivado, por todas as dicas e “truques”! A sua ajuda foi fundamental para terminar esta etapa.
Aos meus colegas e amigos de mestrado, licenciatura e do Porto!
À Isabel Duarte por fazer da sua casa um segundo lar para mim, pelos conselhos e pelos abanões dados ao longo do meu percurso académico.
Ao Pedro Lima, pela amizade e amor. Por se dedicar e ter sido fundamental para manter a minha cabeça fria, ajudando-me a ver tudo numa perspetiva diferente. Por todos os desabafos, por toda a paciência, por toda a prontidão!
À minha família, por todo o apoio e confiança. Pelo conforto, pelos mimos, por tudo! Aos meus pais, mana e Gui, os melhores do Mundo. Agradeço-lhes por tudo.
Ao Instituto de Biologia Molecular e Celular do Porto o meu muito obrigado pelas fantásticas condições de trabalho e pela oportunidade que me foi dada.
À Universidade de Trás-os-Montes e Alto Douro por me acompanhar nesta caminhada de cinco anos, por tudo o que vivi e aprendi.
VII
CHARACTERIZATION OF IN VITRO MODELS FOR EVALUATION OF MOLECULAR MECHANISMS
INVOLVED IN THE ACTION OF ACETYL-L-CARNITINE
ABSTRACT
The Acetyl-L-carnitine (ALC) is thought to be neuroprotective, mainly by preventing mitochondrial dysfunction both in the central and peripheral nervous system. ALC is well known to affect the brain metabolism and bioenergetics, and therefore is expectable that some of the pathways involved in its action may be related to glucose uptake and metabolisms. However, the mechanisms underlying the action of ALC are still unclear.
Cellular models are useful tools to study cellular signaling pathways. Due to the critical role of astrocytes in brain metabolism, it is essential to study the function of ALC also in this cell type. The present work aimed to characterize an astrocyte cell line, the C8-D1A, under the action of ALC. To evaluate the protective capacity of ALC we used a well-known neurotoxic drug, i.e. methamphetamine (METH). The research of the effects of different doses of ALC and METH in primary hippocampal neurons, PC12 cells and in a bEnd.3 cell line were as well a target of this study, since in future researches it will be essential to use co-cultures of astrocytes with neurons or endothelial cells to better understand the role of ALC and the signaling pathways used. Firstly, the action of five different METH doses (3, 1, 0.5, 0.1 and 0.01 mM) was characterized in the different cell types. Secondly, the suitable METH doses selected were used to characterize the putative protective effects of ALC 0.1 and 1 mM. Several parameters were evaluated, including cell death (LDH release assay), morphology (Phaloidin stain) and mitochondrial activity (MTT reduction assay). In the C8-D1A cells, were also evaluated the levels of ATP production, the rate of apoptotic nuclei (Hoechst 33342 stain) and the expression of selected genes (Igf1 e Rnd2) (RT-PCR). Evaluations were conducted into different time points, 24 h and 72 h.
Results showed that C8-D1A cells were resistant to doses ranging up to 1 mM. The primary hippocampal neurons exposed to METH 1 and 0.5 mM after 24 h presented an increase of neuronal arborization and actin aggregation comparing to controls, whereas cells exposed to METH 3 and 1 mM after 72 h suffered loss of nerve terminals and cellular death. The bEnd.3 cells treated for 72 h with METH 3 and 1 mM lost their typical conformation.
When accessing the effects of an ALC pre-treatment on mitochondrial activity, the results showed that C8-D1A cells under the combined action of METH 1 mM and ALC
VIII
presented an increased number of apoptotic nuclei, although these values did not surpass 3%. Interestingly, this METH dose led to reduced ATP synthesis without affecting mitochondrial viability, and the combined action of METH and ALC did not prevent this effect. In the bEnd.3 cells no effects of ALC could be observed in any condition. Preliminary results seem to indicate that both METH and ALC interferes with the cellular metabolism of the C8-D1A line by affecting the transcript levels of Igf1. The present results allowed selecting a range of METH doses suitable to investigate the pathways involved in the action of ALC, and permitted also to preview how neuronal and endothelial cultures would react to the selected doses in a co-culture with the astrocytes. Future studies will be important in understanding the real potential of ALC as a therapeutic agent.
Key words: acetyl-L-carnitine; cellular death; methamphetamine; mitochondrial activity; C8-D1A cell
IX
CARACTERIZAÇÃO DE MODELOS IN VITRO PARA AVALIAÇÃO DOS MECANISMOS MOLECULARES
ENVOLVIDOS NA AÇÃO DA ACETIL-L-CARNITINA
RESUMO
Pensa-se que a acetil-L-carnitina (ALC) tem ação neuroprotetora, principalmente por prevenir a disfunção mitocondrial no sistema nervoso central e periférico. A ALC é bem conhecida por afetar o metabolismo e a bioenergética do cérebro, e por isso é expetável que alguns dos mecanismos envolvidos na sua ação possam estar relacionados com o metabolismo e absorção de glucose. No entanto, os mecanismos subjacentes à ação da ALC não são ainda bem conhecidos.
Os modelos celulares são ferramentas úteis para estudar vias de sinalização celular. Devido ao papel fundamental dos astrócitos no metabolismo cerebral, é essencial estudar-se a função da ALC também neste tipo celular. Pretende-se com este trabalho caracterizar uma linha celular de astrócitos, a C8-D1A, sob a ação da ALC. Para avaliar a capacidade neuroprotetora da ALC usámos uma droga neurotóxica bem conhecida, a metanfetamina (METH, do inglês “Methamphetamine”). Os efeitos das diferentes doses de ALC e METH nos neurónios primários do hipocampo, linhas celulares PC12 e bEnd.3 foram também alvo deste estudo, uma vez que, em estudos futuros, será essencial a utilização co-culturas de astrócitos e neurônios ou células endoteliais para melhor se entender o papel da ALC e as vias de sinalização utilizadas.Começamos por caracterizar a ação de cinco doses diferentes METH (3, 1, 0,5, 0,1 e 0,01 mM) nos diferentes tipos celulares. Em segundo lugar, as doses apropriadas de METH selecionadas foram utilizadas para caracterizar o suposto efeito protetor da ALC 0,1 e 1 mM. Vários parâmetros foram avaliados, incluindo morte celular (ensaio de LDH), morfologia (marcação com faloidina) e atividade mitocondrial (ensaio de MTT). Nas células C8-D1A, foram avaliados também os níveis de produção de ATP, a taxa morte apoptótica (marcação com Hoechst 33342) e a expressão de genes selecionados (Igf1 e Rnd2) (RT-PCR). As avaliações foram realizadas em diferentes pontos do tempo, 24 h e 72 h.
Os resultados mostraram que as células C8-D1A foram resistentes a doses variadas até 1 mM. Os neurónios primários do hipocampo expostos a METH 1 e 0,5 mM após 24 h apresentaram um aumento na arborização neuronal e na agregação de actina face ao controlo, enquanto as células expostas a METH 3 e 1 mM após 72 h apresentaram perda de terminais
X
nervosos e morte celular. A linha celular bEnd.3 tratada durante 72 h com METH 3 e 1 mM perderam a sua conformação típica.
Ao estudar os efeitos do pré-tratamento com ALC na atividade mitocondrial, os resultados mostraram que as células C8-D1A sob a ação combinada da ALC e METH 1 mM sofreram um aumento do número de núcleos apoptóticos, embora estes valores não ultrapassassem os 3%. Curiosamente, esta dose de METH reduziu a síntese de ATP sem afetar a sua viabilidade mitocondrial, e a ação combinada de ALC e METH não impediu este efeito. Na linha celular bEnd.3, nenhum efeito da ALC foi observado em qualquer uma das condições. Os resultados preliminares parecem indicar que a METH e a ALC interferem com o metabolismo celular da linha C8-D1A, afetando os níveis de transcrição de Igf1. Os resultados deste estudo permitiram fazer a seleção de um intervalo de doses de METH adequado para investigar as vias envolvidas na ação da ALC, e permitiu também pré-visualizar a forma como as culturas neuronais e endoteliais reagiriam às doses selecionadas, em co-cultura com os astrócitos. Estudos futuros serão importantes para a compreensão do real potencial do ALC como agente terapêutico.
