1
Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Specific elimination of HIV-1 infected cells using a
Tat/Rev-dependent circuit
Pedro Ricardo Lucas Perdigão
Mestrado em Biologia Molecular e Genética
2
Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Specific elimination of HIV-1 infected cells using a
Tat/Rev-dependent circuit
Pedro Ricardo Lucas Perdigão
Mestrado em Biologia Molecular e Genética
Dissertação orientada pela Doutora Mariana Santa-Marta
e pela Professora Doutora Maria Filomena Caeiro
i
Agradecimentos (Acknowledgements)
Em primeiro lugar, gostaria de agradecer à Doutora Mariana Santa-Marta por me ter aceitado como seu aluno, por todos os conhecimentos transmitidos, por toda a paciência que foi necessária para me orientar, e por todo o apoio dado mesmo quando o trabalho não corria bem.
Ao Professor Doutor João Gonçalves, por me ter aceitado no seu laboratório, pela sua orientação ao longo deste trabalho, pela confiança que teve em mim e pelo incentivo. Agradeço também ao Professor Doutor Moniz-Pereira, por me ter acolhido no seu departamento.
À Prof. Doutora Filomena Caeiro, pela sua disponibilidade e preocupação demonstrada pelo meu trabalho, pela ajuda na avaliação da tese e também por me ter ajudado a dar os primeiros passos neste mundo que é a Virologia.
Quero agradecer aos meus colegas de laboratório, Soraia, Luís, Catarina, Cátia, Ana Catarina, Lídia, Andreia, Acilino, Inês, Lídia, Paula, e também aos antigos colegas que já não se encontram no grupo, Sylvie, Rita e André. Obrigado por toda a disponibilidade dada e paciência que tiveram para me transmitir conhecimentos, e pelo companheirismo que foram essenciais para ajudar a minha integração no grupo.
Ao Fede, pela ajuda na correcção da tese e pelas dicas dadas e que me ajudam a evoluir na arte da escrita científica.
Aos meus amigos, por todas as loucuras e grandes momentos que passámos, em que belas histórias ficaram para recordar. Eles sabem quem são. Obrigado por todo o apoio que sempre me deram.
Quero agradecer à minha família, pais e irmão, por todo apoio dado e por me incentivarem a querer ser sempre melhor. Por último, quero agradecer à minha avó, a quem eu dedico este trabalho. Obrigado por teres estado sempre presente na minha vida, e por me ajudares a tornar na pessoa que sou hoje. Espero que tenhas ficado e continues a ficar orgulhosa de mim.
ii
Abbreviations
Abs Absorbance
AIDS Acquired Immunodeficiency Syndrome
ALA Alanine
BSA Bovine serum albumine
CCR5 CC Chemokine Receptor 5
CD3 Cluster Differentiation 3
CD4 Cluster Designation 4
Cdk9 Cluster Designation 3
cDNA Complemenatary DNA
CMV Cytomegalovirus
CNS Central Nervous System
CPRG Chlorophenol red-ƒÀ-D-galactopyranoside CXCR4 CXC Chemokine Receptor 4
DC Dendritic Cells
DMEM Dulbecco's modified Eagle's medium
DNA Deoxyribonucleic acid
DsRed Discosoma sp. Red fluorescent protein
DTA Diphtheria toxin
E. Coli Escherichia coli
ECL Electrochemiluminescence
EDTA Ethylenediaminetetraacetic acid
ELISA Enzyme-Linked Imunosorbent Assay
Env Envelope protein
ER Endoplasmatic reticulum
FBS Fetal Bovine Serum
FDCs Follicular dendritic cells
FITC Fluorescein isothiocyanate
Gag Group-specific-antigen protein
GAPDH Glyceraldehyde-3-phosphate dehydrogenase
GCV Ganciclovir
GFP Green fluorescent protein
iii
gp41 Glycoprotein 41
HÁ Hemagglutinin
HAART Highly Active Antiretroviral Therapy
HEK Human Embryonic Kidney
HeLa-P4 HeLa-CD4-LTR-β-gal
HIV Human Immunodeficiency Virus
HIV-1 Human Immunodeficiency Virus type 1
HIV-2 Human Immunodeficiency Virus type 2
HLTR HIV-1 LTR target site 1
HRP Horseradish peroxidase
HSV1-TK Herpes simplex virus type 1 thymidine kinase
HSV-TK Herpes simplex virus thymidine kinase
IL-2 Interleukin-2
IL-7 Interleukin-7
IN Integrase
IRES Internal rybossome entry site
KRAB Kruppel-associated box domain
LB Lysogeny Broth
LTR Long-terminal repeats
Lys Lysine
MA Matrix
MOI Multiplicity of infection
mRNA Messenger RNA
NC Nucleocapsid
Nef HIV negative factor
NF-κB Nuclear factor kappa B
NLS Nuclear localization signal
PBS Primer-binding site
PBS Phosphate-Buffered Saline solution
PBS3 Primer-binding site 3
PCR Polymerase chain reaction
PE Phycoerythrin
PIC Pre-integration complex
Pol Polymerase
PR Protease
iv
Rev Regulator of Virion Protein
RNA Ribonucleic Acid
RRE Rev responsive element
RT Reverse transcriptase
SDS Sodium dodecyl sulfate
Sp1 Specificity protein 1
ssRNA Single-stranded RNA
SU Surface
SV40 Simian virus 40
T.A. Transcription activator
TAR Trans-activating response element
Tat Trans-activator of Transcription
TBP TATA-binding protein
TM Transmembrane
TNF-α Tumor necrosis factor-alpha
tRNA Transfer RNA
TU transduction units
Vif Viral Infectivity Factor
VP16 Herpes simplex virus protein 16
VP64 Herpes simplex virus protein 64
Vpr Viral protein R
Vpu Viral protein U
VSV-G Vesicular Stomatitis Virus glycoprotein
ZF Zinc-finger
v
Abstract
Despite the success of antiretroviral cocktails, a cure for HIV-1 remains elusive. This is mainly due to the existence of persistent cellular reservoirs infected with non-transcriptional, latent HIV-1. An effective treatment against HIV-1 would target both active and latent HIV-1-infected cells, and eliminate them without harming non-infected cells. In order to achieve this, we have developed a therapeutic plasmid with a Tat/Rev-dependent toxin expression. This vector contains a HIV-1 transcriptional activator to specifically activate latent infected cells. We confirmed the expression of all vector components necessary to achieve our goal. We then assessed the potential of this therapeutic plasmid to specifically eliminate infected cells by transduction of a T-lymphoma cell line that has a HIV-1 latent provirus integrated in its genome. We demonstrated that our therapeutic plasmid is not only capable of reactivating latent HIV-1 expression by itself, but it also can induce cell death efficiently and specifically in HIV-1 infected cells. Cell death was 60% higher in infected cells than in non-infected cells, indicating that our therapeutic vector was able to fulfill the objective proposed in this work. Therefore, the data presented here represents a promising approach for the development of a gene therapy that could become a viable and successful treatment against HIV-1.
Key-Words: Human immunodeficiency virus type 1 (HIV-1); Latency; Gene therapy; Tat;
vi
Resumo
O vírus da imunodeficiência humana (VIH) é o agente responsável pelo síndroma de imunodeficiência adquirida (SIDA), caracterizado por uma supressão do sistema imunitário levando à ocorrência de doença oportunistas. Desde a sua descoberta em 1983, o VIH terá sido responsável mais de 25 milhões de mortes em todo o mundo, sendo um dos temas mais investigados actualmente. O VIH é um lentivírus pertencente à família
Retroviridae. É um vírus de cadeia de RNA positiva que se encontra encapsidado e que
sofre transcrição reversa no interior da célula, originando um intermediário de DNA que é integrado na forma de um provírus no genoma da célula hospedeira. O genoma proviral do VIH codifica nove grelhas de leitura abertas das quais 3 codificam poliproteínas estruturais (Gag, GagPol e Env). Sendo o VIH um retrovírus complexo, este codifica ainda 6 proteínas acessórias responsáveis pela regulação e coordenação do seu fenótipo (Tat, Rev, Nef, Vpr, Vpx e Vif). A proteína Tat (transactivador da transcrição viral) é responsável pelo aumento da expressão do mRNA viral acima de níveis basais. A Tat liga-se a uma estrutura secundária presente no mRNA nascente, recrutando factores de transcrição que vão aumentar o elongamento do mesmo. A proteína Rev (proteína reguladora viral) tem um papel determinante no splicing e no transporte dos mRNAs virais para o citoplasma.
