UNIVERSIDADE DE TRÁS-OS-MONTES E ALTO DOURO
Cancerização experimental
do
urotélio do rato
Orientador Científico:
Professor Doutor Carlos Alberto da Silva Lopes
Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto Co-Orientador Científico:
Professora Doutora Aura Antunes Colaço Universidade da Trás-os-Montes e Alto Douro
Este trabalho foi elaborado como dissertação original de candidatura ao grau de Doutor em Ciências Veterinárias, de acordo com o disposto no número 1 do artigo 17º do Decreto-Lei número 216/92 de 13 de Outubro.
Trabalho maioritariamente financiado pela
Fundação para a Ciência e Tecnologia; Ministério da Ciência e do Ensino Superior; Bolsa de
Aos meus Pais, por tudo o que me deram,
Aos meus rebentos Filipe e Emília pela cedência do tempo que por direito lhes pertence,
Ao Mário, por todo o seu apoio e por todo o amor, Aos meus avós Isaura e Manuel Martins (In memoriam).
Ao Professor Doutor Carlos Alberto da Silva Lopes À Professora Doutora Aura Antunes Colaço
uma extensa lista, da qual apenas citarei algumas. Outras, embora não nomeadas, desempenharam um importante papel pelo seu apoio, carinho e incentivo nos momentos mais difíceis.
À Universidade de Trás-os-Montes e Alto Douro, na pessoa do seu Magnífico Reitor, Professor Doutor Armando Mascarenhas Ferreira, manifesto o meu reconhecimento pelos apoios concedidos para a realização desta tese de doutoramento.
Agradeço ao Professor Doutor Carlos Alberto da Silva Lopes, Orientador Científico desta tese de doutoramento, o investigador mais brilhante que um dia tive o privilégio de conhecer. Jamais poderei expressar o meu reconhecimento ao meu Querido Professor, por me ter encaminhado para a investigação do Cancro da Bexiga em animais de experimentação. Além dos valiosos ensinamentos agradeço-lhe a lição de vida que me tem transmitido. Espero poder aplicar nos meus futuros trabalhos de investigação científica a metodologia, a precisão, o empenho, o entusiasmo e a dedicação que o Professor Doutor Carlos Lopes me transmitiu.
À Professora Doutora Aura Antunes Colaço, Co-Orintadora Científica desta tese de doutoramento, manifesto o meu sincero agradecimento pelo apoio e pela Amizade incondicional que recebi. O seu incentivo e encorajamento estiveram sempre presentes desde o período de preparação da minha tese de mestrado até à fase final de preparação desta tese de doutoramento. Agradeço-lhe a prontidão e rigor que colocou na revisão crítica de todos os trabalhos que conduziram à concepção desta tese.
Ao Professor Doutor Luís Filipe de La Cruz Palomino, pela oportunidade e pelo apoio que me deu na Faculdade de Veterinária de Lugo, Universidade de Santiago de Compostela. Dele recebi o apoio imprescindível para a execução das mais diversas técnicas realizadas neste trabalho.
Agradeço ao Professor Doutor Jorge de Almeida Rodrigues, digníssimo Coordenador do Departamento de Ciências Veterinárias da UTAD, pelos apoios prestados durante a execução desta tese com que sempre me distinguiu.
Ao Mestre Carlos Alberto Palmeira não posso deixar de reconhecer a simpatia e a amizade que nos unem e a sua preciosa colaboração na realização da técnica laboratorial de Citometria de Imagem, por todas as suas sugestões sempre tão pertinentes.
como meu orientador de Mestrado.
À Professora Doutora Maria dos Anjos Clemente Pires, responsável do laboratório de histopatologia da UTAD, manifesto o meu agradecimento pela disponibilização de um laboratório montado com meios para a execução de algumas técnicas executadas nesta tese de doutoramento.
À Mestre Fernanda Seixas Travassos pela forma desinteressada e amiga com que me ajudou nos últimos anos, e por todas as sugestões sempre apresentadas.
À técnica de Anatomia Patológica Lígia Bento pela disponibilidade demonstrada no tratamento do material histopatológico utilizado neste trabalho de doutoramento.
À Sr.ª Dª. Ana Maria, pela preciosa ajuda e colaboração na limpeza do material utilizado durante a fase experimental.
À minha Amiga Maria João Pires por ouvir os meus desabafos e desesperos, num momento tão difícil como foi a parte experimental deste doutoramento, quando ela passava pelos mesmos tormentos.
Com muita amizade às minhas colegas Maria de Lurdes Pinto e Adelina Gama.
Como todo o trabalho de investigação depende de auxílio financeiro, tenho de incluir nestes agradecimentos uma palavra de apreço para com a Fundação da Ciência e Tecnologia, pela confiança depositada na atribuição de uma bolsa de doutoramento, sem a qual dificilmente teria sido possível concretizar este trabalho.
RESUMO
A revisão bibliográfica desta tese de doutoramento descreve os aspectos gerais da carcinogénese química e enumera os diversos modelos experimentais, com roedores, que se podem utilizar no estudo da carcinogénese química do urotélio. Na parte experimental são apresentados quatro trabalhos experimentais diferentes. No primeiro trabalho caracterizámos as lesões histopatológicas identificadas em ratos Fisher 344 após a exposição oral durante 20 semanas à N-butil-N-(4-hidroxibutil) nitrosamina e instiladas intravesicalmente com a mitomicina C e o Bacillus de Calmette-Guérin. Estudámos o conteúdo em ADN destas lesões e verificámos que os animais instilados com a mitomicina C e o Bacillus de Calmette-Guérin apresentaram menor frequência de aneuploidia de ADN. Nos carcinomas papilares de baixo e de alto grau tratados com a mitomicina C ou o Bacillus de Calmette-Guérin o índice de proliferação diminuiu e o índice de apoptose aumentou. Observámos correlações positivas entre o índice de proliferação e o índice de apoptose (r=0,438, p<0,01), entre o índice de proliferação e a lesão urotelial (r=0,425, p<0,05), e o índice de apoptose e a lesão urotelial (r=0,275, p<0,01). Uma correlação positiva foi encontrada entre a ploidia e o índice de proliferação (r=0,245, p<0,05). No segundo trabalho descrevemos as lesões observadas no urotélio normal do rato após instilações intravesicais repetidas com a mitomicina C ou o Bacillus de Calmette-Guérin, e estudámos o seu conteúdo de ADN e o seu índice de proliferação. Apenas foram identificadas lesões uroteliais nos grupos instilados com a mitomicina C. O índice de proliferação apresentou valores de 11,73%, 22,43%, 31,46% (p<0,05) nas hiperplasias simples, displasias, e carcinomas in situ, respectivamente. A frequência de aneuploidia de ADN foi de 25% nas hiperplasias simples e de 85,2% nas displasias (p=0,041). Todos os carcinomas in situ apresentaram conteúdo multiplóide de ADN e todas as metaplasias epidermóides exibiram conteúdo diplóide de ADN. No terceiro trabalho estudámos a expressividade da caderina E durante a carcinogénese química do urotélio do rato, exposto à N-butil-N-(4-hidroxibutil) nitrosamina durante 10, 15 e 20 semanas. A hiperplasia simples e a metaplasia epidermóide apresentaram um padrão de marcação idêntico ao urotélio normal, com a expressividade confinada à membrana citoplasmática das células uroteliais. A caderina E apresentou uma expressividade diminuída em 38,1% das hiperplasias nodulares, 41,4% das displasias e 100% dos papilomas. Todos os tumores
papilares de baixo potencial maligno, os carcinomas papilares de baixo e alto grau, e os carcinomas invasivos revelaram um padrão de marcação anormal com marcação citoplasmática e descontinuidade na positividade membranar. A perda de expressividade do carcinoma papilar de baixo grau relativamente à hiperplasia simples, hiperplasia nodular e displasia apresentou diferenças estatisticamente significativas (p=0,0001; p=0,007 e p=0,008, respectivamente). Observou-se uma diminuição idêntica na expressividade da caderina E entre o tumor papilar de baixo potencial maligno relativamente à hiperplasia simples, hiperplasia nodular e displasia (p=0,0001; p=0,001 e p=0,0001, respectivamente). No último trabalho experimental, utilizando em conjunto a citometria de imagem e a citogenética, avaliámos o conteúdo de ADN nos carcinomas papilares uroteliais do rato induzidos pela N-butil-N-(4-hidroxibutil) nitrosamina. Para este estudo apenas foram utilizadas as neoplasias com crescimento exofítico classificadas como carcinomas papilares de baixo e de alto grau. Os estudos da ploidia de ADN foram realizados em 28 carcinomas papilares de baixo grau e 21 carcinomas papilares de alto grau, 28,6% dos carcinomas papilares de baixo grau e 100% dos carcinomas papilares de alto grau apresentaram conteúdo aneuplóide de ADN (p<0,005). Para a cultura de células foram seleccionadas amostras de um carcinoma papilar de baixo grau e de um carcinoma papilar de alto grau. As populações de células analisadas em ambas as neoplasias demonstraram existir múltiplas alterações numéricas, alterações cromossómicas e a presença vários cromossomas marcadores.