Palavras-chave: acetil-L-carnitina; morte celular; metanfetamina; viabilidade mitocondrial; linha
XI
INDEX
ACKNOWLEDGEMENTS ... V ABSTRACT ... VII RESUMO ... IX INDEX ... XI FIGURE INDEX ... XV TABLE INDEX ... XVII ABBREVIATIONS ... XIXCHAPTER I – INTRODUCTION ... 1
1.1. The fundamental role of carnitine in the acylcarnitines biosynthesis ... 3
1.2. Acetyl-L-carnitine ... 5
1.2.1. Acetyl-L-carnitine biosynthesis ... 6
1.2.2. Acetyl-L-carnitine supplementation ... 7
1.2.3. Acetyl-L-carnitine applications ... 9
1.2.3.1. Methamphetamine effects and the protective role of acetyl-L-carnitine ... 12
1.3. Models of study ... 16
1.4. Cellular viability evaluation ... 18
CHAPTER II – OBJECTIVES ... 21
CHAPTER III – MATERIAL AND METHODS... 25
3.1. Primary hippocampal cultures ... 27
XII
3.3. Drug treatments ... 28
3.5. Cell viability assay ... 29
3.6. Immunofluorescence ... 30
3.8. Assessment of C8-D1A Igf1 and Rnd2 transcript levels by Real Time Polymerase Chain Reaction (RT-PCR) ... 31
3.9. Data analysis ... 32
CHAPTER IV – RESULTS ... 34
4.1. Cell viability and morphology under METH action ... 36
4.1.2. Effects of METH in the C8-D1A cell line ... 36
4.1.2.1. Effects of METH in cell death ... 36
4.1.2.2. Morphological effects of METH exposure ... 37
4.1.2.3. Effects of METH in cell viability ... 39
4.1.3. Effect of METH in the primary hippocampal neurons ... 40
4.1.3.1. Effects of METH in cell death ... 40
4.1.3.2. Morphologic effects of METH exposure ... 42
4.1.4. Effect of METH in the PC12 cell line ... 44
4.1.4.1. Effects of METH in cell death ... 44
4.1.4.2. Morphological effects of METH exposure ... 45
4.1.5. Effects of METH in the bEnd.3 cell line ... 47
4.1.5.1. Effects of METH in cell death ... 47
4.1.5.2. Morphological effects of METH exposure ... 48
4.2. Cell viability under ALC action after METH exposure ... 50
XIII
4.2.1.1. Effects of ALC over METH-induced apoptotic cell death ... 50
4.2.1.2. Morphology effects of ALC on C8-D1A exposed to METH ... 52
4.2.1.3. ATP production in C8-D1A cells exposed to different ALC and/or METH doses 53 4.2.2. Effects of ALC in the viability of PC12 cells exposed to METH ... 54
4.2.3. Effects of ALC in the viability of bEnd.3 cell line exposed to METH ... 56
4.3. Analysis of Igf1 and Rnd2 genes expression in C8-D1A cells ... 58
CHAPTER V – DISCUSSION ... 60
CHAPTER VI – CONCLUSIONS AND FUTURE PERSPECTIVES ... 72
XV
FIGURE INDEX
Figure 1. L-Carnitine structure. Adapted from (Jones et al., 2010). ... 3 Figure 2. Examples of acylcarnitine structures. Adapted from (Jones et al., 2010). ... 5 Figure 3. Acetyl-L-carnitine structure. Adapted from (Jones et al., 2010). ... 6 Figure 4. Acetyl-L-carnitine in overall energy metabolism. Adapted from (Jones et al., 2010).
... 7
Figure 5. Metabolic pathways of external supplementation of Acetyl-L-carnitine. Adapted
from (Rosca et al., 2009). ... 8
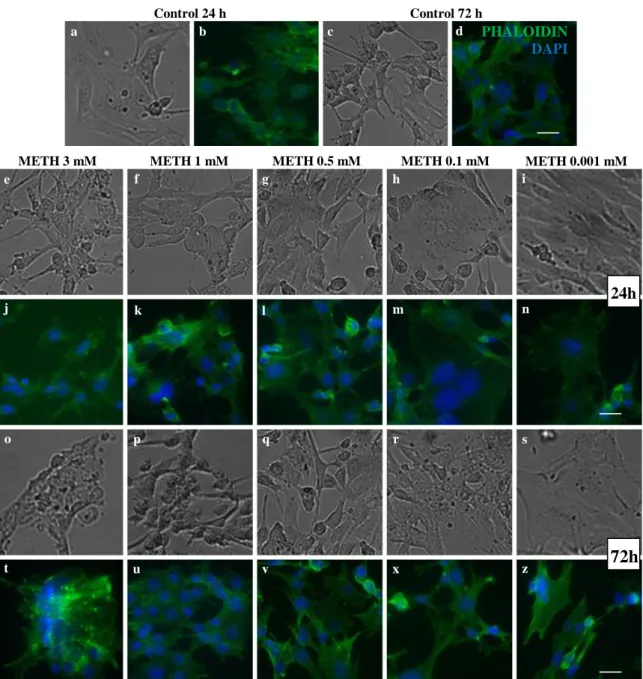
Figure 6.Percent of death in C8-D1A cells exposed to decreasing doses of METH at 24 h and
72 h. ... 37
Figure 7. Morphology of C8-D1A cells after exposure to decreasing doses of METH at 24 h
and 72 h. ... 38
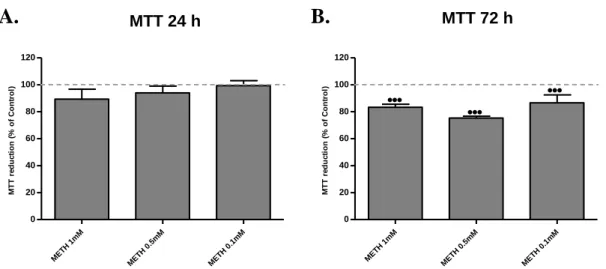
Figure 8. Percent of C8-D1A viability after being exposed to decreasing doses of METH at
24 h and 72 h. ... 39
Figure 9. Percent of death in primary hippocampal neurons exposed to decreasing doses of
METH at 24 h and 72 h.. ... 41
Figure 10. Morphology of primary hippocampal neurons (14 DIV) after exposure to
decreasing doses of METH at 24 h and 72 h.. ... 43
Figure 11. Percent of death on PC12 cells exposed to decreasing doses of METH at 24 h and
72 h. ... 44
Figure 12. Morphology of PC12 cells after exposure to decreasing doses of METH at 24 h
and 72 h.. ... 46
Figure 13.Percent of death in bEnd.3 cells exposed to decreasing doses of METH at 24 h and
XVI
Figure 14. Morphology of bEnd.3 cells after exposure to decreasing doses of METH at 24 h
and 72 h. ... 49
Figure 15. Percent of apoptotic C8-D1A cells when exposed to different doses of ALC and/or
METH at 24 h and 72 h. ... 51
Figure 16. Morphology of C8-D1A cells after treatments with ALC 0.1 and METH 1mM at
24 h and 72 h. ... 52
Figure 17. ATP production of C8-D1A cells after exposure to different doses of ALC and/or
METH at 24 h.. ... 53
Figure 18. Percent of cell viability of PC12 cells exposed to different doses of ALC and/or
METH at 24 h and 72 h. ... 55
Figure 19. Effects of different doses of ALC and/or METH on bEnd.3 cells viability at 24 h
and 72 h. ... 57
Figure 20. qPCR fold changes of Igf1 and Rnd2 in cells treated with different doses of ALC
and/or METH, at 24 h.. ... 59
XVII
TABLE INDEX
Table 1. Studies with ALC in central and peripheral nervous system. Adapted from (Virmani and Binienda, 2004). ... 10
XIX
ABBREVIATIONS
Aβ – Amyloid beta peptide
Acyl-CoA – Acyl-Coenzyme A
AD – Alzheimer’s disease
ADHD – Attention Deficit Hyperactivity Disorder
ALC – Acetyl-L-Carnitine
AMD – Age-related macular degeneration
AMPH – Amphetamine
APP – Amyloid Precursor Protein
ATP – Adenosine Triphosphate
BBB – Blood-brain barrier
BCCL – Bilateral Common Carotid Arteries
C – Carnitine
CACT – Carnitine Acylcarnitine Translocase
CAT – Carnitine acetyltransferase
CoA – Coenzyme A
CPT1 – Carnitine palmitoyltransferase 1
CPT2 – Carnitine palmitoyltransferase 2
CNS – Central Nervous System
DA – Dopamine
DAPI – 4’, 6-diamidino-2-phenylindole
DIV – Days in vitro
dsDNA – double-stranded Deoxyribonucleic Acid
EDTA – EthyleneDiamine Tetraacetic Acid
ETC – Electron transport chain
Gapdh – Glyceraldehyde-3-phosphate dehydrogenase
GLUT1 – Glucose Transporter 1
GLUT3 – Glucose Transporter 3
HBSS – Hank’s Balanced Salt Solution
Igf-1 – Insulin-like Growth Factor 1
XX LA – R-α-lipoic acid LC – L-Carnitine LDH – Lactate dehydrogenase MDMA – 3,4-methylenedioxymethamphetamine METH – Methamphetamine MTT – 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
mRNA – messenger Ribonucleic Acid