Apesar dos avanços no desenvolvimento de terapias contra o VIH-1 através do uso de fármacos antiretrovirais, estes não são capazes de eliminar completamente o vírus do organismo. O maior obstáculo à erradicação do VIH-1 é a existência de reservatórios virais persistentes no hospedeiro. Reservatórios virais são todas as células infectadas com VIH-1 que não são eliminadas do hospedeiro, principalmente devido à capacidade do VIH-1 estabelecer um estado de latência no interior da célula. A latência viral do VIH-1 é caracterizada como um estado de infecção não expressivo que permite a evasão do vírus à resposta do sistema imune, e à acção de fármacos antiretrovirais. Estes reservatórios podem servir como fonte de produção viral. Os principais reservatórios de VIH-1 são os linfócitos T CD4+ de memória, que possuem um tempo de meia-vida prolongado. Deste modo eliminar estes reservatórios virais é extremamente importante.
vii Um tratamento eficaz contra o VIH-1 deve ter como base o desenvolvimento de uma terapia que seja capaz de atingir todas as células infectadas com VIH-1, incluindo as células com VIH latente, sem causar danos a células não infectadas. Deste modo o desenvolvimento de terapias alternativas tal como a terapia génica, mostra especial interesse.
A terapia génica consiste na transferência de transgenes para células alvo promovendo a alteração do seu fenótipo. Até ao momento vários tipos de transgenes foram testadas no combate ao VIH-1 (toxinas, anticorpos, interferência de RNA, etc.). O uso de lentivírus como veículos para a transferência e expressão destes transgenes, apresentam diversas vantagens. Os lentivírus são capazes de infectar células que não se encontram activamente a dividir, e apresentam baixa imunogenecidade. No entanto o aparecimento de estirpes replicativamente competentes e mutantes de inserção são um problema. Deste modo, o uso de vectores lentivirais de terceira geração, em que os componentes lentivirais encontram-se divididos por 4 plasmídeos diferentes e a introdução de sinais específicos do tipo celular, permitem aumentar a especificidade e segurança dos vectores lentivirais.
O objectivo proposto para este trabalho é o desenvolvimento de uma nova estratégia terapêutica que nos permita eliminar especificamente células infectadas com VIH-1, incluindo os reservatórios latentes. Para atingir este objectivo foi construído um plasmídeo lentiviral que transporta um activador de transcrição (proteínas dedos de zinco e Tat) e uma toxina capaz de induzir a morte celular de células na presença da proteína viral Rev. Uma vez que pretendemos atacar células latentes, na qual a expressão de Rev é inexistente, colocámos no mesmo vector, um activador de transcrição. Este activará a expressão viral, forçando a saída de latência e expondo a célula latente à acção da toxina e consequente morte celular. Esta estratégia pretende promover a morte de células infectadas tomando partido do mecanismo de replicação do VIH-1.
De modo a testar esta estratégia, foi inicialmente construída uma proteína de fusão composta pelo domínio activador VP64 e uma proteína dedos de zinco. Foram seleccionadas duas proteínas de dedos de zinco com afinidade para o genoma viral. Estas construções foram testadas para avaliar a sua capacidade de activar a transcrição do VIH-1. Observámos que as proteínas dedos de zinco utilizadas não activaram a transcrição viral, chegando mesmo a inibi-la. Deste modo, determinámos que estas construções não tinham a capacidade de activar a transcrição do VIH-1 e decidimos utilizar a proteína Tat para assumir esta função. Uma vez que a Tat é o activador da transcrição viral do VIH, passámos directamente para o desenvolvimento do vector lentiviral. Utilizámos um vector lentiviral que possui um local de ligação da proteína Rev (RRE) como base para as nossas construções.
viii Este vector tem a particularidade de que, ao iniciar a sua transcrição, os seus mRNAs só são transportados para o citoplasma para tradução na presença de Rev, garantindo que células não infectadas não são afectadas pela expressão dos transgenes. Este vector possui ainda 2 splicing sites do VIH entre os quais foram clonados os genes das toxinas, em fusão com uma proteína repórter fluorescente verde. Estes só são expressos na presença de Rev que se liga aos locais de splicing do mRNA evitando a remoção desta região. Para a construção dos nossos vectores utilizámos a toxina da diphteria (DTA) e a timidina kinase do vírus herpes simplex (HSV-TK), como toxinas de eleição. A DTA promove a morte celular directamente, enquanto a HSV-TK precisa de presença do pró-fármaco Ganciclovir para matar a célula. Clonámos também a proteína Tat em conjunto com uma proteína repórter vermelha sob o controle de um promotor constitutivo, para activar a transcrição dos VIHs latentes e marcar células transduzidas com o nosso vector lentiviral.
Todos os componentes do vector encontram-se funcionais, com excepção da, proteína repórter vermelha. Ao testarmos a capacidade do nosso vector eliminar células infectadas num contexto replicativo e de latência, verificámos um aumento de quase 60 % de morte específica de células infectadas. Deste modo concluímos que este trabalho apresenta uma abordagem promissora para o desenvolvimento de uma nova estratégia de terapia genética contra o VIH-1.
Palavras-chave: Vírus da imunodeficiência humana (VIH-1); Latência; Terapia genica; Tat;
ix
Table of contents
Agradecimentos (Acknowledgements) ... i Abbreviations ...ii Abstract ... v Resumo ... vi Table of contents ... ix1. State of the art ... 1
1.1. Human Immunodeficiency Virus ... 1
1.1.1. HIV-1 genome and structure ... 2
1.1.2. HIV-1 replication cycle ... 3
1.1.2.1. The role of regulatory proteins Tat and Rev in HIV-1 replication ... 4
1.2. HIV-1 latency state ... 5
1.3. Therapeutic strategies against latent HIV-1 and its cellular reservoirs ... 5
1.3.1. Pharmacological strategies ... 5
1.3.2. Gene therapy strategies ... 6
2. Objectives ... 8
2.1. Outlined strategy ... 8
3. Methods ... 9
3.1. Bacteria strains ... 9
3.2. Plasmid DNA Propagation and Extraction ... 9
3.3. Plasmids and Constructions ... 9
3.4. Cell lines and culture conditions ... 11
3.5. Cell transfection ... 12
3.6. Western Blot ... 12
3.7. CPRG assay ... 13
x
3.9. Cell Transduction ... 13
3.10. Cell death and viability assay ... 14
3.11. Flow cytometry ... 14
4. Results and Discussion ... 15
4.1. ZF-VP64 potential for latent HIV-1 reactivation... 15
4.2. Development of a therapeutic lentiviral vector Tat/Rev-dependent ... 17
4.3. Reactivation of a HIV-1 latent cell line by transactivator Tat ... 24
4.4. Specific elimination of HIV-1 latent cells ... 25
5. Conclusions and future perspectives ... 28
6. References ... 30
1
1. State of the art
1.1.
Human Immunodeficiency Virus
The human immunodeficiency virus (HIV) is the causative agent of the acquired immunodeficiency syndrome (AIDS). AIDS is characterized by the suppression of the immune system in otherwise healthy individuals, leading to the emergence of opportunistic diseases that eventually kill the patients [1, 2, 3]. AIDS is currently one of the leading causes of death worldwide (World Health Organization Database, 2009). The syndrome was first described in 1981 but HIV was identified as the causative agent only in 1983 [4]. Two types of HIV have been identified thus far: HIV-1 and HIV-2. HIV-2 is considered less pathogenic than HIV-1, being associated with a slower rate of disease progression. While HIV-1 transmission is the main responsible for AIDS pandemic, HIV-2 is mainly restricted to West Africa, due to its lower capacity of transmission [5, 6, 7].
CD4+ T-helper lymphocytes and macrophages are the main targets of HIV-1 infection [15], which causes their destruction in less than two days [8, 9, 10]. HIV-1 is also capable of infecting dendritic cells (DC) [11], natural killer-T cells [12] and hematopoietic progenitor cells [13]. The central nervous system (CNS) is also affected by HIV-1 invasion, as the virus is capable of surpassing the blood-brain barrier and infecting microglial cells [14]. Cellular tropism of HIV-1 is mostly determined by the presence of specific cell surface receptors such as CD4 and CCR5 and CXCR4 that allows binding and entrance of the virus into target cells [15].