SUMMARY
The bibliographical revision of this PhD thesis describes the general features of chemical carcinogenesis and lists the various experimental models with rodents that can be used to study chemical carcinogenesis of the urothelium. Four different experiments are described in the experimental section. In the first, we characterize histopathological lesions identified in Fisher 344 rats after oral exposure to N-butyl-N-(4-hydroxybutyl) nitrosamine for 20 weeks and having been intravesically instilled with mytomycin C and Bacillus Calmette-Guérin. We studied the DNA content of these lesions and we proved that those animals instilled with mytomycin C and Bacillus Calmette-Guérin dsplayed fewer DNA aneuploidy. The proliferation index for low- and high-grade papillary tumours treated with mytomycin C and Bacillus Calmette-Guérin decreased and apoptotic index increased. We observed a positive correlation between the proliferation index and the apoptotic index (r=0.438, p<0.01), between the proliferation index and the urothelial lesions (r=0.425, p<0.05) and the apoptotic index and urothelial lesions (r=0.275, p<0.01). We also found a positive correlation between DNA content and the proliferation index. In the second experiment, we describe histopathological lesions observed in the normal rat urothelium after repeat intravesical instillations of mytomycin C and Bacillus Calmette-Guérin, and studied its DNA content and proliferation index. Only urothelial lesions from the groups instilled with MMC were identified. The proliferation index gave values of 11.73%, 22.43%, 31.46% (p<0.05) in the simple hyperplasias, dysplasias and carcinomas in situ, respectively. The frequency of DNA aneuploidy was 25% in simple hyperplasia and 85.2% in dysplasia (p=0.041). All the carcinomas in situ displayed multiploid DNA content and all squamous metaplasia displayed diploid DNA content. In the third experiment, we studied the expressiveness of the E-cadherin during the chemical carcinogenesis of the rat urothelium exposed to N-butyl-N-(4-hydroxybutyl) nitrosamine for 10, 15 and 20 weeks. Simple hyperplasia and squamous metaplasia showed normal expressivity pattern, which was identical to normal urothelium, with the expressivity confined to cytoplasmatic membrane of urothelial cells. The expressiveness of E-cadherin was reduced by 38.1% in nodular hyperplasia, 41.4% in dysplasias and 100% papilomas. All the papillary tumours of low malignant potential, the high-and low-grade papillary
carcinomas, as well as invasive carcinoma revealed an unusual expressiveness pattern which included citoplasmatic expression and a lack of continuity in membranal positivity. A lack of expressivity in low-grade papillary carcinoma compared with simple hyperplasia, nodular hyperplasia and dysplasia demonstrated significant statistical differences (p=0.0001; p=0.007; and p=0.008, respectively). A lack of expressivity in low-grade papillary carcinoma compared with simple hyperplasia, nodular hyperplasia and dysplasia showed significant statistical differences (p=0.0001; p=0.001 and p=0.0001, respectively). In the final experiment, in which we used a combination of image analysis and cytogenetic analysis together, we analysed the DNA content of papillary carcinomas in the rat urothelium induced by N-butil-N-(4-hidroxibutil) nitrosamine. Only tumours with exophitic growth classified as low-and high-grade papillary carcinomas were used in this study. DNA ploidy studies were carried out on 28 low-grade papillary carcinomas and 21 high-grade papillary carcinoma, 28.2% of the low-grade papillary carcinoma and 100% of high-grade papillary carcinomas exhibited aneuploid DNA content (p<0.005). Samples from a low-grade papillary carcinoma as well as from a high-low-grade papillary carcinoma were selected for cell culture. The cell populations in both tumours displayed multiple numerical and chromossomal alterations as well as the presence of several marker chromosomes.
PUBLICAÇÕES DECORRENTES E RELACIONADAS COM O TRABALHO EXPERIMENTAL
SOB A FORMA DE ARTIGO
P. A. Oliveira, A. Colaço, L. F. De la Cruz P. and C. Lopes. (2006). Experimental
bladder carcinogenesis-rodent models. Experimental Oncology 28: 1-11.
P. A. Oliveira, A. Colaço, L. F. De la Cruz P. and C. Lopes. (2006). Abnormal
E-Cadherin expression in rat urothelial lesions induced by N-butyl-N-(4-hydroxybutyl) nitrosamine. Journal of Experimental and Clinical Cancer Research
(artigo aceite para publicação).
P. A. Oliveira, A. Colaço, C. A. Palmeira, L. F. De la Cruz P. and C. Lopes. DNA
content analysis, expression of Ki-67 and p53 in rat urothelial lesions induced by N-butyl-N-(4-hydroxybutyl) nitrosamine and treated with Mytomicin C and Bacillus Calmette-Guérin. Anticancer Research (artigo aceite para publicação).
P. A. Oliveira, A. Colaço, C. A. Palmeira, L. F. De la Cruz P. and C. Lopes. Phyllodes
tumour of the urinary bladder in a rat. Veterinary Pathology (artigo em
reapreciação).
SOB A FORMA DE POSTER
P. A. Oliveira, A. Colaço, C. A. Palmeira, L. F. De la Cruz P. e C. Lopes. Estudo do conteúdo de ADN dos carcinomas papilares induzidos pela N-butil-N-(4-hidroxibutil) nitrosamina em ratos. XI Congresso da Sociedade Portuguesa de Anatomia Patológica, Luso 20 a 22 de Abril de 2006.
ÍNDICE
Dedicatória………. v
Agradecimentos……….. ix
Resumo………... xi
Summary……….... xiii
Publicações decorrentes e relacionadas com o trabalho experimental………... xv
Índice de figuras e quadros………. xxiii
Lista de abreviaturas……….……….. xxvii
Capítulo 1 Revisão bibliográfica 1.1. Introdução………..……….……….…….…… 3
1.2. Aspectos gerais da carcinogénese química………....…………..…. 4
1.2.1. Perspectiva histórica dos estudos de carcinogénese química………..… 4
1.2.2. Etapas da carcinogénese……….……….….... 6
1.2.2.1. Iniciação………... 8
1.2.2.2. Promoção……….……….…. 9
1.2.2.3. Progressão……….……….… 10
1.2.3. Absorção e vias metabólicas dos compostos carcinogénicos………..… 11
1.2.4. Classificação dos compostos carcinogénicos……….…….… 14
1.2.5. Alvos moleculares dos compostos carcinogénicos químicos………….. 19
1.2.6. Avaliação da carcinogenecidade……….………..………... 20
1.2.6.1. Ensaios in vitro de transformação celular………..……… 21
1.2.6.2. Ensaios in vivo de carcinogénese………... 22
1.2.6.3. Estudos epidemiológicos……….…….. 24
1.2.6.4. Outros métodos………... 25
1.3. Modelos experimentais para o estudo do cancro da bexiga-Roedores…... 26
1.3.1. A bexiga dos roedores…….………...………….……… 27
1.3.2. Neoplasias espontâneas……….………..……… 28
1.3.3. Requisitos de um modelo para o estudo da carcinogénese química da bexiga………..………... 29
1.3.4.1. Compostos carcinogénicos químicos………... 31
1.3.4.2. Implantação de pellets e indução de cálculos urinários……... 36
1.3.4.3. Agentes promotores……….………...….... 37
1.3.4.4. Radiação…………...………... 38
1.3.5. Cultura de células………...…….. 38
1.3.6. Transplante de tumores……….…………... 39
1.3.7. Modelos transgénicos desenvolvidos para o estudo das neoplasias da bexiga………... 40
1.3.8. Avaliação do desenvolvimento neoplásico………..………... 41
1.4. Referências bibliográficas………….………... 42
1.5. Objectivos………..………...… 59
Capítulo 2 Estudo morfológico e molecular das neoplasias uroteliais induzidas pela N-butil-N-(4-hidroxibutil) nitrosamina e tratadas por instilação intravesical com a Mitomicina C ou o Bacillus de Calmette-Guérin em ratos 2.1. Introdução…………....………...………..…… 63