NGS – Normal Goat Serum
OCTN – Organic carnitine transporter
OCTN2 – Organic carnitine transporter 2
PBS – Phosphate Buffered Saline
PCR – Polymerase Chain Reaction
PLC – Propionyl-L-carnitine
Rnd2 – Rho family GTPase 2
ROCK – Rho-associated Kinase
ROS – Reactive Oxygen Species
Rpl29 – Ribosomal protein l29
rpm – Rotations per minute
Rps18 – Ribosomal protein s18
TCA –Tricarboxylic acid cycle
TBI – Traumatic brain injury
1
3
1. I
NTRODUCTION1.1. The fundamental role of carnitine in the acylcarnitines biosynthesis
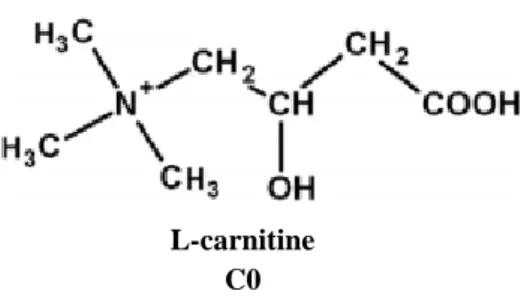
Carnitine is primarily available from food (75%), predominantly from meat and dairy with the remaining 25% coming from endogenous synthesis (Jones et al., 2010; Virmani and Binienda, 2004). The active form of carnitine is L-carnitine (Figure 1), and it is synthesized in the liver, kidneys and brain from two essential amino acids, L-lysine and L-methionine (Flanagan et al., 2010; Sharma and Black, 2009; Virmani et al., 2004). The amphiphilic structure of L-Carnitine (LC) makes it very mobile throughout the cells (Jones et al., 2010). This molecule has several functions in cells, being a vital cofactor for the transport of fatty acids into mitochondria and peroxisomes for β-oxidation, controlling the mitochondrial acyl-CoA/CoA ratio (acyl-coenzyme A/coenzyme A) and the production of ketone bodies (Jones et al., 2010; Virmani and Binienda, 2004). Due to this complex interaction with bioenergetic processes, LC plays a fundamental role in mitochondrial-related disorders.
Figure 1. L-Carnitine structure. Adapted from (Jones et al., 2010).
The LC supplementation has different effects on the mitochondrial metabolism. Although LC restores normal mitochondrial functions by keeping the equilibrium between acyl-CoA and free CoA, it also increases the fatty acids metabolism, by improving CoA recycling and increasing the mitochondrial-free CoA levels, moving the short-chain acyl groups from mitochondria to the cytosol (Sharma and Black, 2009). LC supplementation may be beneficial in epilepsy, attention deficit hyperactivity disorder (ADHD), and other disorders of the nervous system including drug abuse (Virmani and Binienda, 2004). Furthermore studies on the mechanism(s) underlying the effects of LC in cardiovascular diseases have demonstrated that the administration of this compound reduces blood pressure, attenuates the
L-carnitine C0
4
inflammatory process associated with arterial hypertension and increases the blood flow after ischemia in patients with peripheral vascular disease, being capable of restoring endothelial function in spontaneously hypertensive Rats (SHR) (Virmani and Binienda, 2004). In this context, LC has been shown to prevent long-chain acyl-CoA accumulation and consequent production of ROS (Reactive Oxygen Species) by damaged mitochondria, and improved some repair mechanisms against oxidative stress-induced damage to membrane phospholipids (Virmani and Binienda, 2004).
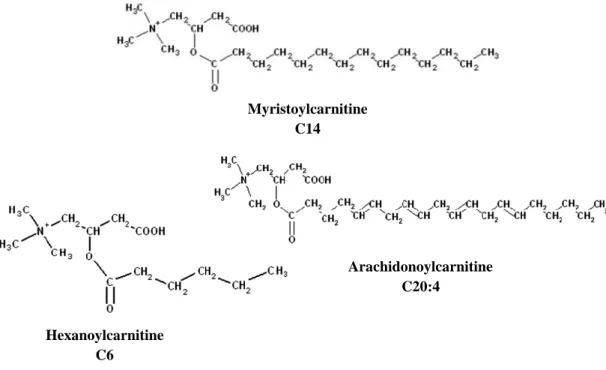
The free form of LC has a major occurrence in the body, but esters are also present (acylcarnitines) (Virmani and Binienda, 2004). The free hydroxyl group of LC can be enzymatically esterified to form acylcarnitines (Figure 2), like acetyl-L-carnitine, myristoylcarnitine, hexanoylcarnitine, arachidonoylcarnitine and propionyl-L-carnitine (Malaguarnera, 2012). The ability of LC to esterify and transport metabolites suggests that acylcarnitine profile may be a useful indicator of altered metabolic states especially related to pathological conditions. Therefore, monitoring specific acylcarnitines will potentially lead to a better understanding of disease mechanisms and contribute to the development of novel therapeutic regimens (Jones et al., 2010).
As described for LC, the supplementation of specific acylcarnitines can have beneficial effects on particular disease states. For instance, supplementation with propionyl-L-carnitine (PLC) has positive effects on general fatigue. Due essentially to the amphiphillic nature of PLC, it can react with the surface of membranes and influence membrane fluidity, membrane enzymes, transporters activity and its therapeutically application is focused on the prevention and treatment of ischemic and congestive heart diseases (Jones et al., 2010; Malaguarnera, 2012). The present work will focus on characterizing cellular models for the disclosure of the possible mechanisms of acetyl-L-carnitine (ALC) action as a neuroprotective compound, preventing toxicity induced by a drug of abuse, methamphetamine (METH) (Muneer et al., 2011b; Muneer et al., 2011c; Summavielle et al., 2011).
5
1.2.Acetyl-L-carnitine
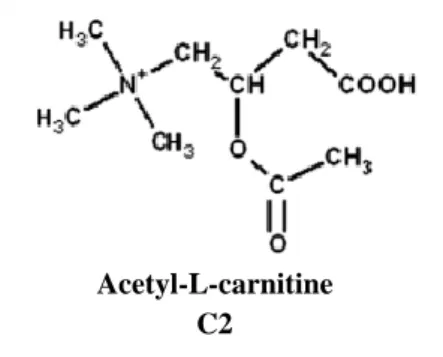
The role of ALC as a therapeutic agent in aging and brain disorders is a subject of interest since the early 1990s (Jones et al., 2010). However, much is still unknown regarding its mechanism of action (Summavielle et al., 2011). ALC is a small water-soluble molecule (Figure 3) that is easily transportable throughout organism, and constitutes the short-chain ester of LC most extensively distributed in the body (Virmani et al., 2004). The two organelles responsible for ALC production are mitochondria and peroxisomes. This molecule is responsible for the transport of acetyl-moieties and acetyl groups from these organelles to other cells (Jones et al., 2010; Smeland et al., 2012).
ALC is present at relatively high levels in the brain, particularly in the hypothalamus. Due to its high distribution in almost all cells and presence in a huge variety of events, ALC can affect lipid, carbohydrate and amino acid, as well as energy metabolism (Smeland et al., 2012).
Figure 2. Examples of acylcarnitine structures. Adapted from (Jones et al., 2010). Myristoylcarnitine C14 Arachidonoylcarnitine C20:4 Hexanoylcarnitine C6
6
Figure 3. Acetyl-L-carnitine structure. Adapted from (Jones et al., 2010).