Since the finding of HIV-1 as the main cause of AIDS, several therapies have been developed in order to suppress the replication of the virus and eliminate it. Currently, the most efficient therapy is the highly active anti-retroviral therapy (HAART), a selected combination of three or more antiretroviral drugs that target several steps of the viral replication cycle. HAART causes a striking decrease in plasma virus levels below the limit of detection of clinical assays, improving significantly the quality of life and the life span of infected patients [16, 17]. However, HAART fails to eradicate HIV-1 from the organism and cure the infection. This is mainly due to the appearance of resistant HIV-1 strains, and most importantly to the fact that anti-retrovirals target only actively replicating virus. Latent (non-replicative) HIV-1 hidden in cellular reservoirs escape from anti-retroviral therapy and become an ilimited source of new replicating viruses.
2
1.1.1. HIV-1 genome and structure
HIV is a member of the Lentivirus group, being part of the Retroviridae family. Viruses from this group are characterized by the presence of a reverse transcriptase (RT) and an integrase (IN). The RT catalyzes the transcription of the genomic RNA into a proviral DNA, and the integrase promotes the integration of the viral DNA into the host cell genome [18].
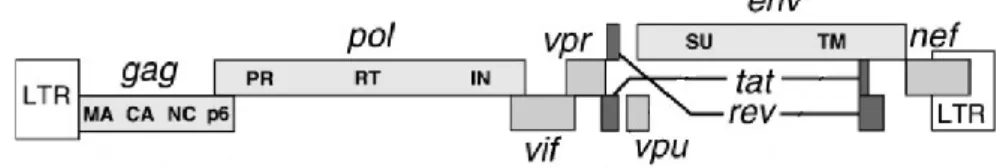
HIV-1 genome consists in a 9.8 Kb single-stranded positive RNA molecule, flanked terminal repeats that are converted to 5’ and 3’ long-terminal repeats (LTRs), after the RNA is reverse transcribed to cDNA. The 5’ LTR contains the viral promoter for transcription. The HIV-1 genome encodes for open reading frames (Figure 1). These can be classified into essential genes (gag, pol and env) and accessory genes (tat, rev, vpu, vpr, vif and nef). The
gag, pol and env genes encode for polyproteins which are then proteolyzed into individual
proteins. gag (group-specific antigen) region codes for the structural proteins – MA (matrix), CA (capsid), NC (nucleocapsid), and p6. pol (polymerase) region encodes for essential enzymes participating in the HIV replication cycle – Protease (PR), Reverse Transcriptase (RT) and Integrase (IN). env (envelope) region encode glycoproteins present in the outer membrane of the virion that mediate binding and entrance into host cells – SU surface/gp120 (SU) and transmembranar/gp41 (TM) [19, 20].
Figure 1- Organization of the HIV-1 genome. Source: Frankel, A. D. and Young, J. A. (1998) Annu Rev
Biochem,67
The mature HIV-1 virion structure (Figure 2) is constituted by a capsid particle, encapsidating 2 copies of RNA genome (complexed with several units of nucleocapsid protein), and also the essential enzymes protease, reverse-transcriptase and integrase. The capsid is stabilized by a shell structure consisted of various copies of the matrix protein. This shelllines up the capsid with the viral envelope, a lipid bilayer containing the surface glycoprotein gp120 anchored to the capsid by the transmembranar protein gp41. Accessory proteins Vif, Nef and Vpr are
3
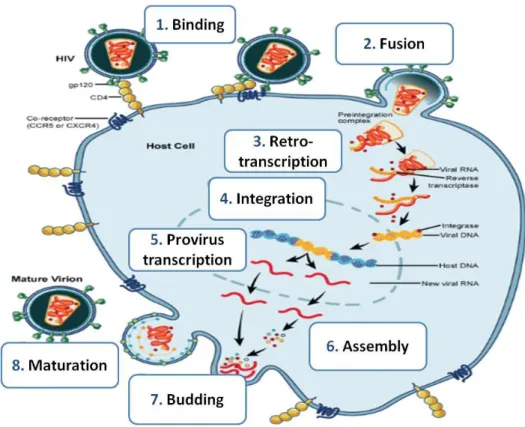
1.1.2. HIV-1 replication cycle
HIV-1 replication events (Figure 3) can be divided into 2 distinct stages: early and late phase. The early phase of HIV-1 replication begins with the binding of the mature virion to the cell surface. Binding occurs by specific interactions between the viral glycoprotein gp120 and the amino-terminal immunoglobulin domain of the cellular CD4 molecule, exposing the CCR5 or CXCR4 co-receptors, which are natural receptors for host´s cytokines [21,22]. As mentioned above, these co-receptors determine the cell tropism of HIV-1 [23]. The interaction of gp120 with these co-receptors causes conformational changes in the transmembranar protein gp41 that, in turn, exposes an N-terminal hydrophobic region (“fusion peptide”). Exposure of this region induces the fusion of viral and cellular membranes, and the subsequent entry of the virion core into the cell cytosol [20,21,24].
Once inside the cell, capsid proteins are dissociated from the viral core, which is converted to a reverse-transcription complex by a process called “uncoating”. In this complex the RNA genome remains associated with the nucleocapsid, matrix, reverse-transcriptase, integrase and the accessory protein Vpr[20,21]. Following uncoating, the single-stranded RNA(+) genome is converted in the cytosol to a double-stranded linear DNA molecule by the reverse-transcriptase [18,21]. Reverse-transcription of the RNA genome is initiated after the binding of a cellular tRNALys primer [22].
The newly formed cDNA molecule is transported to the nucleus as part of the pre-integrative complex containing the RT, IN, MA and Vpr proteins [22]. This nuclear import is assisted by Vpr, which connects the complex to the nuclear import machinery [25]. The viral cDNA is then integrated into the host cell chromosome by the viral integrase, thus concluding the early phase of infection.
The late phase of infection begins with the transcription of the provirus by the host RNA Polymerase II. Tat has an essential role in this stage, activating the 5’ LTR promoter and thereby enhancing the rate of HIV-1 transcription and elongation [20]. Only Tat, Rev and Nef are initially expressed by means of alternative multiple splicing of the HIV-1 transcript. Rev regulates this step and promotes export of single-spliced and unspliced RNA to the cell cytosol [20,21]. Once Rev is expressed, single-spliced RNAs (env, vif, vpu and vpr) and unspliced RNAs (gag and gagpol) are translated in the cytoplasm [21].
Gag and GagPol polyproteins are transported to the cell membrane where assembly of new viral particles occurs. Envelope proteins gp120 and gp41 are directed to the membrane through the ER-Golgi pathway. HIV-1 full-transcript RNA is encapsidated and immature virions are released from the cell. The viral protease triggers subsequently the
4 maturation of virions, promoting the reorganization of their core and enabling them to infect new cells [21, 22].
Figure 3- HIV-1 replication cycle. Adapted from NAID, NIH.
1.1.2.1. The role of regulatory proteins Tat and Rev in HIV-1 replication
Several accessory proteins contribute to the efficiency of HIV-1 infection, replication and spread [21]. However, Tat and Rev are especially relevant for HIV-1 replication and the present work.
The 5’ LTR HIV-1 promoter contains binding sites for several transcription factors, such as NF-κB, Sp1 and TBP. This contributes to the existence of a basal transcriptional activity of the viral promoter. However, the transcriptional transactivator Tat is the actual responsible for the bulk of viral transcription, more than two orders of magnitude higher than basal transcription [20, 21]. Tat binds the trans-activating response element (TAR) stem-loop located at 5’ end of the nascent RNA chain and recruits host proteins that promote the assembly of the transcriptional complex and stimulate the elongation catalyzed by the RNA polymerase II [22, 26].
Rev also has an essential role in HIV-1 replication, regulating the RNA export of single-spliced and unspliced transcripts from the nucleus to the cytoplasm. Rev binds to a loop structure named Rev-responsive element (RRE) located at the 3’ of RNA transcripts and
5 interacts with nucleoporin-like proteins at the nuclear pores [20, 21, 27]. These interactions allow Rev to export the RNA transcripts to the cytosol without being spliced. Besides promoting export of single-spliced and unspliced RNA, Rev can also directly inhibit splicing by preventing spliceosome assembly [20, 28].
1.2.
HIV-1 latency state
HIV-1 latency is defined as a stably integrated silent provirus that does not replicate in normal conditions but can serve as a source of new replicating virus [29]. Since viral expression is abolished, latent HIV-1 is able to evade the host immune response and the anti-retroviral drugs, leading to the development of persistent cellular reservoirs hosting HIV-1 [HIV-16, HIV-17, 29].