2.2. Materiais e Métodos….……….………..….…... 64
2.2.1. Animais………... 64
2.2.2. Protocolo experimental………..……….……… 64
2.2.3. Classificação histopatológica……..……….…... 67
2.2.4. Análise do conteúdo de ADN por citometria de imagem…... 67
2.2.4.1. Procedimento………..….………... 67
2.2.4.2. Quantificação do conteúdo de ADN nuclear…………...………...… 68
2.2.5. Estudo de imunohistoquímica……….………….………..……... 69
2.2.5.1. Metodologia…………...………..…….………. 69
2.2.5.2. Análise da imunorreactividade ao Ki-67 e à p53………..……….… 70
2.2.6. Análise estatística………... 71
2.3. Resultados……….………...………. 71
2.3.1. Variação do peso corporal, do consumo de comida e de água……..….. 71
2.3.2. Peso relativo da bexiga……...…..………..………. 72
2.3.3. Exame macroscópico………..……….…….………... 73
2.3.5. Análise de citometria de imagem………...………….………. 84
2.3.6. Imunorrectividade ao Ki-67 e à p53………..……….. 86
2.4. Discussão………...………... 88
2.5. Referências bibliográficas….……… 91
Capítulo 3 Avaliação do índice de proliferação e do conteúdo em ADN nas lesões uroteliais identificadas após instilações intravesicais repetidas com a Mitomicina C e o Bacillus de Calmette-Guérin em ratos 3.1. Introdução……….……… 97 3.2. Materiais e Métodos……….….… 98 3.2.1. Animais………...… 98 3.2.2. Protocolo experimental………...……….… 98 3.2.3. Classificação histopatológica……….….. 100 3.2.4. Estudo de imunohistoquímica..………..……….…. 100 3.2.4.1. Metodologia……… 101
3.2.4.2. Análise da imunorreactividade ao Ki-67-Índice de proliferação….... 101
3.2.5. Análise do conteúdo de ADN por citometria de imagem…………....… 101
3.2.6. Análise estatística……….… 101
3.3. Resultados……….……… 102
3.3.1. Variação do peso corporal, consumo de comida e água……….…. 102
3.3.2. Exame macroscópico………...… 103
3.3.3. Estudo histopatológico………..……….. 103
3.3.4. Características imunohistoquímicas……… 107
3.3.5. Análise de citometria de imagem……….…… 108
3.4. Discussão……….…….… 111
3.5. Referências bibliográficas……….…… 113
Capítulo 4 Expressão da caderina E nas lesões uroteliais induzidas pela N-butil-N-(4-hidroxibutil) nitrosamina em ratos 4.1. Introdução……….……… 119
4.2. Materiais e Métodos………..…… 120 4.2.1. Animais……….... 120 4.2.2. Protocolo experimental…………...………... 120 4.2.3. Classificação histopatológica……….……….…. 121 4.2.4. Estudo de imunohistoquímica……….…. 121 4.2.4.1. Metodologia………... 122 4.2.4.2. Avaliação da imunorreactividade……… 123 4.2.5. Análise estatística………..…….….. 123 4.3. Resultados……….……….... 123
4.3.1. Variação do peso corporal, consumo de comida e de água……... 123
4.3.2. Exame macroscópico………... 124 4.3.3. Estudo histológico……….……... 124 4.3.4. Características imunohistoquímicas……….…… 126 4.4. Discussão……….……….… 134 4.5. Referências bibliográficas……….…… 135 Capítulo 5 Estudo do conteúdo de ADN dos carcinomas uroteliais papilares induzidos pela N-butil-N-(4-hidroxibutil) nitrosamina em ratos 5.1. Introdução……….…… 141
5.2. Materiais e Métodos……….……….… 142
5.2.1. Animais……….…... 142
5.2.2. Protocolo experimental………...……….… 142
5.2.3. Classificação histopatológica………... 143
5.2.4. Análise do conteúdo de ADN por citometria de imagem………….…... 144
5.2.5. Análise de citogenética……...……….… 144
5.2.6. Análise estatística……….……… 145
5.3. Resultados……….…… 145
5.3.1. Exame macroscópico………... 145
5.3.2 Exame microscópico……….… 146
5.3.3. Análise de citometria de imagem……… 147
5.4. Discussão……….………. 151 5.5. Referências bibliográficas……….……… 153
Capítulo 6
Discussão global e Conclusões
6.1. Discussão global e conclusões……….…. 157
ÍNDICE DE FIGURAS
Figura 1.1 Etapas da carcinogénese química, acontecimentos envolvidos em cada uma
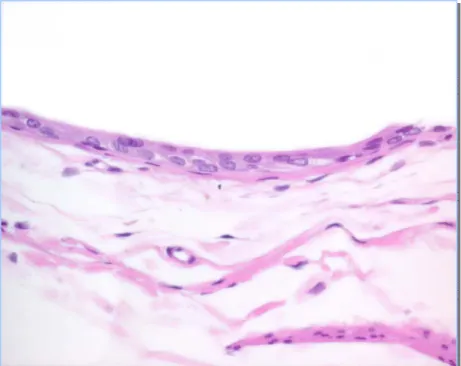
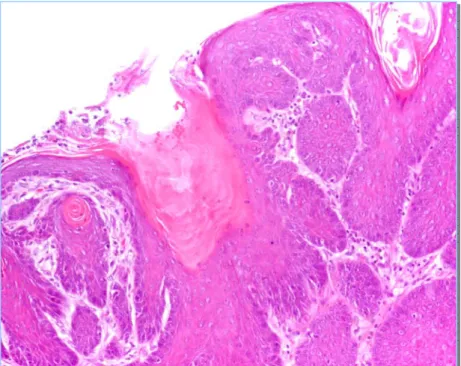
delas……….…………... 7 Figura 1.2 Fases da activação metabólica dos compostos carcinogénicos…….……… 13 Figura 1.3 Nova proposta para a classificação dos compostos carcinogénicos químicos…….. 18 Figura 1.4 Patogenia da carcinogénese urotelial no rato e no murganho………... 26 Figura 1.5 Possíveis aplicações das amostras de bexiga obtidas de trabalhos experimentais.... 27 Figura 2.1 Protocolo experimental…..………...………. 65 Figura 2.2 Peso médio das bexigas nos diferentes grupos……….. 72 Figura 2.3 Urotélio normal do rato, grupo controlo (H&E, 400X)………... 75 Figura 2.4 Hiperplasia simples (H&E, 400X)……….………..…...…... 76 Figura 2.5 Hiperplasia nodular (H&E, 400X)………... 76 Figura 2.6 Displasia (H&E, 600X)………... 77 Figura 2.7 Carcinoma in situ (H&E, 400X)………... 78 Figura 2.8 Papiloma (H&E, 200X)………..……….…... 78 Figura 2.9 Tumor papilar de baixo potencial maligno (H&E, 200X)………...….…………... 79 Figura 2.10 Carcinoma papilar de baixo grau (H&E, 400X)……….……..….….. 80 Figura 2.11 Carcinoma papilar de alto grau (H&E, 600X)………..…... 80 Figura 2.12 Carcinoma invasivo (H&E, 400X)………... 81 Figura 2.13 Carcinoma espinocelular (H&E, 200X)……….…….….………... 82 Figura 2.14 Metaplasia epidermóide (H&E, 200X)………….…….……….………...….. 82 Figura 2.15 Tumor filóide do urotélio (H&E, 40X)………... 83 Figura 2.16 Tumor filóide, observar a atipia citoplasmática (H&E, 400X)………... 83 Figura 2.17 Histograma de ADN com conteúdo de ADN diplóide……….……...…… 85 Figura 2.18 Histograma de ADN com conteúdo de ADN aneuplóide…………..………...…... 85 Figura 2.19 Ploidia de ADN nas diferentes lesões identificadas……….………... 85 Figura 2.20 Expressividade do Ki-67 (400X)………...…………... 86 Figura 2.21 Expressividade da p53 (400X)………... 87 Figura 2.22 Valores médios do LI e AI nas lesões avaliadas………. 88 Figura 3.1 Protocolo experimental………..……….……..……. 99 Figura 3.2 Percentagem de lesões uroteliais identificadas após instilação intravesical com
SF, BCG e MMC em fêmeas de rato Fisher 344………... 103 Figura 3.3 Urotélio normal de um animal do grupo controlo (H&E, 400X)…….………….… 104 Figura 3.4 Hiperplasia simples observada após instilação com MMC (H&E,600X)……….… 105 Figura 3.5 Displasia identificada após instilação com MMC (H&E, 600X)……….. 105
Figura 3.6 Carcinoma in situ observado após instilação com MMC (H&E, 400X)…………... 106 Figura 3.7 Metaplasia epidermóide identificada após instilação com MMC (H&E, 400X)….. 106 Figura 3.8 Expressividade do Ki-67 numa lesão de displasia observada após instilação
intravesical com MMC (600X)………..……….. 107 Figura 3.9 Valores médios do índice de proliferação nas diferentes lesões identificadas após instilação intravesical com MMC……….…... 108 Figura 3.10 Histograma de ADN com uma população diplóide de células analisada numa
lesão de hiperplasia……….……. 110 Figura 3.11 Histograma de ADN com uma população aneuplóide de células analisadas numa lesão de displasia……….. 110 Figura 3.12 Histograma de ADN com uma população multiplóide de células analisadas
numa lesão de carcinoma in situ………...………... 110 Figura 4.1 Expressividade da caderina E no urotélio normal (600X)……….… 127 Figura 4.2 Expressividade da caderina E na hiperplasia simples (600X)………... 127 Figura 4.3 Expressividade da caderina E na hiperplasia nodular (600X)………... 128 Figura 4.4 Expressividade da caderina E na displasia (600X)………...………….… 128 Figura 4.5 Expressividade da caderina E no papiloma (600X)………... 129 Figura 4.6 Expressividade da caderina E no tumor papilar de baixo potencial maligno