1.2.1. Acetyl-L-carnitine biosynthesis
Under physiological conditions (Figure 4), carnitine palmitoyltransferase 1 (CPT1) catalyzes the transfer of acyl groups from acyl-CoA to carnitine to produce acylcarnitines and free CoA. The carnitine palmitoyltransferase 2 (CPT2) catalyzes the transfer of acyl groups from acylcarnitines to free CoA present in the mitochondria, producing acyl-CoA and free carnitine. The free carnitine formed is transported to the citosol and it will be used again to transport acyl groups to the mitochondrial matrix. The acyl-CoA is metabolized through β-oxidation cycle, which the final product is actyl-CoA (Virmani and Binienda, 2004). The actyl-CoA recently formed can enter the Krebs cycle or can be used to synthesize ALC. ALC is synthesized in a reaction catalyzed by carnitine acetyltransferase (CAT), which reversibly converts acetyl-CoA (derived from pyruvate, amino acids, and fatty acids) and free carnitine to ALC and CoA without ATP consumption (Virmani and Binienda, 2004). This process of acetyl-CoA conversion regenerates free CoA, required for fatty acid oxidation and Krebs cycle to proceed (Rosca et al., 2009). CAT is a mitochondrial matrix enzyme attached to the inner membrane and its activity was reported to be present in almost all tissues (Virmani and Binienda, 2004). Particularly in the liver, it was found in peroxisomes, mitochondria, and microsomes (Aureli et al., 1999). In the brain, the levels of CAT are high in the hippocampus, colliculi and basal ganglia (Virmani and Binienda, 2004).
Physiological concentrations of total carnitine and ALC in the human plasma of adults are maintained within 23–73 µmol/L and 3–14 µmol/L, respectively. The degree of acetyl-CoA synthesis is observed in the plasma concentration and urinary excretion of ALC, which vary according to physiological and pathological states (Rosca et al., 2009).
Acetyl-L-carnitine C2
7
Figure 4. Acetyl-L-carnitine in overall energy metabolism. (Acetylcarnitine = Acetyl-L-carnitine, according to NCBI website, Pubchem Compound - http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=1, 22.12.12, 4 pm); CoASH = Coenzyme A reduced thiol). Adapted from (Jones et al., 2010).
1.2.2. Acetyl-L-carnitine supplementation
In humans, orally-supplemented ALC is absorbed at the gastrointestinal tract and pass through a process of deacetylation during or immediately after its uptake into intestinal cells. The portion of the free carnitine formed intracellular recently is re-acetylated (Gross et al., 1986; Rosca et al., 2009).
Although the drug absorption is poor, the concentration of ALC in plasma and in the cerebrospinal fluid increases after oral administration, and the compound is transported across the blood-brain barrier (BBB) by the organic cation/carnitine transporter OCTN2 (organic carnitine transporter 2), a saturable sodium-dependent system (Rosca et al., 2009; Smeland et al., 2012). Carnitine transporters from the OCTN family are present on both neurons and astrocytes (Smeland et al., 2012). Exogenous administered ALC (Figure 5) is transported into the mitochondria matrix via the inner membrane CACT (carnitine acylcarnitine translocase) in the change with carnitine, thus providing acetyl groups for the synthesis of sterols, fatty acids and ketone bodies. The ALC transported to the mitochondria citosol is exposed to CAT, which reversibly converts ALC into acetyl-CoA and carnitine. The formation of acetyl-CoA by CAT or newly synthesized from acetate is indispensable to the process of Krebs cycle, the synthesis of lipoic acid and the protein acetylation within mitochondria (Rosca et al., 2009).
8
In the mitochondria, the equilibrium between acetyl-CoA and CoA (acetyl-CoA/CoA ratio) is crucial for mitochondrial metabolism, and an increase of this ratio has negative effects on mitochondria, affecting the activity of pyruvate dehydrogenase, inhibiting the mitochondrial β-oxidation and the Krebs cycle. During ALC supplementation, the mitochondrial content of acetyl-CoA is the result of acetyl-CoA formation, due to the mass-action of CAT, and utilization of the former. Since the acetyl-CoA formed it is used for synthesis of lipoic acid and citrate, two irreversible pathways, the process drives the acetyl-CoA values to a safe range. The mitochondrial content of endogenous ALC is an indicator of mitochondrial metabolism of acetyl-CoA (Rosca et al., 2009). Diagram represented in Figure 5 shows the metabolic pathways of ALC after exogenous administration.
Figure 5. Metabolic pathways of external supplementation of Acetyl-L-carnitine (AcetylC = Acetyl-L-carnitine; C = carnitine). Adapted from (Rosca et al., 2009).
9
1.2.3. Acetyl-L-carnitine applications
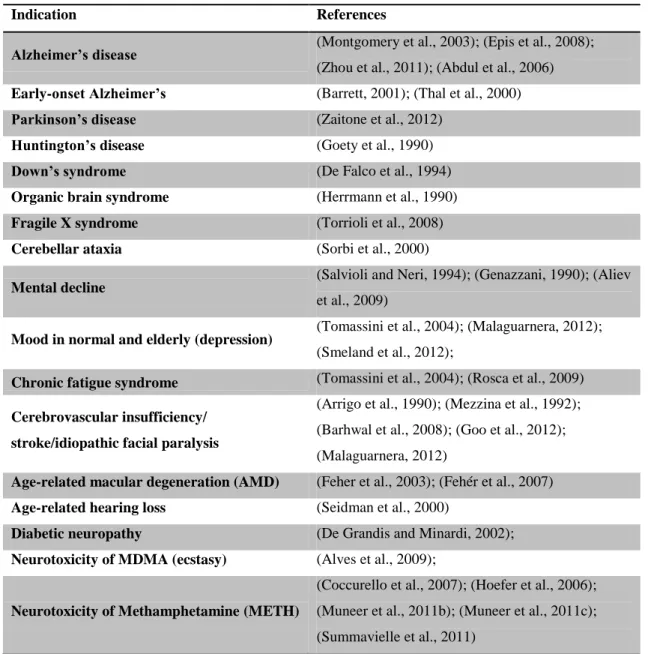
The supplementation with ALC could feasibly affect brain metabolism and be used as a therapeutic agent in aging and in neurological disorders (Jones et al., 2010). ALC has been considered a “mitochondrial nutrient” that reverses mitochondrial dysfunction, having neurophysiological and neuroprotective actions on cellular processes and against neurotoxic damages in central and peripheral nervous system (Malaguarnera, 2012; Rosca et al., 2009; Smeland et al., 2012; Virmani and Binienda, 2004). Moreover, ALC is reported to alter metabolic pathways in diseases correlated with mitochondrial-related disorders (Dhitavat et al., 2002; Di Cesare Mannelli et al., 2007; Virmani and Binienda, 2004). In addition, ALC is reported to increase the endogenous pool of antioxidants enzymes (El-Awady et al., 2011; Hao et al., 2011), helping maintaining the cellular redox system components (NAD+/NADH and NADP+/NADPH) (Elanchezhian et al., 2009) and the ionic homeostasis (Calabrese et al., 2006). This subsequent stabilization of the membrane ensures an optimal mitochondrial enzyme activity (Aureli et al., 2000; Hao et al., 2011). ALC also decreased the expression of iNOS and stimulate protein and membrane phospholipids synthesis leading to increase resistance to neurotoxic events (Abdul et al., 2006; Calabrese et al., 2010; Virmani and Binienda, 2004). Former studies reported that ALC enhanced acetylcholine synthesis (Summavielle et al., 2011; Virmani and Binienda, 2004) and affected the serotonergic, glutamatergic, dopaminergic and GABAergic systems (Tolu et al., 2002). Many studies show that ALC action can have positive effects in several pathologies (Table 1). Moreover, ALC is well tolerated, with few adverse effects found at similar frequency in both ALC and placebo groups (Virmani and Binienda, 2004).
10
Table 1. Studies with ALC in central and peripheral nervous system. Adapted from (Virmani and Binienda, 2004).