The main reservoirs of latent HIV-1 are resting memory CD4+ T-cells, established early during primary infection. Their frequency is rare, in the order of 1 in 106 resting CD4+ cells [16, 17, 29]. Latency state is generally achieved when an infected CD4+ lymphoblast reverts back to a resting memory state and HIV-1 expression is extinguished [30]. Latent integrated proviral DNA is highly persistent in resting memory T-cells, being able to induce viral replication after T-cell activation [31]. Other cell lineages such as monocytes-macrophages, monocytes-macrophages, astrocytes, follicular dendritic cells (FDCs) and hematopoietic progenitor cells can also harbor stably integrated latent HIV-1 [32].
The estimated half-life of HIV latent reservoirs is longer than the average life span of the patients [30], pointing out the fact that HAART must be maintained for life. Treatment interruptions result in the recovery of viral replication and consequently disease progression. HIV-1 latency is considered as the main barrier for complete eradication of the virus from the infected host. HAART only targets actively replicating virus, and therefore there is a need for development of a therapy that can target latent HIV and its cellular reservoirs.
1.3.
Therapeutic strategies against latent HIV-1 and its cellular reservoirs
1.3.1. Pharmacological strategies
The mechanisms involved in HIV-1 latency have been extensively studied [29, 31]. At least in theory, these mechanisms could be manipulated pharmacologically in order to reactivate latent HIV-1 and expose it to anti-retroviral treatments. Three categories of agents have been tested: T-cell activators, such as the combination of interleukin-2 (IL-2) with antibodies against CD3, Interleukin-7 (IL-7) and Prostatin [33-35]; inhibitors of histone-modifying enzymes, such as valproic acid [36]; and inhibitors of DNA methylation, such as 5-azacytidine [31, 37, 38, 39]. Despite the evidence that these drugs could induce latent viral expression, none of the strategies mentioned above eradicated latent HIV from cellular
6 reservoirs, and some of them were associated with undesirable generalized immune activation [29, 31, 39]. These limitations highlight the need for alternative strategies against latent HIV-1 and its cellular reservoirs.
1.3.2. Gene therapy strategies
Gene-based therapy is a promising alternative to treat HIV-1 infection. Gene therapy strategies against HIV-1 consisted in delivering transgenes into HIV-1-susceptible cells in order to inhibit HIV-1 replication, turn them resistant to infection or immunize against HIV-1 antigens [40]. Other potential therapeutic strategies based on gene targeting have been developed to date, such as protein-based therapies (transdominant negative mutants, toxins, single-chain antibodies, zinc-fingers proteins, etc.), and RNA-based strategies (antisense, ribozyme, RNA aptamers, RNA interference) [40, 41]. However, these strategies failed to confer complete inhibition of HIV-1 replication for a sustained period of time, due to the emergence of resistant mutants and various efficacy and safety issues.
Zinc-finger chimeras are of particular interest in gene therapy treatment, due to their capability to bind DNA in a specific and efficient manner. A zinc-finger consists of a ββα fold coordinated by a zinc ion, with the α-helix making specific contact with 3-4 nucleotides [42]. Critical amino acids of a zinc-finger can be mutated to enable the altered protein domain to recognize any given combination of nucleotide sequences. Artificial zinc-fingers can therefore be designed to target any DNA sequence, possessing a great potential for gene targeting and regulation [43, 44].
Other interesting approach for gene therapy is the delivery of toxins or suicidal genes to target cells, an approach regularly used to target cancer cells [45]. Diphtheria toxin (DTA) and thymidine kinase from herpes simplex virus (HSV-TK) are examples of these killing agents. While diphtheria toxin kills cells directly, by inhibiting protein expression and inducing cell apoptosis. HSV-TK induces cell death only in the presence of the pro-drug Ganciclovir (GCV). HSV-TK converts ganciclovir in highly-toxic triphosphates that inhibit the DNA Polymerase and leads to apoptosis [46]. The high efficiency of these toxins makes them useful tools for the elimination of unwanted cells targeted by gene delivery.
Nevertheless, the main key to the successful application of gene therapy is the use of an efficient, specific and non-toxic gene delivery system. Currently the most efficient gene delivery system is cell transduction by viruses. Viral particles can be manipulated to express therapeutic genes or to infect and replicate specifically in certain cells. Furthermore, last generation viruses incorporate various properties that make them remarkably safe and cause relatively low immune activation. The choice of virus depends largely on the target cell and on the requirement for either transient or long-term gene expression [47].
7 Lentivirus-based delivery systems are of particular interest in gene therapy. Lentivirus based therapies have the advantage of being capable to efficiently modify and deliver genes into dividing and non-dividing cells. Lentiviral vectors have been successfully used as vehicle for therapy in lymphocytes, monocytes, stem cells and cells in the central nervous system [43], all of them important reservoirs of HIV-1. HIV-1-based vectors have the advantage over other vectors by containing HIV-1 cis-acting elements within the vector. These elements could enhance an anti-HIV effect by combination of the vector transgene effect and competition with wild-type HIV-1. However they present two main drawbacks, namely the potential occurrence of insertional mutagenesis and production of emergent replication-competent lentivirus (RCL), that could generate new human pathogens [40, 48, 49].
In order to avoid these obstacles, several modifications have been included in the third generation of lentivirus packaging systems (Figure 5).
Figure 4- Third generation of lentiviral packaging system. Sinn, P.L. et al. Gene Therapy (2005), 12
Third generation systems consist of a split-genome packaging system in which the
rev gene is expressed from a separate plasmid and the 5’ LTR from the transfer vector is
replaced by a strong Tat-independent constitutive promoter. Packaging signal (ψ) and unnecessary accessory genes are deleted from the packaging vector. These modifications reduce the probability of RCL generation, since only three HIV-1 encoded proteins (Gag, Pol, and Rev) are required for vector production [40, 48, 49, 50].
8
2. Objectives
The overall aim of the present work is to develop a strategy to eliminate specifically HIV-1-infected cells, including latent reservoirs. Such strategy will be based on the delivery of toxins into cells using a non-integrative lentiviral vector. The vector will be designed in order to ensure that the toxins will kill only those cells where Rev is present, i.e. those cells infected by HIV-1. In other words, we will take advantage of the replication mechanism of HIV-1 to kill specifically infected cells, without harming non-infected cells.
2.1.
Outlined strategy
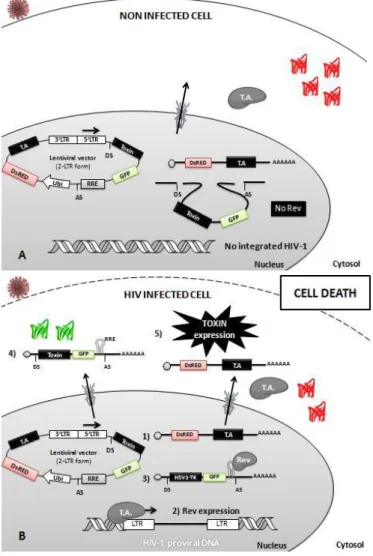
The strategy outlined for this work is based on a toxin delivery system promoted by a non-integrative lentiviral vector expressing a toxin. We will take advantage of the replication mechanism of HIV-1, by exploit a Tat/Rev-dependent circuit for specific elimination of infected cells, without harming non-infected cells. Proposed strategy is presented in figure 6.
Figure 5- Schematic representation of the strategy proposed for cell-specific elimination of HIV-1 infected cells. Cells will be transduced with a
nonintegrative lentivirus. This vector will contain the Rev Responsive element-RRE, along with a toxin and GFP genes, between two HIV-1 splicing sites under the control of HIV-1 Tat-responsive LTR. A transcription activator (T.A.) will be placed downstream of HIV-1 splicing sites along with a DsRed reporter protein. A) schematic representation of the transduction outcome in uninfected cells. Transduced cells will express
constitutively DsRed and T.A. In the absence of Rev, the toxin and the GFP genes will be excised by splicing due to the presence of HIV-1 splicing sites. B) schematic representation of the transduction outcome in HIV infected cells. In HIV-1
infected cells, T.A. expression will promote HIV-1 transcription leading to Tat and Rev expression. Rev will allow the export of the unspliced mRNA to the cytoplasm, avoiding the removal of the Toxin-IRES-GFP cassette positioned between the HIV-1 splicing sites. Therefore, toxin is expressed promoting the killing of infected cells.