(600X) 129
Figura 4.7 Expressividade da caderina E no carcinoma papilar de baixo grau (600X)……….. 130 Figura 4.8 Expressividade citoplasmática da caderina E no carcinoma papilar de baixo grau (60X)………..….. 130 Figura 4.9 Expressividade da caderina E no carcinoma papilar de alto grau (600X)……….… 131 Figura 4.10 Expressividade da caderina E no carcinoma invasivo (600X)…………... 131 Figura 4.11 Expressividade da caderina E na metaplasia epidermóide (600X)……….. 132 Figura 5.1 Protocolo experimental………..……….………..…. 143 Figura 5.2 Carcinoma papilar de baixo grau (H&E, 600X)………...………….… 146 Figura 5.3 Carcinoma papilar de alto grau (H&E, 600X)………..……... 147 Figura 5.4 Preparações metafásicas representativas de várias populações de células
neoplásicas encontradas no carcinoma papilar de baixo grau.………... 149 Figura 5.5 Preparações metafásicas representativas das várias populações de células
tumorais encontradas no carcinoma papilar de alto grau ……… 150 Figura 5.6 Preparação metafásica do carcinoma papilar de alto grau, observar os
ÍNDICE DE QUADROS
Quadro 1.1 Factores que condicionam e/ou modificam a carcinogénese química………. 14 Quadro 1.2 Requisitos de um modelo para o estudo da carcinogénese química da
bexiga………... 29 Quadro 1.3 Tumores quimicamente induzidos na bexiga dos roedores……….… 35 Quadro 1.4 Classificação dos agentes promotores para a bexiga………... 38 Quadro 1.5 Aplicabilidade dos diferentes métodos utilizados no estudo das neoplasias
vesicais………. 41 Quadro 2.1 Média do peso corporal, consumo de água e de comida das fêmeas de ratos Fisher 344 expostas à BBN e tratadas com instilações intravesicais de MMC, BCG e SF. 72 Quadro 2.2 Percentagem de lesões uroteliais nas fêmeas de rato Fisher 344 expostas à
BBN e instiladas intravesicalmente com MMC, BCG e SF………... 74 Quadro 2.3 Conteúdo de ADN nas diferentes lesões histológicas identificadas e nos
diferentes grupos………...…………. 84 Quadro 2.4 Associação ente o LI e o AI nas diferentes lesões uroteliais e nos diferentes grupos………...… 87 Quadro 3.1 Dados relativos ao peso corporal, consumo de água e de comida...………… 102 Quadro 3.2 Ploidia de ADN nas diferentes lesões uroteliais…………..……....………… 109 Quadro 3.3 Análise da ploidia de ADN entre hiperplasias simples e displasias………… 111 Quadro 4.1 Média do peso corporal, consumo de água e de comida das fêmeas de ratos Fisher 344 expostas à BBN durante 10, 15 e 20 semanas………... 124 Quadro 4.2 Percentagem das lesões uroteliais identificadas em fêmeas de rato Fisher
344 após 10, 15 e 20 semanas de exposição à BBN……… 125 Quadro 4.3 Imunorreactividade das lesões/tumores uroteliais à caderina E, e a sua
relação com a classificação histológica………..…………. 133 Quadro 5.1 Percentagem das lesões uroteliais identificadas após exposição à BBN……. 146 Quadro 5.2 Ploidia de ADN no urotélio normal e nos carcinomas papilares uroteliais…. 147 Quadro 5.3 Lesões histológicas e ploidia de ADN nos carcinomas papilares avaliados
LISTA DE ABREVIATURAS E SÍMBOLOS ADN ácido desoxirribonucleico
AI índice apoptótico
ANOVA análise de variância ARN ácido ribonucleico
BBN N-butil-N-(4-hidroxibutil) nitrosamina BCG Bacillus de Calmette-Guérin
BCPN N-butil-N-(3-carboxibutil) nitrosamina CIS carcinoma in situ
CV coeficiente de variação DBN dibutilnitrosamina DP desvio padrão E eutanásia
ENS Etilsulfonilnaftaleno-1-sulfonamida
FANFT N-[4-(5-nitro-2-furil)-2-tiazolil] formamida G Grupo
GTG bandeamento produzido pela tripsina e corado por Giemsa H&E hematoxilina e eosina
H2O2 peróxido de hidrogénio
ID índice médio de ADN LI índice de proliferação MMC mitomicina C MNU N-metil-N-nitrosueia n número
p probabilidade
PBS tampão fosfato salino
RLN radicais livres de nitrogénio RLO radicais livres de oxigénio S semanas
SCC carcinoma espinocelular SF solução salina fisiológica
TPBPM tumor papilar de baixo potencial maligno X vezes
X média ♀ fêmea ♂ macho
CAPÍTULO 1
1.1. INTRODUÇÃO
Em alguns países industrializados aproximadamente 30% da mortalidade deve-se a neoplasias malignas. A incidência e a mortalidade por neoplasias malignas têm aumentado independentemente do envelhecimento da população. O cancro é a doença mais temida para a maioria da população suplantando o temor da tuberculose no século passado, da peste Bubónica na Idade Média ou da lepra nos tempos bíblicos.
Registos da civilização Egípcia a documentarem a ocorrência de urina de cor escura nas pessoas idosas, presumivelmente hematúria, são os primeiros arquivos históricos que sugerem a ocorrência de doenças da bexiga. O primeiro caso de cancro da bexiga foi descrito por Ferri em 1552 (Ito et al., 1989). O cancro da bexiga representa aproximadamente 2 a 5% de todas as neoplasias malignas, é a quarta neoplasia maligna mais comum no homem e a oitava na mulher (Stamouli et al., 2004). Apesar das evoluções registadas no diagnóstico e nas aproximações terapêuticas as taxas de mortalidade associadas a esta doença têm-se mantido constantes. A sua relação com a carcinogénese química é reconhecida desde que Rhen sugeriu em 1895 que os compostos químicos utilizados como matéria-prima na indústria das tintas, posteriormente identificados como anilinas, eram responsáveis pela elevada incidência de cancro da bexiga nos trabalhadores destas empresas. Actualmente, considera-se o consumo do tabaco como o factor de risco mais importante para o desenvolvimento desta neoplasia (Claude et al., 1986; Shirai, 1993). Existem referências sobre a relação entre a exposição a agentes terapêuticos e o desenvolvimento do cancro da bexiga, são exemplos a fenacetina, a ciclofosfamida e a clornafazina (Mostafa et al., 1999). Consideram-se factores de risco suspeitos o café, as bebidas alcoólicas, os adoçantes e a utilização de tintas para o cabelo (Silverman et al., 1992; Cordon-Cardo et al., 1994).
O cancro da bexiga é a neoplasia do trato urinário mais frequente no cão e a segunda mais comum no gato. Os tumores da bexiga no cão correspondem a 0,5 a 1% de todas as neoplasias caninas, aproximadamente 90% destas neoplasias são de origem epitelial e são malignas, e 75 a 90% são carcinomas das células uroteliais. Cerca de 25% dos bovinos que vivem em áreas endémicas a onde cresce o feto Pteridium spp. desenvolvem tumores da bexiga associados a um síndrome designado de Hematúria Enzoótica Crónica. Fora destas áreas geográficas a incidência de cancro da bexiga
nestes animais é muito inferior (Meuten, 2002). No cão, incluem-se como factores de risco para o desenvolvimento de cancro da bexiga a exposição às nitrosaminas, os banhos com insecticidas, a obesidade e os tratamentos quimioterápicos com a ciclofosfamida. Os cães residentes em zonas industrializadas apresentam uma maior prevalência de cancro da bexiga, daí que este animal possa ser utilizado como sentinela na detecção de compostos químicos carcinogénicos para a bexiga (Phillips, 1999).
Há quase um século que os modelos experimentais com roedores são utilizados quer na identificação de compostos carcinogénicos químicos quer no estudo dos mecanismos pelos quais estas substâncias causam cancro. Os modelos experimentais de carcinogénese química para o estudo do cancro da bexiga são utilizados para estudar a biopatologia tumoral e investigar o efeito de diferentes agentes terapêuticos.