Indication References
Alzheimer’s disease (Montgomery et al., 2003); (Epis et al., 2008);
(Zhou et al., 2011); (Abdul et al., 2006)
Early-onset Alzheimer’s (Barrett, 2001); (Thal et al., 2000)
Parkinson’s disease (Zaitone et al., 2012)
Huntington’s disease (Goety et al., 1990)
Down’s syndrome (De Falco et al., 1994)
Organic brain syndrome (Herrmann et al., 1990)
Fragile X syndrome (Torrioli et al., 2008)
Cerebellar ataxia (Sorbi et al., 2000)
Mental decline
(Salvioli and Neri, 1994); (Genazzani, 1990); (Aliev et al., 2009)
Mood in normal and elderly (depression) (Tomassini et al., 2004); (Malaguarnera, 2012); (Smeland et al., 2012);
Chronic fatigue syndrome (Tomassini et al., 2004); (Rosca et al., 2009) Cerebrovascular insufficiency/
stroke/idiopathic facial paralysis
(Arrigo et al., 1990); (Mezzina et al., 1992); (Barhwal et al., 2008); (Goo et al., 2012); (Malaguarnera, 2012)
Age-related macular degeneration (AMD) (Feher et al., 2003); (Fehér et al., 2007)
Age-related hearing loss (Seidman et al., 2000)
Diabetic neuropathy (De Grandis and Minardi, 2002);
Neurotoxicity of MDMA (ecstasy) (Alves et al., 2009);
Neurotoxicity of Methamphetamine (METH)
(Coccurello et al., 2007); (Hoefer et al., 2006); (Muneer et al., 2011b); (Muneer et al., 2011c); (Summavielle et al., 2011)
In rat models, the supplement of ALC could improve the homocysteine-induced memory deficits remarkably, diminishing the tau hyperphosphorylation and accumulation of Aβ, suggesting that ALC can have beneficial effects on homocystine-induced AD-like pathological and behavioral impairments (Zhou et al., 2011). Epis and coworkers (2008) demonstrated that ALC can directly influence the primary event in Alzheimer's disease pathogenesis, the β-amyloid cascade, by interfering with the production of the non-amyloidogenic metabolite, using in vitro models, specifically a neuroblastoma cell line and primary cultures of hippocampal neurons (Epis et al., 2008). Parkinson’s disease can be seen
11
as a mitochondrial dysfunction related to oxidative stress and environmental toxicants. Some studies revealed that ALC acts in combination with α-lipoic acid through pathways that promote neuroprotection in Parkinson's disease (Zaitone et al., 2012). In the campus of mental decline not associated with this two diseases, studies with young and aged animals suggest that ALC and R-α-lipoic acid (LA) oral supplementation may ameliorate age-associated mitochondrial ultrastructural decay, improving mitochondria proliferation and reducing the vacuoles in the hippocampus (Aliev et al., 2009). In neurological diseases like depression, ALC supplementation increased the levels of serotonin in cortex, consistent with ALC’s potential efficacy for depressive symptoms (Smeland et al., 2012). In cancer patients that pass to chemotherapy cycles, and also on chronic fatigue in patients with multiple sclerosis, ALC has proved to be effective in the treatment of fatigue syndrome (Tomassini et al., 2004). ALC is reported to improve neuronal energy and to increase the synthesis of the neurotransmitter acetylcholine in the cholinergic system (Rosca et al., 2009).
Goo and coworkers (2012) investigated the effects of ALC in secondarily-induced cerebral chronic ischemia models using rats with permanent ligation of bilateral common carotid arteries (BCCL). Chronic ALC administration significantly attenuated neurodegenerative changes and has shown beneficial effects after cerebral ischemia, as well as in spontaneous aging-related cerebral dysfunction via hippocampal protection (Goo et al., 2012). Barhwal and coworkers (2008) demonstrated that ALC may be an effective therapeutic agent for hypoxic stress and related neurodegenerative disorders, since it attenuates cytotoxicity induced by hypoxia and increases cellular glutathione levels and cytochrome c oxidase activity (Barhwal et al., 2008).
ALC is already used as a metabolic therapy for age-related macular degeneration (AMD). A combination of ALC, omega-3, fatty acids and coenzyme Q10 has been introduced for the early treating of AMD. This therapy improved the mitochondrial metabolism, specifically rising the metabolism of lipids and ATP production in the retinal pigment epithelium, which decreased ROS generation and the photoreceptor turnover. Interestingly, this is the only combination of ingredients corresponding to recommended daily allowance, and at the same time, that showed clinically proved efficacy (Fehér et al., 2007).
12
1.2.3.1.Methamphetamine effects and the protective role of acetyl-L-carnitine
Beyond the protective effect in neurodegenerative diseases, ALC has been reported to have a neuroprotective action on toxicity induced by amphetamines, such as methamphetamine (METH) (Muneer et al., 2011b; Muneer et al., 2011c; Summavielle et al., 2011).
In terms of consume, it was estimated that there are 15–16 million addicted users of METH worldwide, being this addiction considered an international public health problem (Martin et al., 2012).
Historically, METH use in Europe has been concentrated in the Czech Republic, where it is estimated that the number of problem users of METH correspond to twice (20.300) of opioid users (9.700). In recent years, METH has become the most frequently cited primary drug among people seeking treatment for the first time in Slovakia. There are also increased levels of METH-use among some subpopulations in Hungary. In the Reitox reports, seven countries (Denmark, France, Latvia, Slovenia, UK, Bulgaria and Norway) were reported to display increased use of the METH, especially among the regulars of nightclubs and parties (EMCDDA, 2009 ). In Portugal, the substances detected in cases of overdose were predominantly opiates (includes heroin, morphine and codeine), followed by cocaine, however in 2008 and 2009, METH was responsible for 1% of the overdoses (EMCDDA, 2011).
METH is a potent stimulant of the central nervous system (CNS) highly addictive, that cause behavioral and psychological disturbances during intoxication and after chronic use produces long-term health complications (Kiyatkin and Sharma, 2009) due to extensive neural damage (Barr et al., 2006; Scott et al., 2007). As a result of its lipophilicity, METH is extensively distributed in the brain (Fowler et al., 2008; Segal et al., 2005). The METH chronic users have common brain structural abnormalities, more specifically white matter hypertrophy and loss of grey matter in specific regions like prefrontal cortex (responsible for deliberative and executive functions), striatum, hippocampus (responsible for learning and memory, as well as relapse to drug seeking) and amygdala (which is involved in the detection of emotional salience in environmental stimuli) (Davidson et al., 2000; Deng et al., 2001; Nelson and Trainor, 2007; Recinto et al., 2012; Siever, 2008; Thompson et al., 2004).
13
METH induces the release of monoamine neurotransmitters, such as dopamine (DA), serotonin and norepinephrine (Barr et al., 2006; Kish, 2008; Sulzer et al., 2005). This acute monoamine release leads to a various range of symptoms, like euphoria, increase of energy and hypersexuality (Barr et al., 2006). The release of epinephrine and norepinepherine by adrenal glands cause problems related to increase of blood pressure and cardiac pathologies such as stroke and cardiac arrhythmia (Barr et al., 2006; Ho et al., 2009). Moreover it causes physiologic side effects, including anxiety, aggressive behavior and paranoia (Barr et al., 2006). It has been described that, during withdrawal, the synaptic levels of these type of neurotransmitters suffer a decrease in various brain regions (Cadet et al., 2011) and the subsequent administrations of METH do not increase the neurotransmitters levels to values similar to control (Barr et al., 2002; Cadet et al., 2011). When the acute administration is interrupted, the withdrawal symptoms include fatigue, anxiety, depression, suicide thoughts and intense craving for METH (Barr et al., 2006; Zorick et al., 2010). There are some mechanisms that were already been described to explain how METH induces neurotoxicity, such as oxidative stress (Barr et al., 2006; Cadet et al., 1994; Cadet et al., 2007; Sun et al., 2011; Volz et al., 2007), mitochondrial dysfunction (Potula et al., 2010), inflammatory process associated with the microglia activation (Bento et al., 2011; Snider et al., 2013) and alterations of the BBB (Clark et al., 2013; Martins et al., 2011).