9
3. Methods
3.1. Bacteria strains
Escherichia Coli (E Coli) JM109 (New England Biolabs, USA) or E. Coli STBL3
bacteria (Invitrogen, USA) were used for all plasmid propagation and cloning. Both strains are recA- preventing unwanted recombination between plasmid and chromosomal bacterial DNA. Genotypes are shown in table I (annexes).
3.2. Plasmid DNA Propagation and Extraction
All plasmids were transformed in bacteria by electroporation (V= 1.80 Volt, R= 200 Ω, Cap= 25 µFD), in 1 mm cuvettes. Transformed bacteria were cultured in Lysogeny broth (LB) medium supplemented with 100 μg/mL ampicillin (USB, Cleveland, USA). Lentiviral vectors and plasmids used for virus production were grown at 30 ˚C with agitation at 220 r.p.m., with the exception of lentiviral vectors derived from pNL-GFP-RRE, which were grown at 25 ˚C and 125 r.p.m., to avoid unwanted recombination. All other plasmids were grown at 37 ˚C with agitation (220 r.p.m.). Plasmids were extracted by miniprep using the ZR Plasmid MiniprepTM-Classic Kit (Zymo Research, USA), or by midiprep using the JETStar 2.0 Plasmid Purification MIDI Kit (Genomed, Portugal) or Invisorb Plasmid Midi Kit (Invitek, Berlin). Extraction of plasmids for clone screening was carried out by miniprep preparation by alkaline lysis, following the Maniatis protocol [80].
3.3. Plasmids and Constructions
pNL4-3 plasmid (Figure 17, Annexes) contains a full-length HIV-1 genome for the production of infectious and replicative viral particles [51]. pMtat(-)is a Tat deficient HIV-1 plasmid.it contains a full length HXB2 proviral HIV-1 genome with a mutation on the initiation codon of tat [52]. pNL4-3 and pMtat(-) were provided by NIH AIDS Research and Reference Reagent Program, USA.
Plasmids used for lentivirus production such as packaging plasmid pCMV-GagPol [69] expressing HIV-1 GagPol proteins including fully functional Integrase, and Rev-expressing pCMV-Rev (Figure 22, Annexes) [53] were provided by NIH AIDS Research and Reference Reagent Program, USA. pCMV-VSVG (Addgene, USA) [54] expresses vesicular stomatitis virus (VSV-G) envelope glycoprotein.
All plasmid constructions were carried out by DNA digestion with enzymes from Fermentas (Canada). T4 DNA Ligase (Fermentas, Canada) was used in all vector-insert ligations. Polymerase Chain Reaction (PCR) was performed in Doppio thermocycler (VWR International, USA) using Phusion High-Fidelity DNA Polymerase (Finnzymews, Finland),
10 according to the manufacture’s instruction. All primers used for PCR reactions are presented in table 1 in annexes. All PCR conditions programs used are presented in table 2 in annexes. Clones were screened by digestion with appropriate restriction enzymes and resolved by agarose gel electrophoresis. Positive clones sequence was confirmed by DNA standard sequencing (Macrogen, Netherland).
pALA-VP64 is a pcDNA 3.1 backbone vector expressing a 5 finger zinc-finger protein bearing Alanines in the recognizing alpha-helix fused to a VP64 activator domain protein [55]. For the construction of the pHLTR3-VP64 and pPBS3-VP64, HLTR3 and PBS3 were removed from pSIN-KRAB-HLTR3 [56] and pSIN-SKD-PBS3 [42], by digestion with SfiI, respectively. The resulting fragments were gel extracted and cloned in pALA-VP64 SfiI digested, substituting the ALA zinc finger. pALA-VP64, pSIN-KRAB-HLTR3 and pSIN-SKD-PBS3 were kindly provided by Dr. Carlos Barbas III.
pNL-GFP-RRE(SA) (Fig. 24, Annexes), a Rev-dependent expression vector was used as the backbone vector for the cloning of DTA-GFP-RRE (pNLGR-DTA) and pNL-TK-GFP-RRE (pNLGR-TK), and was kindly provided by Dr. Jon W. Marsh [27]. pNLGR-DTA and pNLGR-TK were constructed by insertion of a DTA and a HSV1-TK fragments PCR amplified in pNL-GFP-RRE, respectively. DTA was PCR amplified from FuW-DTA [57] using the primers N-DTA-BamHI F and N-DTA-SalI R (PCR Conditions: Program B); and HSV1-TK was PCR amplified from FuW DsRed-TK [57] using the primers BamHI F and N-TK-SalI R) (PCR Conditions: Program B). PCR fragments were gel purified, digested with BamHI and SalI, and inserted into the BamHI and SalI sites of the pNL-GFP-RRE(SA), multiple cloning site (MCS).
FuW DsRed-IRES-Tat (FuW DsRed-Tat) was constructed by insertion of a DdRed-IRES-Tat cassette into the lentiviral vector FugW (FuW-GFP) (Figure 20, Annexes) from AddGene (USA) [58]. DsRed-IRES-Tat cassette was generated by overlap-extension PCR, a PCR variant method that allows the construction of a fusion protein by PCR. The DsRed-IRES and the tat fragment were initially amplified separately, with overlapping primers. DsRed-IRES fragment was amplified from pIRES2 DsRed-Express2 vector (Figure 19, Annexes) (BD Biosciences Clontech, USA) using the primers DsRed-BamHI F and N-IRES-Tat R (PCR Conditions: Program A). tat gene was amplified from pCMV-Tat from (Figure 21, Annexes) NIH AIDS Research and Reference Reagent Program (USA) using primers N-Tat-F and Reverse N-Tat-EcoRI R (PCR Conditions: Program A). Fragments were purified and used as substrate for the second PCR reaction, were DsRes-IRES and Tat are joined together during 9 cycles of amplification in the absence of primers. This first round of PCR that allows the hybridization of the complementary sequences were performed using Program D. We then added a pair of external primers (N-DsRed-BamHI F and N-IRES-Tat
11 R) and performed another 30 cycles of PCR to amplify the final product(PCR Conditions: Program B). PCR product was purified, digested with (BamHI/EcoRI) and cloned into FugW.
For the construction of the lentiviral vectors, pNLGR-DTA-DsRed-IRES-Tat (pNLGR-DTA-Tat) and pNLGR-TK-DsRed-IRES-Tat (pNLGR-TK-Tat), we performed a ligation of three fragment: the pNLGR vector BamHI and EcoRI digested; The (pUb)-DsRed-IRES-Tat fragment PCR amplified from FuW DsRed-Tat using primers pUb-NheI F and N-Tat-EcoRI R (PCR Conditions: Program C); and digested with NheI and EcoRI; and the DTA-GFP-RRE fragment amplified from pNLGR-DTA using the primers DTA-BamHI F and N-RRE-BamHI R (PCR Conditions: Program C); or the TK-GFP-RRE amplified from pNLGR-TK using the primers N-pNLGR-TK-BamHI F and N-RRE-BamHI R (PCR Conditions: Program C). digested with BamHI and NheI.
pCMV-GagPol D116N is a vector expressing HIV-1 GagPol proteins with a defective mutant Integrase, that unables the lentiviral vector to integrate in the cell genome. Construction of pCMV-GagPol D116N was achieved by cloning a PCR fragment of the GagPol region of NLENG1-ES-IRES D116N [59] provided by Dr. David Levy using the primers N-GagPol-NotI F and N-GagPol-XhoI R, into the NotI-XhoI sites of the pcDNA 3.1 Zeo(+) (Figure 18, Annexes) from Invitrogen (USA), possessing the Cytomegalovirus (CMV) promoter.
3.4. Cell lines and culture conditions
Human embryonic kidney (HEK) 293T (ATCC, USA) is a highly transfectable cell line derived of the 293 cell line into which the temperature sensitive gene for SV40 T-antigen was inserted. Human cervical epithelial carcinoma derived cell line HeLa-P4 (HeLa-CD4-LTR-β-gal, AIDS Reagent, MD, USA,) contains the β-gal gene under the control of HIV Tat-responsive LTR promoter, allowing expression of β-gal in presence of Tat or other LTR-specific transcription activator [60]. HEK 293T and HeLa-P4 cell lines were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10 % (v/v) heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/mL Penicillin, 100 μg/mL Streptomycin, and 0.25 μg/mL Amphotericin B (DMEM-FBS10).