1.2. ASPECTOS GERAIS DA CARCINOGÉNESE QUÍMICA
Vários compostos químicos são referidos como responsáveis por diversas neoplasias no Homem. A definição de carcinogénese química é bastante complexa e inclui: a indução por compostos químicos de neoplasias que não são normalmente observadas; a indução por compostos químicos, num período de tempo mais curto, de neoplasias que são normalmente observadas; a indução por compostos químicos de mais neoplasias do que aquelas que são normalmente encontradas (Butterworth et al., 1992).
1.2.1. Perspectiva histórica dos estudos de carcinogénese química
O cancro é uma doença conhecida há milénios e foi descrita pela primeira vez por Hipócrates no século IV, antes de Cristo, como “karkinos”. Galeno, no século II, introduziu a palavra neoplasia, definindo-a como o crescimento excessivo de uma área do corpo. O papiro de Edwin Smith, datado do século XVII descreve pela primeira vez uma neoplasia da glândula mamária (Guttiérrez e Salsamendi, 2001).
Em 1761, John Hill observou uma incidência elevada de cancro nasal nos indivíduos que inalavam rapé. Segundo Hayes (1995), Percival Pott em 1775 estabeleceu, pela primeira vez, a relação causal entre a exposição a substâncias químicas e o desenvolvimento neoplásico. Este autor referiu a elevada susceptibilidade que os
limpadores de chaminés Londrinos tinham em desenvolver cancro do escroto, em consequência da sua exposição crónica à fuligem e ao alcatrão do carvão. Alguns anos mais tarde, e com base nestas observações, o guia do grupo de limpadores de chaminés Dinamarquês recomendava o banho diário para estes profissionais (Hayes, 1995; Gutiérrez e Salsamendi, 2001). Em 1895, Rhen relacionou pela primeira a exposição às anilinas com o desenvolvimento de cancro da bexiga e desde então esta é uma das neoplasias mais associada à exposição ambiental, e ocupacional, a compostos químicos (Shirai, 1993). Estudos epidemiológicos realizados posteriormente permitiram relacionar o desenvolvimento das neoplasias da bexiga com a exposição ocupacional à borracha, tintas e alumínio (Cohen et al., 1991; Siemiatycki et al., 1994; Gomes-Carneiro et al., 1997; Garner, 1998; Dybdahl et al., 1999; Bertram, 2000). Ainda no século XIX, associou-se a exposição a compostos químicos presentes na cadeia alimentar, na atmosfera, no local de trabalho e no lar com a carcinogénese. Segundo Guttiérrez e Salsamendi (2001) o aparecimento do microscópio permitiu identificar e estudar a célula neoplásica, e relacioná-la com o cancro. O início da utilização, no século XX, de modelos experimentais com animais, permitiu estudar a carcinogenecidade de determinados compostos e dar início aos primeiros tratamentos oncológicos.
O primeiro trabalho experimental de carcinogénese química foi realizado por Yamagiwa e Ishikawa em 1918. Estes investigadores friccionaram as orelhas do coelho com carvão e constataram que se desenvolviam papilomas e carcinomas cutâneos. Mais tarde os hidrocarbonetos aromáticos policíclicos presentes no carvão foram classificados e considerados como agentes carcinogénicos (Huff, 1999; Toth, 2001). O sucesso destes dois investigadores criou os alicerces para a utilização experimental de animais no estudo das doenças humanas (Toth; 2001). O primeiro trabalho experimental para indução de neoplasias da bexiga foi executado em cães por Hueper, em 1938, este investigador utilizou como composto carcinogénico a 2-naftiloamina. Em 1944, Amstrong e Bonser administraram por via oral, a murganhos da estirpe CBA o 2-acetiloamonofluoreno e observaram o desenvolvimento de papilomas e carcinomas uroteliais.
Posteriormente Berenblum e Shubik (1947) através da utilização conjunta dos hidrocarbonetos aromáticos policíclicos e do óleo de cróton, no modelo experimental da
carcinogénese da pele com o murganho, demonstraram que o desenvolvimento neoplásico decorria em várias etapas. Estes compostos quando aplicados em doses baixas, só por si, não tinham propriedades carcinogénicas, no entanto quando associados, e na mesma dose, induziam o desenvolvimento neoplásico. A ordem de aplicação destas substâncias era importante para a carcinogénese, só se desenvolviam neoplasias quando primeiro se aplicava os hidrocarbonetos e só depois o óleo de cróton, nunca na ordem inversa. Estes autores consideraram que a conversão das células normais em neoplásicas era desencadeada pela presença destes compostos e concluíram que a carcinogénese era um processo complexo que incluía uma fase de iniciação e outra de promoção, sendo necessário mais do que uma alteração genética para o desenvolvimento do cancro (Berenblum e Shubik, 1947). Na década seguinte Foulds (1954) ao estudar o adenocarcinoma da glândula mamária em murganhos, introduziu o termo progressão. Entre 1960 e 1970 foram descobertos vários compostos químicos com propriedades carcinogénicas para a bexiga, a sua aplicação a modelos experimentais proporcionou a concepção de modelos reproduzíveis para o estudo detalhado dos mecanismos bioquímicos, patológicos e imunológicos envolvidos na etiologia e patogenia do cancro da bexiga (Cohen, 1998a).
Após a descrição da estrutura do ácido desoxirribonucleico (ADN) por Watson e Crick em 1953, as interacções entre diversos compostos químicos e esta molécula foram gradualmente caracterizadas. Nas últimas décadas, os avanços na genética molecular têm permitido reconhecer o envolvimento e as funções de diferentes genes na carcinogénese química.
1.2.2. Etapas da carcinogénese
Os estudos realizados em modelos animais, em ensaios in vitro e em estudos epidemiológicos permitiram concluir que o desenvolvimento neoplásico decorre através de um processo complexo, dividido sob o ponto de vista operacional em três etapas distintas: iniciação, promoção e progressão (Berenblum e Shubik, 1947; Foulds, 1954; Grisham et al., 1984; Cohen, 1991; Hasegawa et al., 1998). Considera-se que na primeira e na última etapa deste processo, ou seja, na iniciação e na progressão, ocorrem alterações na estrutura do genoma (Simons, 1995; Pitot, 2001). Na etapa intermédia,
promoção, ocorre uma alteração na expressão dos genes, com a proliferação selectiva das células iniciadas e o aparecimento de células pré-neoplásicas (Grisham et al., 1984; Gutiérrez e Salsamendi, 2001; Pitot, 2001). Na iniciação e na promoção a apoptose e a proliferação celular ocorrem em taxas diferentes, mas mantêm-se em equilíbrio; na progressão o equilíbrio é alterado e surge a neoplasia (Mehta, 1995; Kanojia e Vaidya, 2006) (Figura 1.1).
Figura 1.1-Etapas da carcinogénese química, acontecimentos envolvidos em cada uma delas.
No Homem, a vida desenvolve-se sob condições diferentes das experimentais. Apesar da essência do processo de carcinogénese ser o mesmo, entre o Homem e os animais de experimentação, os diferentes compostos químicos a que o primeiro é exposto, ao longo da vida, modificam a velocidade do processo e alteram a frequência de mutações, o ritmo de crescimento celular e a expressão fenotípica dos genes alterados. Por outro lado, a susceptibilidade individual e os mecanismos de defesa interagem entre si modificando cada uma das etapas do processo (Gutiérrez e Salsamendi, 2001).
Iniciação Promoção
Aductos no ADN Células iniciadas
Células normais
Reparação do ADN Proliferação celular Proliferação
celular Progressão CANCRO Apoptose Toxicidade celular Proliferação celular Q Quuíímmiiccooss
1.2.2.1. Iniciação
Desde 1947 que a primeira fase da carcinogénese é designada de iniciação (Beremblum e Shubik, 1947). Os resultados obtidos a partir de distintos trabalhos experimentais permitiram concluir que esta etapa resulta de alterações genéticas irreversíveis e predispõe a célula normal à evolução maligna e à imortalidade (Beremblum e Shubik, 1947; Stenbäck, 1981; Mehta, 1995; Dybing e Sanner 1999; Trosko, 2001, Trosko, 2003). A célula iniciada não é uma célula neoplásica, deu o primeiro passo para nela se transformar, depois de ocorrerem sucessivas alterações genotípicas e fenotípicas (Trosko, 2003). A célula iniciada do ponto de vista fenotípico é idêntica às restantes células, no entanto sofreu mutações que favorecem a sua proliferação, mas não a diferenciação (Trosko, 2001).
A ocorrência de danos no ADN é o primeiro evento da carcinogénese química (Santala et al., 2005). Os danos no ADN podem ser corrigidos por mecanismos de reparação enzimática (Bertram, 2000). No entanto, as células em proliferação têm menos tempo disponível para os reparar (Richardson et al., 1986; Frowein, 2000).