As it was referred above, METH increases release of dopamine (Barr et al., 2006; Kish, 2008; Sulzer et al., 2005) and damages the dopaminergic neuronal system (Thomas et al., 2010) in the striatum (Beauvais et al., 2011; Cadet et al., 2011). METH acts like a substrate for the DA transporter, allowing the drug to access the dopaminergic neurons, where it inhibits the vesicular monoamine transporter, increasing DA in the synaptic cleft (O'Dell et al., 1993; Sanchez-Alavez et al., 2013) and subsequent stimulation of DA receptors in the brain (Krasnova and Cadet, 2009; O'Dell et al., 1993; Xu et al., 2005). Moreover, DA in the presence of high doses of METH has a greater propensity to oxidize and form ROS, which damages dopamine terminals. Furthermore, both superoxide and nitric oxide can generate the most damaging radical, the peroxynitrite (ONOO−) (Barr et al., 2006). The mechanism of the serotonergic neurotoxicity is not as well characterized, but the way of action of METH appears to be similar to what occurs in the DA terminals, where the serotonergic terminals suffer depletion when there is an administration of high METH doses in animal models (Kuczenski et al., 1995). The extracellular glutamate levels are also affected by METH
14
(Stephans and Yamamoto, 1994). The excessive generation of ROS and oxidative stress can damage the cellular structures and result in mitochondrial impairment. Oxidative stress also inhibits the electron transport chain, severely disrupting the mitochondrial respiration (Kuczenski et al., 1995), therefore causing energy deficits, followed by secondary excitotoxicity (Krasnova and Cadet, 2009; Potula et al., 2010; Virmani et al., 2003). The accumulation of DA and the neuronal degeneration induced by METH is often associated with activation of astroglia cells (astrocytes and microglia) (Kitamura et al., 2010; Snider et al., 2013) in the striatum (Friend and Keefe, 2013). Reactive astrogliosis and glial scar formation occurs at the injury location, and astrogliosis can hinder the recovery process of the neurons (Sofroniew, 2009). Manipulation of reactive astrogliosis might create a favorable environment for neuronal regeneration and thereby serve as a promising intervention to decrease the damaging effect of reactive astrogliosis in brain injury (Hamby and Sofroniew, 2010; Snider et al., 2013). METH has also been connected to changes in the gene expression (Cadet et al., 2009), and study the up or downregulation of these genes could lead to a better understanding of the metabolic pathways of METH action. The Rho-associated Kinase (ROCK) has been reported to play an important role in gap junction formation, morphology, and migration of astrocytes (Yu et al., 2012). Moreover, its known that ROCK activity influences the stability of RND proteins (Goh and Manser, 2012). There are several of other members of Rho family that regulate actin dynamics, cytoskeleton, transcriptional regulation, vesicle trafficking, morphogenesis, phagocytosis, mitogenesis, apoptosis and tumorogenesis (Boureux et al., 2007; Oleksy et al., 2006; Ridley, 2006) and can have an important role on the behavior of astrocytes and neurons exposed to METH and ALC. There are other systems that influence the astrocytes behavior, like the Insulin-like growth factor-I (IGF-I), which exerts neuroprotective actions in the central nervous system, activating the astrocytes (Bellini et al., 2011).
The activation of these neurotoxic pathways by the exposure of cells to METH creates a great model for the study of ALC action and their neuroprotective effects on these cells.
Muneer and his coworkers (2011c) studied the effects of METH-induced disruption of glucose uptake, leading to BBB dysfunction, and the protective effects of ALC in glucose transporter protein-1 (GLUT1) in primary human brain endothelial cell (the main component of BBB). Authors conclude that ALC has protective effects against damage on glucose transport induced by METH. Moreover, mice treated only with METH were inactive and
15
lethargic by past 5 weeks, whereas mice treated with the conjugation of METH and ALC were still active comparing to the control mice. These authors suggest that ALC protects against METH-induced disruption of BBB permeability both in acute and chronic conditions. They observed that METH produced a significant reduction in electrical resistance, which was prevented by ALC. The stabilization induced by ALC exposure on the glucose uptake and GLUT1 protein levels may be a lead for the involvement of ALC in glucose metabolism (Muneer et al., 2011c).
Muneer and his coworkers (2011b) also hypothesized that METH exposure affects the glucose uptake and metabolism in astrocytes and human neurons. Authors found that METH exposure inhibited the glucose uptake by neurons and astrocytes, and that ALC protected these cells from METH-induced effects, suggesting the activation of fatty acid oxidation. ALC is the primary cofactor of mitochondrial β-oxidation of fatty acids for ATP production. Astrocytes can have an alternative survival mechanism, and, therefore, blockage of glucose uptake did not significantly affect astrocytic cell death. The decrease in neuronal glucose uptake caused by METH was associated with reduction of glucose transporter protein-3 (GLUT3) expression. ALC prevented the decrease in glucose uptake induced by METH in neurons by stabilizing the protein levels of GLUT1 and GLUT3 (Muneer et al., 2011b).
Summavielle and her coworkers (2011) showed in PC12 cells, an in vitro model to mimic DA neurons (Piga et al., 2005; Schlegel et al., 2004), that the pre-exposure to ALC appears to prevent the release of DA induced by METH (Summavielle et al., 2011).
Repeated systemic treatment in rats with ALC, during METH withdrawal demonstrated positive effects in the depressive state, suggesting that ALC may represent a potential therapeutic strategy for the treatment of METH addiction during the withdrawal phase (Hoefer et al., 2006). Results also showed that both the acute and repeated co-administration of METH with ALC inhibited hyperactivity induced by METH. The in vivo protective action of ALC against METH consumption evoke the possibility of beneficial effects of ALC in abuse behavior (Coccurello et al., 2007).
16
1.3.Models of study
There are a large number of models to study the effects of psychostimulants drugs, like METH. The central objective of current neurobiological in the field of addiction research is to understand the molecular, neuropharmacological and neurocircuitry changes that occur with abuse of psychostimulant drugs and the effective pharmacotherapies (Koob et al., 2009).
The animal models of addiction recapitulate aspects of the conditions in humans and have excellent face validity (for instance, to mimic the intravenous self-administration). They also have validity to study the stress response during drug withdrawal, having functional similarity with the conditions in humans. Even though no animal model fully mimics addiction in humans, such models do permit exploration of elements of the drug addiction process (Koob et al., 2009). Studies related to the understanding of the effects of ALC in sensitization of mouse and withdrawal from METH concludes that ALC it is effective in the control or reduction of these symptoms (Coccurello et al., 2007; Hoefer et al., 2006).
Cell death caused by METH involves not only the striatum and cortex, but also the hippocampus (Venkatesan et al., 2011). The mammalian hippocampus retains its ability to generate neurons throughout life. However, psychostimulant drugs, like METH caused decrease in hippocampal volume and cell density. The orientation, shape and several cellular markers were also affected (Yuan et al., 2011).
Cell lines are often used in place of primary cells to study biological processes (Kaur and Dufour, 2012). For instance, the PC12 cell line is commonly used as an in vitro model to understand the biochemical mechanisms underlying the physiology and metabolism of central dopaminergic neurons (Piga et al., 2005; Schlegel et al., 2004). A number of factors contribute to the wide use of PC12 cells, they are not expensive, easy to handle, and mimic many features of central DA neurons (Fornai et al., 2007). PC12 cell line represents a model to predict the neurotoxicity of a variety of compounds acting on central dopaminergic neurons (Fornai et al., 2007; Piga et al., 2005; Schlegel et al., 2004).
The BBB has a crucial role on the maintenance of brain homeostasis, it is permeable to nutrients and vitamins, protecting against toxic molecules and organisms. If METH disrupts the BBB, by affecting the endothelial cells (Martins et al., 2011), it will be important to understand the changes in the morphology of these cells, contributing to get a step closer to understand the neurotoxic mechanisms of METH and beneficial effects of ALC in the BBB.