Jurkat E6-1 (NIH AIDS Research and Reference Reagent Program, USA) is a human CD4+ T leukemia cell line [61]. J-Lat Full Length Clone 10.6 (J-Lat 10.6) is a Jurkat derived cell line that poorly expresses a stably integrated HIV-1deltaEnv-GFP provirus. In the presence of TNF-α (10 ng/mL) or other specific T cell activators EGFP and viral expression is induced [79]. Both Jurkat and J-Lat 10.6 cell lines were provided by NIH AIDS Research and Reference Reagent Program, USA and were cultured at a density of 1 x 106 cells/ml of RPMI-1640 medium supplemented with 10 % (v/v) heat-inactivated fetal bovine
12 serum, 2 mM L-glutamine, 100 U/mL Penicillin, 100 μg/mL Streptomycin, and 0,25 μg/mL Amphotericin B (RPMI-FBS10).
All cell lines were cultivated in T75 tissue culture flasks (75 cm3) (Orange Scientific, Belgium), at 37° C with 5 % CO2. Every cell culture media and reagents, otherwise indicated, were from Lonza (Basel, Switzerland).
3.5. Cell transfection
HEK293T cells were transfected by the calcium phosphate method [62]. 5 x 105 cells were seeded in each well of 6-well plates (34,7 mm Ø) (Orange Scientific, Belgium). Twenty-four hours after, cells were transfected with 5 µg of total DNA according to protocol. Cell medium was changed the next day and 48 hours after transfection cells were analyzed.
For the transfection of HeLa-P4 cells, 8 x 104 cells per well were seeded in 24-well plates (14,5 mm Ø) (Orange Scientific, Belgium). The next day cells were transfected with 2 ug of total DNA, using the FuGENE HD reagent (Promega) and 48 hours after transfection cells were analyzed.
3.6. Western Blot
HEK293T transfected cells were washed twice with phosphate-buffer saline (PBS) solution and lysed with a RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1 % NP-40, 1 % Sodium Deoxycholate, 0.1 % SDS, H2O) supplemented with complete EDTA-free
Protease Inhibitor Cocktail Tablets (Roche, USA) on ice. Thirty minutes after, cells were centrifuged (14 800 rpm, 30 min., 4 ˚C) and cell supernatant was recovered. Total protein was quantified by the Bradford method [63], using the Quick Start™ Bradford Protein Assay (Bio-Rad, USA). The “Mini-PROTEAN Tetra Electrophoresis System” (Bio-Rad, USA) was used for running and transfer. Fifty µg of total protein of each sample denatured at 95 ˚C for 10 min, was resolved in a 4-12% SDS-polyacrylamide gel, at 120 V. Proteins were electrotransferred into a nitrocellulose membrane (250 mA, 60 min.). Membrane was blocked with a 5 % milk-PBS 0.2 % Tween20 solution for 60 min and proteins were detected as follow. HLTR3-VP64 and PBS3-VP64 proteins were detected using a Rat α-HA monoclonal antibody conjugated with HRP (Clone 3F10, Roche Applied Science, Germany), diluted 1:5000 in 3 % milk-PBS 0.2 % Tween20, for 60 min. at room temperature with agitation. HIV-1 GagPol capsid p24 precursors proteins was detected with a mouse α-p24 (Clone HIV-183-HHIV-12- 183-H12-5C, NIH AIDS Research and Reference Reagent Program, USA), diluted 1:10000 in 3 % milk-PBS 0.2 % Tween20 overnight at 4 ˚C. Endogenous control GAPDH was detected using a a mouse α-GADPH (Clone 6c5, Santa Cruz Biotechnology, USA) (diluted 1:5000 in 3 % milk-PBS 0.2 % Tween20) overnight at 4 ˚C. Primary antibody was washed, and incubated with a goat α-mouse IgG conjugated with HRP (clone Bio-Rad, USA) secondary antibody
13 (diluted 1:40000 in 3 % milk-PBS 0.2 % Tween20) for 60 min. at room temperature. Membrane was washed with PBS 0.2 % Tween20. Antibody detection was made with ImmobilonTM Western Chemiluminescent HRP substrate (Millipore, USA). Membranes were incubated with HRP substrate for 3 min. at room temperature and then revealed in a chemiluminescence film Amersham HyperfilmTM ECL (GE Healthcare, UK).
3.7. CPRG assay
Detection of β-galactosidase (β-gal) expression in HeLa-P4 cells was carried out by a Chlorophenol red-β-D-galactopyranoside (CPRG) colorimetric assay (Roche, Germany). Once expressed, β-gal catalyzes the hydrolysis of the yellow-orange CPRG, converting it into the chromophore chlorophenol red. Chlorophenol red production yields a dark red solution, and can be detected by reading its absorbance at 550 nm. 48 hours after transfection of HeLa P4 cells, cells were incubated with a CPRG lysis buffer (1 % NP-40, 5 mM MgCl2,
PBS) at room temperature for 15 min. CPRG substrate (6 mM in lysis buffer) was added to the samples, and β-gal expression was measured in an Infinite M200 plate reader (Tecan).
3.8. Virus production
Viruses were generated by transfection of HEK 293T cells by the calcium phosphate method [62]. 3 x 106 cells were seeded in 10 cm plates (Orange Scientific, Belgium) 24 hours prior to transfection. Cells were transfected with 10 μg of lentiviral vector, 7 μg of packaging plasmid pCMV-GagPol (Functional Integrase) or pCMV-GagPol D116N (Non-functional Integrase), 3 μg of pCMV-Rev for nuclear exportation of the RRE-bearing unspliced lentiviral mRNA to the cytosol, and 2 μg of pseudo-typed envelope expressing plasmid pCMV-VSVG. Next day cell medium was replaced and, 48 hours after transfection, cell supernatant was collected. Viral suspension was clarified by centrifugation at 1500 rpm for 10 min and viruses were concentrated by Lenti-X concentrator reagent (Clontech).
Viral production was quantified by a p24 capture ELISA assay, (NCI-Frederick Cancer Research and Development Center – AIDS Vaccine Program kit (NIH, USA)) according to manufacturer’s instructions. Lentiviral transduction units (TU) were determined using the 1 pg of p24 protein 10 TU relation.
3.9. Cell Transduction
For the transduction of 2,5 x 105 of Jurkat cells we used 2 TU/cell (50 ng of p24) in 500 µL of RPMI-FBS10 medium. Cells were transduced by centrifuging the plates at 220 rpm
for 60 min at 22 ˚C, in order to deposit virions on the cell surface and facilitate adsorption. Plates were incubated at 37 ˚C for 6 hours, after what medium was changed.
14
3.10. Cell death and viability assay
Cell death analysis was carried out using LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen, USA). This assay uses a fluorescent non-permeable dye that reacts with cellular amines. Since these dyes cannot penetrate intact cell membranes, only cell surface proteins react with the dye, resulting in a relatively dim staining. In dead cells, the compromised membrane, allows the dye to enter and stain the interior of the cell as well as the cell surface, increasing the cell staining and allowing discrimination between live and dead cell populations. Since these dyes react covalently with proteins, this discrimination is maintained after fixation of the cells.
To measure cell death and viability, approximately 1 x 106 cells were washed with PBS and ressuspended in 1 mL of PBS with the fluorescent reactive dye (1:1000) at 4 ˚C for 30 min. Cells were washed twice with PBS-1 % BSA, and fixated with PBS-1 % Paraformaldehyde. Cell death was assessed by analyzing the cell population fraction that was highly stained with the violet fluorescent dye using flow cytometry (λ Ex. 405 nm/λ Em. 450 nm).
3.11. Flow cytometry
Cells were washed twice with PBS and then fixated with PBS-1 % Paraformaldehyde. Unstained and single-stained cells were used for parameters adjustment and compensation between fluorochrome signals. GFP and DsRed expression was detected by excitation with blue laser (488 nm) and detection in FITC (530/30 nm) and PE (585/42 nm) filter, respectively. Dead cells were detected by excitation with violet laser (405 nm) and detection with Pacific Blue Filter (450/40 nm). Flow cytometry analysis was performed in a LRS Fortessa (BD Biosciences, USA), by acquirement of 10,000-gated events from each sample. Data were analyzed using FlowJo software (Tree Star, USA).
15
4. Results and Discussion
4.1.