As células iniciadas podem permanecer latentes durante semanas, meses ou anos, ou então podem crescer de forma clonal e autónoma (Scott et al., 1984; Dybing e Sanner, 1999; Player et al., 2004). A iniciação favorece a divisão celular simétrica, originando duas novas células iniciadas (Trosko, 2003). A expansão clonal das células iniciadas resulta de um processo mitogénico que aumenta o número de células novas e impede, por inibição da apoptose, a morte das células iniciadas (Trosko, 2001).
A acumulação de danos no ADN tem particular importância nas células estaminais, porque persistem durante muito tempo e estão distribuídas pelos vários tecidos (Potter, 1978; Simons, 1999; Williams, 2001; Player et al., 2004). Em 1978, Potter afirmou que as células neoplásicas podiam apresentar um fenótipo compreendido entre o aspecto embrionário e a diferenciação terminal, e que todas elas tinham origem monoclonal a partir de uma célula estaminal. As células estaminais são, por definição, células imortais até ser induzida a sua diferenciação ou morte; se impedirmos a sua diferenciação tornam-se iniciadas e acumulam-se nos tecidos como clones de células anormais (Trosko, 2003).
A iniciação é um fenómeno rápido, irreversível e hereditário. A proliferação celular é fundamental durante esta etapa, se a divisão celular ocorrer antes do ADN ser reparado, o dano torna-se permanente e passa a estar “fixo”. A iniciação é um processo aditivo, o desenvolvimento neoplásico depende da dose do carcinogénico, aumentando a dose aumenta a incidência, a multiplicidade das neoplasias e diminui o período de tempo para se manifestar clinicamente. Nem todas as células expostas a um agente iniciador serão iniciadas, mesmo que tenham sofrido mutações, porque a célula iniciada tem de possuir potencial proliferativo e sofrer mutações nos genes que regulam a sua diferenciação terminal (Farber, 1984; Yuspa e Poirier, 1988; Klaunig et al., 2000; Trosko, 2001). Células iniciadas espontaneamente existem em todos os organismos vivos. A iniciação pode surgir por mutações espontâneas desencadeadas por acontecimentos normais como são a depurinação e a desaminação do ADN (Gomes-Carneiro et al., 1997; Trosko, 2001). Os radicais livres de oxigénio (RLO) sintetizados durante o metabolismo celular e os erros que ocorrem na replicação do ADN são associados à iniciação. Embora a iniciação espontânea seja mais rara do que a induzida a sua ocorrência é corroborada pelo desenvolvimento espontâneo de neoplasias nos animais de laboratório (Pitot e Dragan, 1991; Gomes-Carneiro et al., 1997).
1.2.2.2. Promoção
O conceito de promoção foi introduzido quando se identificaram substâncias químicas que carecendo de actividade carcinogénica apreciável conseguiam estimular o desenvolvimento neoplásico (Beremblum e Shubik, 1947). A promoção é uma etapa reversível, após o desaparecimento do agente promotor pode ocorrer, possivelmente por apoptose, uma regressão na proliferação celular. É uma etapa modelada por factores fisiológicos e condiciona a duração da carcinogénese experimental, ou seja, determina o período de tempo necessário para a identificação da neoplasia.
Os compostos promotores não interagem directamente com o ADN e para actuarem não precisam de activação metabólica (Yuspa et al., 1983; Butterworth et al., 1992; Weisburger, 1999; Williams, 2001). Por outro lado, e de forma indirecta, os promotores podem, por oxidação, danificar o ADN (Gutiérrez e Salsamendi, 2001). Se de início se pensava que estes eventos estavam associados a mecanismos epigenéticos,
actualmente considera-se que a promoção também envolve alterações genéticas (Simons, 1995; Hanahan e Weinberg, 2000). A eficácia dos agentes promotores depende da sua concentração no tecido alvo e do tempo de contacto (Butterworth et al., 1992). Alguns agentes promotores são específicos para um tecido, mas outros pelo contrário actuam em simultâneo sobre vários tecidos (Yuspa e Hennings, 1983; Scott et al., 1984; Yuspa e Poirier, 1988; Gutiérrez e Salsamendi, 2001).
Nos trabalhos experimentais de carcinogénese química com exposição prolongada, e doses elevadas, praticamente todos os agentes promotores induzem o desenvolvimento neoplásico sem ter ocorrido iniciação. A exposição aos agentes promotores fenobarbital, 12-Ο-tetradecanoilforbol-13-acetato, benzeno, asbestos e arsénico, sem a prévia aplicação de agentes iniciadores, conduz só por si, ao desenvolvimento neoplásico (Melnick et al., 1996; Trosko, 2001). Esta contradição aparente tem duas explicações possíveis: ou o efeito genotóxico não foi identificado nos ensaios de mutagenecidade e genotoxicidade disponíveis; ou as células iniciadas surgiram espontaneamente. Neste último caso pode pensar-se num efeito indirecto do agente promotor que ao aumentar as divisões celulares favoreça a ocorrência de mutações e de erros na replicação do ADN. Nem todas as células expostas a agentes promotores participam na promoção, só as células estimuladas a dividir-se, indiferenciadas, e resistentes à apoptose, podem contribuir para o desequilíbrio entre o crescimento e a morte celular, e conduzirem ao aparecimento de um conjunto de células que por fim origina uma neoplasia maligna (Trosko, 2001).
1.2.2.3. Progressão
As lesões identificados durante a iniciação e a promoção designam-se em histopatologia por lesões pré-neoplásicas e/ou neoplasias benignas (Gutiérrez e Salsamendi, 2001). A sua transformação, através de mecanismos genéticos e epigenéticos, em lesões malignas é a última das etapas da carcinogénese, e a mais prolongada, designa-se por progressão (Klaunig et al., 2000; Williams, 2001).
A progressão caracteriza-se pela irreversibilidade, instabilidade genética, proliferação celular, invasibilidade, metastização, e modificações nas características bioquímicas, metabólicas e morfológicas das células (Pitot e Dragan, 1991; Butterworth
et al., 1992; Klaunig et al., 2000; Dixon e Kopras, 2004). Na progressão, a proliferação celular é independente da presença do composto carcinogénico e resulta da associação entre a mutação e a selecção clonal. A mutação actua de forma aleatória, por sua vez a selecção favorece o crescimento das células com maior autonomia, ou seja, com o ciclo celular menos controlado e com maior potencial metastático. É necessário mais do que uma alteração genética para se atingir a malignidade, mas não é evidente que o caminho percorrido pelas células seja sempre o mesmo (Lutz, 2000; Guttiérrez e Salsamendi, 2001). A angiogénese, como acontecimento epigenético, é fundamental durante a progressão neoplásica. A aquisição do fenótipo angiogénico precede o desenvolvimento das características que contribuem para a malignidade e a sua inibição impede o desenvolvimento neoplásico (Hawighorts et al., 2001).
1.2.3. Absorção e vias metabólicas dos compostos carcinogénicos
Após exposição a um agente carcinogénico químico este pode ser absorvido por diferentes vias (oral, inalatória, cutânea ou injectável) e distribuído pelos diversos tecidos (Clonnolly et al., 1988). As substâncias absorvidas por via oral passam pelo fígado e só depois são distribuídas pelo organismo; as absorvidas no pulmão distribuem-se pela circulação sanguínea atingindo posteriormente o parênquima hepático (King et al., 1995; Van Leeuwen e Zonneveld, 2001).
Os compostos carcinogénicos classificados como directos não precisam de activação metabólica, actuam directamente no ADN, mas a maioria requer activação metabólica e designam-se de indirectos ou pró-carcinogénicos (Sarasin e Meunier-Rotival, 1976; Hayes, 1995; Lai e Shields, 1999; Klaunig et al., 2000; Poirier et al., 2000). Na activação metabólica participam dois grupos distintos de reacções enzimáticas designadas como reacções da fase I e reacções da fase II. A activação metabólica dos compostos químicos é efectuada pelas reacções da fase I, enquanto que as reacções da fase II protegem o organismo através do processamento dos compostos activados em produtos inertes facilmente elimináveis do organismo (Figura 1.2) (Hayes, 1995; Bartsch e Hietanen, 1996; Mostafa et al., 1999; Gonzalez e Kimura, 2001; Park et al., 2005). O desempenho das enzimas metabolizadoras é fundamental para compreender a carcinogénese química e perceber as diferenças entre as espécies no que
concerne à susceptibilidade do desenvolvimento neoplásico (Lai e Shields, 1999; Guengerich, 2000; Gonzalez, 2001; Kyrtopoulos, 2006). As enzimas da fase I participam nas reacções de oxidação, de redução e de hidrólise, e são classificadas como oxi-redutases (mono-oxigenases do citocromo P450, mono-oxigenases da flavina, ciclo-oxigenases e álcool desidrogenases) e hidrolases (epoxidohidrolases) (Hayes, 1995; Garner, 1998; Galati et al., 2000; Oesch et al., 2000; Garcea et al., 2003). As enzimas da fase II participam em reacções de conjugação e de inactivação dos compostos carcinogénicos químicos e incluem-se neste grupo as transferases (glutationa S-transferase, acetilS-transferase, UDP-gluocoronosilS-transferase, sulfotransferase e metiltransferase) (Oesch et al., 2000; Guengerich, 2000; Gonzalez, 2001).