17
There are two ways to study the BBB, by in situ perfusion models in animals and by in vitro cultures of endothelial cells from cerebral microvessels or other endothelial cell sources like immortalized cell lines (Brown et al., 2007). Several studies show that bEnd.3 cells are a suitability immortalized mouse brain endothelial cell line that can be used as a model for BBB (Brown et al., 2007; Chen et al., 2011; Kapitulnik et al., 2012; Liu et al., 2012). Unlike the primary cultures, the bEnd.3 do not lose the blood-brain barriers characteristics over repeated passages, like the formation of functional barriers and respond like primary cultures to disrupting stimuli (Brown et al., 2007). Given these characteristics, bEnd.3 monolayer proves to be a favorable model to mimic the BBB. Not only endothelial cells contribute to the BBB, astrocytes also play an essential role in the maintenance of this barrier, regulation of blood flow, regulation of glutamatergic neurotransmission and neurotransmitter metabolism, supplying energy metabolites to neurons and maintenance of the extracellular balance of ions and fluid (Sofroniew, 2009). Similar to what occurs with the brain endothelial cells, there are some cell lines which mimic fibrous astrocytes present in the brain, like for instances the C8-D1A cell line (Kurihara et al., 2011). This cell line is usually used as an alternative to the primary astrocytes (Ellis et al., 2010; Kurihara et al., 2011; Liu et al., 2012). Astrocytes ensure the development and normal function of the brain and spinal cord. Astrogliosis occurs after METH and other toxic insults in the brain, therefore, glial response is considered an indicative of neurotoxicity (Simões et al., 2007). Manipulation of reactive astrogliosis might create a favorable environment for neuronal regeneration and thereby serve as a promising intervention to decrease the damaging effect of reactive astrogliosis in brain injury (Yu et al., 2012). There are also studies that mimic the BBB using bEnd.3 and C8-D1A cell lines together, used to understand the permeability and alteration of trans-endothelial electrical resistance (TEER), providing useful insights about BBB role in CNS disease progression and drug delivery (Booth and Kim, 2012). Is fundamental to mimic the brain matter, due the fact that cells interaction may be critical to the hypothesis being tested, since both primary cells and cell lines are being study in the absence of their local environment (Kaur and Dufour, 2012).
Understanding the role of ALC on cell viability and its mechanisms of action opens up a range of new perspectives for the future use of ALC in the treatment of many clinical conditions.
18
1.4.Cellular viability evaluation
There is an enormous amount of experimental procedures available to study the behavior of cells, cell cultures and/or tissues, from model organisms when exposed to different stimulus (Galluzzi et al., 2009). There are three major types of cell death: necrosis, programmed cell death (or apoptosis) and autophagic cell death (Galluzzi et al., 2009; MacKenzie and Clark, 2008). In an initial screening, it is important to begin with direct methods that allow us to understand in what scale the stimulus induced affect cellular viability and cause cell death. In order to quantify cells that are dying or died is essential to perform multiple and methodologically distinct assays, given the existent difficulties in determining the accurate number of cells that have passed the point-of-no-return of the signaling cascades that leads to cell death (Galluzzi et al., 2009).
The LDH release and MTT reduction assays have been used for the evaluation of cell damage (Gómez-Serranillos et al., 2009) and are considered appropriate for the first rounds of high-throughput studies. These assays are commonly used for cell death research, because they allow the analysis of different cell types (e.g., primary hippocampal neurons, PC12, bEnd.3 and C8-D1A cell lines). These two methods are fast and do not involve preprocessing of samples (e.g., cell lysis) and specific laboratory material and equipment (Galluzzi et al., 2009). LDH release measures the level of the cell membrane compromise (plasma membrane breakdown that lead to necrosis), which in the context of cytotoxicity assays is widely accepted as actual cell death. Membrane bleeding and loss of integrity is actually an early indicator of the death that will ultimately follow (Kroemer et al., 2005). The MTT reduction assay measures the percentage of living cells, through estimation of active mitochondrial dehydrogenase function (Lobner, 2000). The metabolic activity of the mitochondrial respiratory chain is extensively considered as a marker of the number of viable cells. Integrating the LDH release and MTT reduction assays, allows the cross-confirmation of the cytotoxicity and viability datasets (Galluzzi et al., 2009; Lobner, 2000).
Although LDH and MTT assays are useful to the first screening, is also important to understand the morphologic changes caused by the several conditions tested. The quick reorganization of the actin cytoskeleton in response to external stimulus is a crucial property of several motile eukaryotic cells (Westendorf et al., 2013). Actin has a long history of proposed connections with nuclear events as well as its classical role as a cytoskeletal component (Posern et al., 2004). So, understanding the dynamics of the actin filament is
19
essential to a detailed description of their interactions and role in the cells (Scoville et al., 2009). The F-actin filament can be marked by a specific molecular probe, phalloidin. With this specific marker we can evaluate morphologic changes on cell cytoskeleton, using simple immunofluorescence analysis (Alama-Bermejo et al., 2012).
After the first screening, there are several methods for death detection, including death by apoptosis. The Hoechst 33342 and Hoechst 33258 are usually used to distinguish condensed pycnotic nuclei in apoptotic cells. They are similar dyes that when bind to dsDNA (AT-rich regions) emit blue fluorescence. However, Hoechst 33342 is more cell permeant (Galluzzi et al., 2009). Although these dyes can be used to automate counting (e.g. flow cytometry) the apoptotic cells can be analyzed by fluorescence microscopy (Yuan et al., 2013).
Taking into account the possible impact that ALC and METH exposure have in the several brain cells mentioned above, we decided to evaluate the effects of these two substances on 4 different cellular types (primary hippocampal neurons, PC12, bEnd.3 and C8-D1A cell lines). For that, we used appropriate methodologies for the first rounds of high-throughput studies, in order to understand the suitable doses for further tests, which are now ongoing on our lab.
21
23
2. OBJECTIVES
The aim of this work was to characterize cellular models that will be used to disclose possible mechanisms of ALC action, in the nervous system, as a neuroprotective compound. Particular attention was given to astrocytes (C8-D1A cell line), which were a new line in the laboratory relevant to understand the role of ALC at the level of glucose uptake. Other cell types that interact with the astrocytes such as neurons and endothelial cells were also a target of this study. METH was used to induce toxicity and allow the characterization of the potential protective action of ALC in basic parameters. The study was organized as follows:
1. Characterization of the dose-response curve to METH (damage-inducing agent) in the different cell types used: the C8-D1A cell line (astrocytes type I clone), the bEnd.3 cortex endothelial cell line, primary hippocampal neurons and the PC12 cell line. 2. Evaluation of ALC ability to prevent METH-induced changes in the different cell
types.
3. Characterization of ALC and METH action (under the conditions selected in 1 and 2) in key molecular-players involved on important pathways previously selected in astrocytes.
25
CHAPTER III – MATERIAL AND
METHODS
27
3. MATERIAL AND METHODS
3.1.Primary hippocampal cultures
Rats used in this study were maintained under a 12 h dark/light schedule and were given ad libitum food and water access. All experiments were performed respecting the Guidelines of the European Union Council 86/609/EU and the Portuguese law for the care and use of experimental animals (DL nº 129/92) at the Animal Facility of the Instituto de Biologia Molecular e Celular (IBMC), University of Porto. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Cultures of rat primary hippocampal neurons were prepared from E18 Wistar rat embryos (18 days of gestation). Rats and embryos were sacrificed by decapitation, embryo’s brains were dissected out and hippocampi were separated from other brain regions. Briefly, after dissection, the hippocampi were treated with trypsin (0.06%, 15 min, 37ºC; Gibco, Invitrogen) in Ca2+ and Mg2+ free Hank’s Balanced Salt Solution (HBSS, 5.36 mM KCl, 0.44 mM KH2PO4, 137 mM NaCl, 4.16 mM NaHCO3, 0.34 mM Na2HPO4.2H2O, 5 mM glucose, 1
mM sodium pyruvate, 10 mM HEPES and 0.001% phenol red). The hippocampi were washed with HBSS containing 10% fetal bovine serum, to stop trypsin activity, and then washed once in Hank solution to remove serum and avoid glia growth. Finally, the hippocampi were transferred to Neurobasal medium® (Gibco, Invitrogen), supplemented with B27® (1:50 dilution; Gibco, Invitrogen), glutamate (25 μM), glutamine (0.5 mM) and gentamicin (0.12 mg/mL). The cells were dissociated mechanically, and plated on poly-D-lysine coated 24 microwell plates (MW24) at a density of 9104 cells/cm2. The cells were kept at 37°C in a humidified incubator with 5% CO2/95% air, 14 days in vitro (DIV). Half of the culture
medium was discarded when cells reach 7 DIV, and replaced with new culture medium. The preparation of these primary cultures was performed by other researchers within our laboratory.