ZF-VP64 potential for latent HIV-1 reactivationFor the development of our therapeutic plasmid we initially constructed several zinc-finger fusion proteins, designed to target specifically the HIV-1 LTR promoter and reactivate viral transcription. We used HLTR3 and PBS3 zinc finger proteins, which bind specifically to conserved regions of the LTR promoter (figure 14 of Annexes) [42, 56], and VP64 as our activation domain. VP64 is a synthetic tetramer of the herpes simplex virus protein VP16 which have been shown to confer transcriptional control to zinc finger proteins [64]. We initially choose ZF-VP64 as our transcriptional activator instead of HIV-1 viral protein Tat in order to avoid unspecific activation of cells, as we expected ZFs to target more specifically HIV-1 proviral DNA. A short nuclear localization signal (NLS) rich in basic amino acids was added to the ZF-VP64 protein to direct the chimera to the nucleus. Additionally, an a Human influenza hemagglutinin (HA) tag was added at the N-terminus for protein detection and downstream applications [65].
Figure 6- Development of an artificial transcriptional activator to reactivate HIV-1 latent transcription. A)
Schematic representation of ZF-VP64 constructions . ZF-VP64 expression is under the control of the CMV
promoter. ZF-VP64 proteins possess a nuclear localization sequence (NLS) for nuclear targeting and a Human influenza hemagglutinin (HA) tag for protein detection. B) Detection of ZF-VP64 chimera protein by Western Blot. HEK293T cells were transfected with 5 µg of pHLTR3-VP64 (clones 5 and 9) and pPBS3-VP64 (clones 1 and 6). Forty-eight hours after transfection, cellular extracts were run in a 12 % polyacrylamide gel and transferred to a nitrocellulose membrane. HLTR3-VP64 and PBS3-VP64 proteins were detected with an anti-HA antibody. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal loading control.
Figure 6.A shows the constructions developed and used to evaluate the capability of ZF-VP64 to reactivate latent HIV (described in Methods). HLTR3 and PBS3 are zinc-fingers that target the HIV-1 genome. ALA is a non-specific DNA binder zinc-finger protein and was used as a negative control for all the assays. pHLTR3-VP64 and pPBS3-VP64 were constructed from pALA-VP64 (see Methods). After construction of both plasmids, two clones of each were selected and sequenced. HEK293T cells were transfected with HLTR3-VP64
16 clone 5 and 9 and PBS3-VP64 clones 1 and 6 and their expression was confirmed by Western Blot (Figure 6.B).
The ability of ZF-VP64 proteins to reactivate silent HIV-1 viruses was assessed by: 1) transfection of ZF-VP64 plasmids or pCMV-Tat (positive control) on β-Gal-Hela-P4 cells and 2) by co-transfection of HEK 293T cells with ZF-VP64 plasmids and an HIV-1-derived plasmid, pMtat(-). The pMtat(-) plasmid bears a tat mutation that impairs viral expression from the LTR promoter in normal conditions [52]. A pCMV-Tat construct was used as a positive control in both cases. Induction of LTR-mediated transcription by ZF-VP64 plasmids in HeLa P4 cells was measured 48 hours post-transfection by means of the β-Galactosidase (β-Gal) activity assay (Figure 7.A). p24 extracellular levels were measured, as an indicator of HIV-1 expression, 48 hours after co-transfection of HEK293T cells, by means of the p24 ELISA assay from cell supernatants (Figure 7.B.). In both cell lines, ZF-VP64-mediated HIV-1 transcription was normalized versus the values of the positive control pCMV-Tat.
Figure 7- Evaluation of ZF-VP64 potential to reactivate latent HIV-1 transcription. A) HeLa P4 LTR-mediated
transcription induced by ZF-VP64. HeLa P4 cells were transfected with 2 µg of each ZF-VP64 constructions or pCMV-Tat (positive control). β-Gal expression was assessed by CPRG assay (see methods for details) 48 hours after transfection. HeLa P4 LTR-mediated transcription was assessed by normalizing β-Gal expression versus pCMV-Tat values. B) HIV-1 transcription of tat-deficient expressing HIV vector by ZF-VP64. HEK293T cells were co-transfected with pMtat(-) and ZF-VP64 constructs or pCMV-Tat. Viral expression was analyzed by means of the p24 ELISA assay (see methods for details) and HIV-1 LTR-mediated transcription was determined by normalizing p24 levels versus the levels of the positive control pCMV-Tat. Data represent the media values obtained from a single experiment performed in triplicate.
As shown in figure 7.A., none of the transfected ZF-VP64 clones were able to induce β-Gal expression in HeLa P4 cells, when compared to non-transfected cells. In fact,
17 the basal expression of β-Gal seems to be inhibited by ZF-VP64 constructs. PBS3 zinc finger binds to the primer binding site of the HIV-1 genome which is located downstream of the 5’LTR and this site is not present in HeLa P4 cells. Therefore, we did not expect PBS3-VP64 to induce β-Gal expression. Nevertheless, we expected HLTR3-VP64 to be capable of inducing β-Gal expression, since the HeLa-P4 LTR promoter contains the HLTR3 binding site. HeLa P4 cells transfected with the pCMV-Tat positive control induced β-Gal expression, indicating that cell transfection and β-Gal expression assay were carried out successfully.
Co-transfection of the HIV-1 Tat-deficient vector pMtat(-) with pcDNA 3.1 or the DNA non-specific binder pALA-VP64 resulted in basal HIV-1 transcription, as suggested by p24 extracellular levels. On the other hand, the co-transfection of pHLTR3-VP64 and pPBS3-VP64 plasmids with pMtat(-) decreased HIV-1 viral expression bellow the basal levels by approximately 30 % and 10 %, respectively. These results are consistent with those obtained in HeLa-P4 cells.
Contrary to what we initially expected, not only HLTR3-VP64 and PBS3-VP64 were unable of enhancing HIV-1 transcription, but they inhibited it. We studied the binding sites of the zinc-fingers and we hypothesized that they could be preventing cellular transcription factors from enhancing the activity of the LTR promoter. Most of the HLTR3 binding site overlaps with the binding site for the cellular transcription factor Sp1, which is fundamental for basal HIV-1 transcription and Tat-mediated transactivation [66, 67, 68]. Therefore, HLTR3-VP64 could be blocking its access to the HIV-1 promoter abolishing viral transcription. On the other hand, the PBS3 zinc finger binds to the primer-binding region downstream of the LTR promoter transcription start site. Binding of PBS3-VP64 to its binding site might therefore interfere with the elongation of HIV-1 transcription, but not with Sp1 binding. This could explain the less restrictive phenotype of PBS3 on HIV-1 transcription.
Since data gathered from two different assays consistently demonstrated that HLTR3-VP64 and PBS3-VP64 were unable to induce HIV-1 transcription, we decided to abandon this strategy and choose Tat as our transcriptional activator, even though Tat could induce unspecific activation of cells.
4.2. Development of a therapeutic lentiviral vector Tat/Rev-dependent
HIV-dependent expression vectors are mostly based on the use of a Tat-responsive LTR promoter [69, 70]. However, expression of these vectors is not completely dependent on viral expression. There are always some basal levels of LTR-mediated transcription that is independent of Tat. Our assay will use a fluorescent reporter protein in order to detect infected cells, and this unspecific basal transcription could produce some fluorescence background and interfere with the interpretation of the results. Furthermore, unspecific expression of the toxin could induce cell death in non-infected cells. In order to avoid these
18 potential problems and to eliminate specifically HIV-1-infected cells, we developed a toxin-expression therapeutic plasmid based on the pNL-GFP-RRE vector. This vector has been shown to be highly efficient and specific as a reporter for HIV infection [27]. It contains the binding site of Rev (RRE) and a GFP gene reporter, under the control of a Tat-responsive LTR promoter. Both GFP and RRE are present between HIV-1 splicing sites and thus the region between them is removed in the absence of Rev, making their expression highly dependent of an actively replicating HIV.
In order to build our therapeutic lentiviral vector we inserted a diphtheria toxin (DTA) gene or a herpes simplex virus type 1 thymidine kinase (HSV1-TK) gene between the HIV-1 splicing sites of the pNL-GFP-RRE that are under the control of the LTR promoter. These toxins were chosen because of their different mechanisms of action. DTA kills the cells once it is expressed, but HSV1-TK produces a deadly toxin only in the presence of the prodrug GCV. Downstream of the vector splicing sites, we inserted a cassette constitutively expressing a bicistronic mRNA coding for both Tat and DsRed, under the control of the cellular Ubiquitin promoter (pUbi-DsRed-IRES-Tat). This cassette will allow us to identify transduced cells (DsRed positive cells) and to reactivate silent HIV-1 proviral DNA (due to Tat expression) simultaneously. All plasmid constructions are shown in Figure 8.