A activação metabólica ocorre predominantemente no fígado, ao nível do retículo endoplasmático liso, onde o sistema microsomal do citocromo P450 é mais abundante, e em menor extensão na bexiga, na pele, no trato gastrintestinal, no esófago, nos rins e no pulmão (Bartsch e Hietanen, 1996; Mostafa et al., 1999; Guengerich, 2001; Van Leeunwen e Zonneveld, 2001; Oda 2004). Durante esta fase as mono-oxigenases do citocromo P450, reacções de fase I, introduzem um grupo polar reactivo no composto carcinogénico tornando-o lipofílico, o que o converte num produto electrofílico potente e com capacidade para estabelecer ligações covalentes designadas de aductos com o ADN (Straub e Burlingame 1981; Lai e Shields, 1999; Galati et al., 2000; Park et al., 2005). As reacções da fase II são catalisadas por enzimas hepáticas e extra hepáticas, citoplasmáticas e citocrómicas, actuando em separado ou em conjunto. Estas enzimas através das reacções de conjugação decompõem o grupo polar em açúcares, aminoácidos, glutationa e sulfato; metabolitos menos tóxicos, mais solúveis na água e facilmente eliminados pela urina e pela bílis (Galati et al., 2000; Oesch et al., 2000; Gonzalez e Kimura, 2001; Van Leeunwen e Zonneveld, 2001).
Figura 1.2-Fases da activação metabólica dos compostos carcinogénicos.
Paralelamente a estas reacções ocorrem reacções de peroxidação com a síntese de RLO. Os RLO estão associados a várias doenças crónicas e à carcinogénese química (Weisburger, 1999; Klaunig et al., 2000; Ohshima et al., 2005). Os RLO, por intermédio das reacções químicas de oxidação, halogenação e nitrificação, danificam o ADN, o ácido ribonucleico (ARN) e as proteínas; o que se traduz num aumento de mutações e na alteração das funções enzimáticas (Lutz, 2000; Ohshima et al., 2003; Park et al., 2005). Foi demonstrado em vários trabalhos experimentais que os
Exposição ao composto carcinogénico Absorção Distribuição Activação Inactivação EXCREÇÃO (RINS, FÍGADO E PULMÃO) Mecanismos genotóxicos •Aductos no ADN
•Perda de porções cromosómicas, fusão de cromossomas e não disjunção de cromossomas
Mecanismos não genotóxicos •Inflamação
•Imunosupressão •Radicais livres de oxigénio •Activação de receptores •Silêncio epigenético
Danos no genoma Alteração da transdução
•Hipermutabilidade •Instabilidade do genoma •Perda do controlo da proliferação
celular •Resistência à apoptose
CANCRO Biotransformação
compostos químicos que originam RLO em excesso favorecem por genotoxicidade a promoção e a progressão neoplásica (Galati et al., 2000).
Parker et al. (2005) referem que a mesma enzima pode activar um químico e inactivar outro, tudo depende da estrutura química da substância. A especificidade dos sistemas de activação, dos diferentes tecidos, condiciona os locais de desenvolvimento neoplásico e está dependente do polimorfismo genético. Este último factor é responsável pela expressão e distribuição das enzimas envolvidas nas reacções das fases I e II, e consequentemente pela susceptibilidade individual ao desenvolvimento neoplásico (Schut e Castonguay, 1984; Hayes, 1995; Hengstler et al., 1998; Mostafa et al., 1999; Gonzalez, 2001; Lutz, 2000). Indivíduos com grande quantidade de enzimas da fase I, e menor da fase II, têm maior probabilidade de sintetizarem compostos reactivos intermédios e de sofrerem mais danos no ADN (Rojas et al., 2000).
As vias metabólicas atrás descritas assumem a mesma importância no Homem e nos animais, contudo existem entre eles diferenças qualitativas e quantitativas que por vezes conduzem a interpretações erradas quando se utilizam modelos animais na pesquisa e análise das propriedades carcinogénicas dos compostos químicos (Guengerich, 2000; Gonzalez, 2001; Gonzalez e Kimura, 2001).
Vários estudos têm sido desenvolvidos com o objectivo de avaliar a influência dos factores exógenos e endógenos na susceptibilidade individual à carcinogénese (Quadro 1.1) (Barrett e Anderson, 1993; Bartsch e Hietanen, 1996; Maronpot, 1996; Lutz, 1998; Ishikawa et al., 2001; Miller et al., 2001).
Quadro 1.1-Factores que condicionam e/ou modificam a carcinogénese química. Idade
Sexo
Exposição a radiações ionizantes Vírus
Alimentação e estilo de vida Vias metabólicas Espécie animal Sistema hormonal Estado imunitário Trauma Constituição genética Fármacos citostáticos
1.2.4. Classificação dos compostos carcinogénicos
O termo carcinogénico foi inicialmente definido como a capacidade de um composto, sob condições apropriadas, desencadear o desenvolvimento de cancro no Homem e nos animais (Gomes-Carneiro et al., 1997; Huff, 1999). Com a descoberta das
diferentes etapas e mecanismos envolvidos na carcinogénese esta definição tornou-se incompleta e desactualizada (Butterworth e Bogdanffy, 1999). Do ponto de vista experimental considera-se que um composto é carcinogénico quando a sua administração a animais de laboratório induz um aumento, estatisticamente significativo, na incidência de neoplasias, de um ou mais tipos histológicos, em comparação com o identificado no grupo de animais controlo, ou seja, não expostos à substância (Guttiérrez e Salsamendi, 2001).
A incorporação de um composto carcinogénico num determinado grupo não é fácil, porque as informações obtidas de diferentes estudos são complexas e por vezes contraditórias. Alguns autores classificam os compostos carcinogénicos em função da sua intervenção em cada uma das etapas da carcinogénese. Desta forma os compostos carcinogénicos incompletos ou iniciadores são químicos mutagénicos que provocam uma alteração irreversível do ADN desencadeando a iniciação (Mirsalis et al., 1990; Pitot e Dragan, 1991). Os compostos promotores aumentam a proliferação celular nos tecidos susceptíveis, o que contribui para fixar as mutações e favorece modificações na expressão dos genes (Mehta, 1995; Gomes-Carneiro, 1995). Os compostos carcinogénicos completos têm simultaneamente propriedades de iniciadores e promotores, dependendo da dose e do tempo de exposição (Barrett e Shelby, 1986; Pitot e Dragan, 1991; Farmer, 1994; Hasegawa et al., 1998; Trosko, 2001).
Outros autores classificam os compostos carcinogénicos em função do seu mecanismo de acção: genotóxicos e não genotóxicos (mitogénicos e citotóxicos) (Cohen e Ellwein, 1991; Butterworth et al., 1992; Nguyen-ba e Vasseur, 1999; Klaunig et al., 2000; Williams, 2001). Os compostos carcinogénicos genotóxicos são carcinogénicos completos, alteram qualitativa e quantitativamente a informação genética da célula (Trosko, 2001). Apresentam uma relação directa entre a sua estrutura e actividade, são mutagénicos nos ensaios in vitro, e activos em todas as doses, podendo afectar várias espécies de animais, e atingir diferentes órgãos (Klaunig et al., 2000; Guttiérrez e Salsamendi, 2001). Em doses altas causam toxicidade e proliferação celular, aumentam a replicação do ADN e potenciam sua acção carcinogénica (Cohen, 1998c). Após a sua difusão transmembranar são metabolizados em compostos electrofílicos que entram no núcleo e interagem com locais nucleofílicos (ADN, ARN, e proteínas) modificando a sua integridade estrutural por estabelecerem com eles aductos
(Miller e Miller, 1975; Straub e Burlingame, 1981; Hayes e Pulford, 1995; Ashby, 1996; Weisburger, 1999; Frowein, 2000; Bertram, 2000; Baird e Mahadevan, 2004). A formação dos aductos constitui o primeiro passo crítico da carcinogénese e se não forem reparados antes da replicação do ADN podem surgir mutações nos proto-oncogenes e nos genes supressores de tumores, fundamentais para a iniciação (Sobels 1975; Barrett e Wiseman, 1987; Farmer, 1994; Williams, 2001; Li et al., 2005). O número de aductos formado pelos compostos carcinogénicos é variável e cada um deles pode desencadear uma alteração específica no ADN (Straub e Burlingame, 1981; Farmer, 1994; Otteneder e Lutz, 1999). As mutações associadas aos aductos podem surgir por delecção, por frameshift ou pela substituição dos nucleotídeos (Garner, 1998). As mutações desencadeiam um número indefinido de transformações celulares traduzidas pela expressão proteica aberrante e alteram o controlo do ciclo celular (Butterworth e Bogdanffy, 1999; Poirier et al., 2000; Guttiérrez e Salsamendi, 2001). Os aductos assumem grande importância na carcinogénese química, porque distorcem a molécula de ADN, provocando uma transcrição incorrecta; a existência de muitos aductos pode partir a cadeia de ADN provocando mutações e/ou perda do material genético (Cohen, 1995a; Hayes et al., 1995; Trosko, 2001).