3.2.Maintenance of cell lines cultures: PC12, bEnd.3 and C8-D1A cells
In the present study three different types of cell lines were used: PC12 cell line (cell line 1721, ATCC, U.S.A.), an astrocyte type I clone C8-D1A cell line (cell line
CRL-28
2541, ATCC, U.S.A.) and a cerebral cortex endothelial cell line from endothelioma bEnd.3 (cell line CRL-2299, ATCC, U.S.A.). The PC12 cell line is derived from a rat pheochromocytoma. The C8-D1A was derived from explant cultures of 8-day postnatal mouse cerebella after in vitro spontaneous transformation. The bEnd.3 was transformed by infection of endothelial cells from the cerebral cortex of Mus musculus with the NTKmT retrovirus vector that expresses polyomavirus middle T antigen.
Cells were grown normally in flasks coated with poly-L-lysine (PC12 cells) or not coated (bEnd.3 and C8-D1A cells) containing Dulbecco’s Modified Eagle Medium (1x) plus GlutaMAXTM-I (Gibco, Life Technologies, UK) supplemented with 10% of Fetal Bovine Serum (Gibco, Invitrogen, South America) and 1% of penicillin/streptomycin (Gibco, Invitrogen, UK). Cells were kept at 37°C in a humidified incubator with 5% CO2/95% air.
The confluence of cells was monitored and the culture medium was discarded every 3 days. For trypsinization, cells at 80-90% of confluence were washed with PBS (Gibco, Invitrogen, UK) before addition of trypsin 0.05% in EDTA 1x (Gibco, Invitrogen). Trypsin activity was achieved at 37ºC until cell detachment and it was stopped by the addition of culture medium; cells were collected and centrifuged for 5 minutes at 1200 rpm. The supernatant was discarded and culture medium was added to the pellet to perform cells resuspension. A sample was collected and mixed with Tripan Blue in order to estimate the total number of cells using a Neubauer chamber. In order to apply different assays, cells were cultured on 24 well plates at a density of 8104 cells/cm2 for PC12 cells, 7.5104 cells/cm2 for bEnd.3 cells and 8104 cells/cm2 for C8 D1A cells. The same densities of cells were cultured on coverslips when immunofluorescence assays were intended.
3.3.Drug treatments
Cells were treated with two different doses of ALC (0.1 and 1.0 mM) alone or in combination with different doses of METH (0.01 to 3.0 mM), directly dissolved in culture medium for 24 h or 72 h. When ALC and METH were combined, pre-treatment with ALC preceded METH exposure in 30 minutes. ALC was kindly provided by Sigma-Tau, S.p.A. (Pomezia, Italy). METH hydrochloride was obtained from Sigma-Aldrich® (St. Louis, MO).
29
3.4.Cell death assay
The CytoTox 96® Assay (Promega Corporation, U.S.A.) was used to quantify lactate dehydrogenase (LDH). LDH is a stable cytosolic enzyme that is released upon cell lyses (prior to cell death). Released LDH in culture supernatants is measured with a 30 minute coupled enzymatic assay, which results in conversion of a tetrazolium salt (INT) into a red formazan product. The variation of absorbance is proportional to the number of lysed cells. The LDH activity is determinate by NADH oxidation and INT reduction over a defined period of time.
This assay requires a positive control (incubation of cells in a lysis solution during 45 minutes at 37ºC) (Promega Corporation, U.S.A.); a negative control (cells with no treatments); and a blank well containing only the cell medium (culture medium background). After 24 h or 72 h of treatment, the medium of each well was collected (experimental conditions), centrifuged at 14000 rpm, for 10 minutes, and the samples were diluted (1:1 in distillated water) and transferred to a flat-bottom 96-well enzymatic assay plate. The reconstituted substrate mix (Promega Corporation, U.S.A.) was added to each well and the plate was incubated at room temperature for 30 minutes, protected from light. The addition of Stop Solution (Promega Corporation, U.S.A.) stopped the reaction. The absorbance was read at 490 nm and the percentage of dead cells was calculated for each condition. The percentage of cytotoxicity was determined as following:
o itoto i ity Experimental condition – ackground – egative control
Positive Control – egative Control 1
3.5.Cell viability assay
To assess cell viability of PC12, bEnd.3 and C8-D1A cell lines the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used. This is a standard colorimetric assay for measuring the activity of enzymes that reduce MTT to formazan, giving to the medium a purple color. This reaction takes place only when mitochondrial reductase enzymes are active, and therefore conversion can be directly related to the number of viable (living) cells (Mosmann, 1983). After 24 h or 72 h of treatment (experimental conditions), cells medium was removed and replaced by 150 µL of fresh
30
medium containing 0.5 mM of MTT (Sigma Chemical Co., St. Louis, MO) and plates were incubated for 2.5 h at 37°C. A negative control was also used (cells with no treatment). After the incubation, MTT was discarded and 150 µL of dimethyl sulfoxide (DMSO, Sigma Chemical Co., St. Louis, MO) was added to dissolve the produced formazan crystals. Absorbance at wavelength 540 nm was measured in a microplate reader, with a reference filter of 690 nm. The percentage of viability was determined as following:
o ia ility Experimental condition
egative Control 1
3.6.Immunofluorescence
For immunofluorescence assays cells were trypsinized as described above, counted and harvest in a 24 well plate coated with Poly-L-lysine (Sigma-Aldrich, U.S.A.) (PC12 cells), Poly-D-lysine (primary hippocampal neurons) (Sigma-Aldrich, U.S.A.) or non-coated glass coverslips (C8-D1A and bEnd.3 cells). After treatment, cells were washed 3 times (5 minutes) with PBS and fixed with 4% PFA in PBS/MP Buffer (Microtubule Protecting Buffer) for 10 minutes at RT in a shaker platform. Cells were washed again and permeabilized with Triton-X 100 0.1% in PBS, for 10 min at RT with agitation. Cells were washed and blocked (NGS 10% in PBS 1x) for 1h at RT in a shaker and further incubated for 1h with Alexa Fluor 488® phalloidin (1:150 in PBS 1x) (Gibco, Invitrogen), at RT in the dark. DAPI (Gibco, Invitrogen) or Hoechst 33342 (Gibco, Invitrogen) was added to the fluorescent mounting medium (Palex Medical, Spain) in order to visualize nuclei or count apoptotic nuclei, respectively. Images were captured using a Zeiss Axio Imager Z1 fluorescence microscope (Carl Zeiss, Germany).
3.7.Assessment of ATP Synthesis
The C8-D1A cell line ATP synthesis was evaluated by luminescence using the kit CellTiter-Glo® (Promega Corporation, U.S.A.), which signals metabolically active cells through the quantification of the ATP. This assay is based on a luciferase/luciferin reaction that, in the presence of ATP, produces oxyluciferin and releases energy in the form of luminescence. Since the luciferase reaction requires ATP, the luminescence produced is
31
proportional to the amount of ATP present, an indicator of cellular metabolic activity. Cells were incubated for 24 h in culture medium with the respective ALC and METH doses. After C8-D1A cell line treatments, CellTiter-Glo® reagent was added to each well in a 1:1 proportion to the cell culture medium. The mixture was transferred to 96-well opaque-walled plates and readings were performed in the plate reader (BioTek Synergy 2 Multi-mode Microplate Reader, U.S.A.) with an integration time of 1 second/well and values were analyzed with Gen5™ Data Analysis Software. Control wells were used to correct background luminescence interference. A standard curve per assay was generated.
3.8.Assessment of C8-D1A Igf1 and Rnd2 transcript levels by Real Time Polymerase Chain Reaction (RT-PCR)
Cells were seeded on petri dishes 100 mm (Orange Scientific) at a density of 4104 cells/mL. At 80% of confluence, ALC 0.1 or 1 mM alone or in combination with METH 1 mM was added and cells were incubated for 24 h. Total RNA was extracted using RNeasy® Mini Kit (Qiagen, Hilden, Germany) and its quality was confirmed by Experion automated electrophoresis system (Bio-Rad Hercules, CA, USA). Synthesis of cDNA was performed by Qiagen RT2 HT First Strand cDNA kit (Qiagen, Hilden, Germany). A total amount of 2.0 µg of RNA was used to study gene expression through iQ5 multicolor Real-Time PCR detection system (Bio-Rad). Based on mRNA sequences available on NCBI platform, primers were designed by Beacon Designer Software version 7.9 (Primer Biosoft International, Palo Alto, CA) with the following considerations: anneling temperature 60±2ºC; primer length 18-24 bp; final product length 100-200 bp and GC content 30-80%. Three housekeeping genes were tested (Gapdh, Rpl29 and Rps18). Primer sequences of genes studied are summarized in table 2.