Figure 8- Therapeutic plasmids developed for the proposed strategy. The pNL-GFP-RRE (pNLGR) lentiviral
vector was kindly provided by Dr. Jon Marsh. pNLGR contains a green fluorescent protein (GFP) gene and a Rev-responsive element (RRE) between donor (DS) and acceptor (AS) splicing sites of HIV-1. pNLGR-DTA and pNLGR-TK were constructed by inserting the diphtheria toxin (DTA) or the herpes simplex virus type 1 thymidine kinase (HSV1-TK) genes upstream of the GFP gene in pNL-GFP-RRE. An Ubiquitin promoter (Ubi)-DsRed-Internal Ribossome Entry site(IRES)-Tat cassette was placed downstream of the HIV-1 splicing-sites in pNLGR-DTA and pNLGR-TK to construct pNLGR-pNLGR-DTA-Tat and pNLGR-TK-Tat, respectively. All plasmid constructions are described in Methods.
19 The functionality of all individual components of our therapeutic vectors (toxins, Tat and DsReD) were assessed by different methods. DTA and HSV-TK expression was determined indirectly by analyzing cell death in response to transfection of HEK293T cells with the lentiviral vectors containing their genes. We transfected DTA or pNLGR-DTA-Tat lentiviral vectors together with pCMV-Tat in the presence or absence of a Rev-expressing plasmid [pCMV-Rev (Rev+) or pcDNA 3.1 Zeo (Rev-)]. A FugW-DTA construct was used as a positive control. DTA toxicity was assessed 48 hours after transfection. In parallel, we transfected pNLGR-TK-Tat or pNLGR-TK, together with pCMV-Tat, in the presence or absence of the Rev-expressing plasmid. A FugW-DsRed-TK construct was used as a positive control in this case. For the evaluation of HSV-TK toxicity, cells were treated with GCV (1 µg/mL) or left untreated for 5 days [57, 58]. Toxicity was determined by means of the LIVE/DEAD Fixable Dead Cell Stain Kit (described in Methods).
Figure 9- Evaluation of the specific cellular death of HEK293T transfected with our lentiviral vectors. 5 x
105 HEK293T cells were co-transfected with 1 µg of pNLGR-DTA, pNLGR-DTA-Tat,pNLGR-TK or pNLGR-TK-Tat lentiviral vectors, 0.5 µg of pCMV-Tat and 0.5 µg of pCMV-Rev (+Rev) or pcDNA 3.1 Zeo (-Rev). DTA-induced cellular death was analyzed 48 hours post-transfection. HSV1-TK-induced cell death was analyzed 5 days after transfection, in the absence or presence of 1 ug/ml of GCV added to the cells 24 hours after transfection . Fraction of dead cells was determined by flow cytometry using the LIVE/DEAD Fixable Dead Cell Stain Kit (see Methods for details).
As shown in Figure 9, cells transfected with pNLGR-DTA or pNLGR-DTA-Tat show a 5-fold increase in the percentage of dead cells versus non-transfected cells, indicating that DTA was being expressed. Nevertheless, the level of cellular dead was lower than expected. This could be the result of low transfection efficiency, since the positive control showed similar levels of DTA-induced cell death. Transfection of HEK293T cells with pNLGR-TK and
20 pNLGR-TK-Tat also showed an increase of cellular death (more than 6-fold) in the presence of GCV. In the absence of GCV, the levels of cell death were similar in transfected cells and non-transfected cells, supporting the idea that HSV1-TK is not toxic by itself. These results suggest that DTA and HSV1-TK constructs are successfully transfected and that toxins are effectively expressed.
All the constructs tested in this assay were able to promote cell death. Nevertheless, we did not observe any effect dependent on the presence or absence of Rev. This result indicated that Rev was not able to control toxin expression. We hypothesized that the overexpression of the vector overcame Rev-dependence, forcing their way out of the nucleus without being spliced. Therefore, we need to test the dependence of our lentiviral vector on Rev in a more physiological context.
Tat expression was evaluated indirectly by assessing the capability of our lentiviral vectors to induce LTR-mediated β-Gal transcription in HeLa P4 cells. β-Gal expression was measured 48 hours after transfection (Figure 10.A). FuW DsRed-Tat and pCMV-Tat were used as positive controls. Since Tat should be constitutively expressed by the Ubiquitin promoter downstream of HIV-1 splicing sites, there was no need to co-transfect cells with Tat and Rev-expressing plasmids.
Hela-P4 transfected with pNLGR-TK-Tat, FuW DsRed-Tat, or positive control pCMV-Tat induced β-gal expression (Figure 10.A). On the other hand, we were unable to detect β-gal expression in cells transfected with pNL-DTA, pNL-TK and pNL-DTA-Tat. The failure of pNL-DTA-Tat at inducing β-gal expression may be due to defective or inefficient expression of Tat or to a toxic effect of DTA on producer cells. Even though FuW DsRed-Tat showed low levels of β-Gal activity, the CPRG assay is very sensitive to transfection and presents high levels of variation between different assays. Bearing in mind these results, we believe Tat is most probably being expressed. However, further analyses will be needed to identify the cause of these results.
In order to evaluate DsRed expression, HEK293T cells were transfected with our lentiviral constructions and cells were analyzed 48 hours post-transfection by flow cytometry (Figure 10.B). FuW DsRed-Tat was used as a positive control. Tat and Rev-expressing plasmids were unnecessary also in this case, because DsRed expression is controled by the Ubiquitin promoter downstream of HIV-1 splicing sites and should be therefore constitutive.
DsRed expression was detected only in cells transfected with FuW DsRed-Tat meaning that DsRed was not expressed in pNLGR-DTA-Tat and pNLGR-TK-Tat, even in those where we detected Tat expression. In the case of DTA constructs, these results are consistent with the results on Tat expression and they could be explained by a toxic effect of DTA in producer cells. In the case of TK constructs, the DsRed gene may have a defect originated during the cloning of the plasmids. Even though DsRed is very important to
21 monitor transduction, and to evaluate specific elimination of infected cells by our lentiviral plasmids, we decided to proceed with our assays.
Figure 10- Evaluation of Tat and DsRed expression from lentiviral vectors. A) β-Gal detection by CPRG assay 48 hours post-transfection of HeLa P4 cells with our lentiviral vectors. Cells were transfected with 2 µg of our lentiviral constructs. FuW DsRed-Tat and pCMV-Tat were used as positive control. HeLa P4 LTR-mediated transcription was assessed by normalizing β-Gal expression values to pCMV-Tat. B) DsRed expression analysis. HEK293T cells were transfected with 5 µg of our lentiviral constructs. FuW DsRed-Tat was used as a positive control. Cell fraction expressing DsRed (DsRed positive cells) was identified by flow cytometry 48 hours post-transfection.
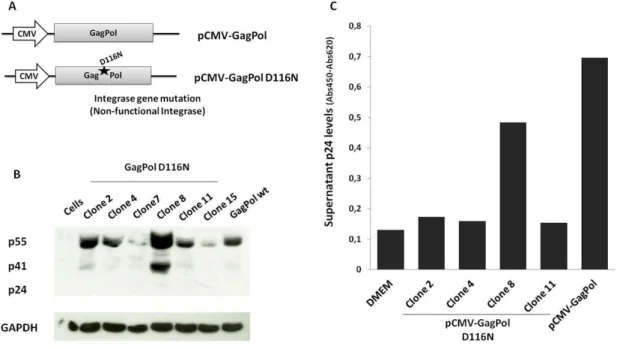
Since one of the major drawbacks of lentiviral gene therapies is insertional mutagenesis, we built an integrase-deficient packaging vector pCMV-GagPol D116N to prevent integration of the lentiviral vector into the cell genome, increasing its safety. This vector has a mutation (D116N) on the gagpol region, in the enzyme active site, eliminating the integration activity but keeping the virus reverse-transcription and Tat-mediated transactivation of gene transcription intact [59, 72, 73](Figure 11.A). This mutation will block lentiviral integration, keeping the viral cDNA in a 2-long terminal repeat (2-LTR) circle. In this state, the vector will be capable of expressing its genes for several days [59], as well as avoid Tat overexpression in non-infected cells.
As described in the Methods section, clones were screened by digestion with the appropriate restriction enzymes, and resolved by agarose gel electrophoresis to identify those containing gagpol fragments (Data not shown). Positive clones were selected and analyzed for: 1) gene expression of Gag proteins by Western blot (Figure 11.B) and 2) assembly of viral particles by p24 ELISA assay (Figure 11.C). These clones were compared to the positive control pCMV-GagPol, a packaging plasmid expressing GagPol proteins with