A reparação dos aductos é um processo coordenado por várias enzimas e controlado por diferentes genes. Pode realizar-se pela excisão de bases, ou de nucleotídeos, por reparação recombinante ou por reparação mismatch (Farmer, 1994; Moustacchi, 1998; Miller et al., 2001; Hanawalt et al., 2003). A identificação de aductos sugere que o composto carcinogénico foi absorvido, metabolizado e distribuído pelos tecidos, escapou aos mecanismos de desintoxificação e de reparação celular (Garner, 1998; Airoldi et al., 1999; Guengerich, 2000). A identificação e a análise dos aductos pode fazer-se através da utilização de carcinogénicos radioactivos marcados, sendo os mais comuns o carbono 14 e o tritium, estes marcadores identificam um aduto por cada 106 ou 107 nucleotídeos (Garner, 1998). Contudo, as técnicas mais utilizadas para detectar os aductos são os imunoensaios com marcação pelo fósforo 32, a cromatografia gasosa associada à espectrometria de massa e por cromatografia líquida de alta precisão associada à espectroscopia fluorescente (Farmer, 1994; Airoldi et al., 1999). Estão disponíveis no mercado anticorpos monoclonais e policlonais que identificam por técnicas de imunohistoquímica os aductos (Santella et al., 2005). Existe
uma correlação positiva entre a quantidade de aductos detectados nos modelos animais e o número de neoplasias desenvolvidas (Yuspa e Poirier, 1988; Williams, 2001; Baird e Mahadevan, 2004).
Os compostos carcinogénicos não genotóxicos actuam como promotores, não requerem activação metabólica, não tem reactividade directa com o ADN, não formam aductos e são negativos nos testes de mutagenecidade realizados in vivo e in vitro (Butterworth et al., 1992; Melnick et al., 1996; Butterworth e Bogdanffy, 1999; Gonzalez, 2001). Estes compostos modulam a proliferação e a morte celular, potenciam os efeitos dos compostos genotóxicos, não apresentam uma relação directa entre a estrutura e a actividade. São específicos para um órgão e para uma espécie animal concreta (Farmer, 1994; Melnick et al., 1996; Gomes-Carneiro et al., 1997; Butterworth e Bogdanffy, 1999; Klaunig et al., 2000). Melnick et al. (1996) referem que a exposição a estes compostos favorece a síntese de outras substâncias responsáveis pelo desenvolvimento neoplásico. Estes compostos promovem nas células alvo efeitos que, indirectamente, desencadeiam a transformação neoplásica ou favorecem o desenvolvimento neoplásico a partir de células geneticamente transformadas (Williams, 2001). Os compostos carcinogénicos não genotóxicos são classificados como
mitogénicos e citotóxicos, em função da sua acção ser, ou não, mediada por um
receptor, respectivamente (Cohen, 1991; Cohen et al., 1992; Butterworth e Bogdanffy, 1999). Os primeiros, como os ésteres do forbol, as dioxinas e o fenobarbital, induzem a proliferação celular no tecido alvo através da interacção com um receptor celular; por sua vez os citotóxicos provocam a morte celular, nos tecidos susceptíveis, seguida de hiperplasia compensatória, é o caso do clorofórmio (Cohen et al., 1991; Cohen et al., 1992; Buttermorth et al., 1992; Klaunig et al., 2000). O aumento da proliferação celular esta relacionado com o agravamento de danos genéticos espontâneos, o que favorece, por diminuição do tempo disponível para reparação do ADN, a conversão das lesões endógenas em mutações (Cohen, 1991; Melnick et al., 1996). Por outro lado, as células necrosadas são destruídas pelo sistema imunitário com a síntese concomitante de RLO, radicais livres de nitrogénio (RLN) e enzimas proteolíticas (Lutz, 1998; Ohshima e tal., 2005; Valko et al., 2006). Quando a produção dos RLO e RNO excede a capacidade anti-oxidante das células podem ocorrer, por oxidação, danos nos lípidos, proteínas, hidratos de carbono e ácidos nucleicos, conduzindo à carcinogénese e à morte celular
(Ohshima et al., 2005). Os compostos não genotóxicos mitogénicos, para exercerem a sua acção, têm de atingir uma determinada concentração no tecido. Em contrapartida, a acção dos compostos citotóxicos é independente da sua concentração (Butterworth et al., 1992; Butterworth e Bogdanffy, 1999). Os conhecimentos disponíveis em relação ao mecanismo de acção dos compostos carcinogénicos não genotóxicos é substancialmente inferior ao dos compostos carcinogénicos genotóxicos.
A classificação dos compostos carcinogénicos em função do seu mecanismo de acção continua a gerar alguma polémica, Bolt et al. (2004) propõem a divisão dos compostos genotóxicos em dois grupos: reactivos com o ADN e genotóxicos ao nível do cromossoma. Os compostos reactivos com o ADN são subdivididos em três subgrupos diferentes: reactivos com o ADN e iniciadores (sem dose limite); genotóxicos fracos (actuam por mecanismos secundários) e compostos borderline (Figura 1.3).
Figura 1.3-Nova proposta para a classificação dos compostos carcinogénicos químicos.
Os compostos carcinogénicos químicos podem, quando associados, por interagirem nas diferentes vias metabólicas, ter efeitos aditivos, sinérgicos ou antagónicos (Lutz, 2000). A associação do álcool com o ferro tem um efeito aditivo obre o carcinoma hepático; o sinergismo entre fumar e a exposição aos asbestos
C CAARRCCIINNOOGGÉÉNNIICCOO Q QUUÍÍMMIICCOO G GEENNOOTTÓÓXXIICCOO NNÃÃOO G GEENNOOTTÓÓXXIICCOO REACTIVO COM O ADN GENOTÓXICOS BORDERLINE INICIADORES ACÇÃO NO CROMOSSOMA
favorece o desenvolvimento do cancro do pulmão. O antagonismo pode exemplificar-se pela acção protectora dos frutos e vegetais na modulação da susceptibilidade individual ao desenvolvimento neoplásico (Lutz, 2000; Peterson, 2005).
1.2.5. Alvos moleculares dos compostos carcinogénicos químicos
Os genes que intervêm na carcinogénese são numerosos e a sua identificação revolucionou o estudo da carcinogénese química e da oncologia (Kinzler e Vogelstein, 1997). De todos eles assumem particular importância os proto-oncogenes, os genes reguladores do ciclo celular e os genes supressores de tumores (Mehta, 1995; Nguyen-ba e Vasseur, 1999; Klaunig et al., 2000).
Os danos no ADN, só por si, não são suficientes para desencadearem o desenvolvimento tumoral. O desenvolvimento tumoral implica transformações nos mecanismos de defesa celular, controlados por checkpoints que podem impedir, antes de ocorrer a replicação do ADN (paragem em G1) e a divisão celular (paragem em G2), a entrada das células com danos no ADN no ciclo celular (Khan et al., 1999; Khan e Dipple, 2000). A capacidade das células escaparem aos mecanismos de defesa celular, quando expostas a um composto carcinogénico, tem um contributo indiscutível durante a carcinogénese (Khan e Dipple, 2000). As proteínas supressoras de tumores p53, p21 e pRb, por favorecerem a paragem das células em G1, desempenham papéis cruciais na protecção celular (Khan et al., 1999). A p53 pode interromper o ciclo celular em G1 para proceder à reparação dos danos no ADN ou para manter a estabilidade do genoma e das células (Melnick et al., 1993; Loeb, 1998; Khan e Dipple, 2000; Pritchard et al., 2003). A perda da função da p53, durante a carcinogénese, pode, pelo bloqueio da resposta apoptótica normal perante danos genéticos, predispor as células pré-neoplásicas à acumulação de mutações adicionais e, por outro lado, activar os oncogenes e inactivar os genes supressores tumorais (Klaunig et al., 2000). A proteína p21 é um dos alvos de acção da proteína p53, e funciona como uma cinase supressora do crescimento celular (Topley et al., 1999). A perda da função da proteína Rb aumenta a taxa de proliferação celular e impede a diferenciação terminal.
A activação e a desregulação do gene ras foram identificadas em várias neoplasias induzidas quimicamente em roedores. As mutações do gene ras existem em cerca de