PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS BIOLÓGICAS
Caracterização das subunidades catalíticas do proteassoma
20S em cepas de
Trypanosoma cruzi
susceptíveis e resistentes
ao benzonidazol
AUTOR: CÁSSIO BARROS DE OLIVEIRA
ORIENTADORA: PROFa. Dra. RENATA GUERRA DE SÁ
CO-ORIENTADORA: DRa SILVANE MARIA FONSECA MURTA
Dissertação submetida ao programa de Pós-Graduação do Departamento de Ciências Biológicas da Universidade Federal de Ouro Preto, como parte integrante dos requisitos para obtenção do título de mestre em Ciências Biológicas, área de concentração: Biologia Molecular.
Catalogação: sisbin@sisbin.ufop.br
O48c Oliveira, Cássio Barros de.
Caracterização das subunidades catalíticas do proteassoma 20S em cepas do Trypanosoma cruzi susceptíveis e resistentes ao benzonidazol
[manuscrito] / Cássio Barros de Oliveira. – 2007. 65 f.: il.; color.; grafs.; tabs.
Orientadora: Profa. Renata Guerra de Sá.
Dissertação (Mestrado) – Universidade Federal de Ouro Preto. Instituto de Ciências Exatas e Biológicas.
Área de concentração: Biologia Molecular.
1.Trypanosoma cruzi - Teses. 2. Drogas - Resistência - Teses.
A Deus;
Aos meus queridos pais e irmãos, pelo amor, carinho e por sempre acreditarem na realização desse trabalho;
À Prof.a Dr.a Renata Guerra de Sá, pelos vários ensinamentos, tanto na parte profissional quanto pessoal. Pelo exemplo de profissionalismo e persistência. E, sobretudo, por mostrar a vida de uma maneira mais simples;
Ao Prof. Dr. Elísio Alberto Evangelista, um verdadeiro pai científico, que incentivou meu estudo na área da Bioquímica e apoiou toda a minha trajetória durante a graduação e mestrado, seus “causos” e exemplo de vida nunca serão esquecidos;
À Prof.a Dr.a Maria Lúcia Pedrosa, pessoa atenciosa carismática, alegre e muito “chique”, sempre disposta a ajudar;
Ao Prof. Dr. Elio Hideo Babá, pelos ensinamentos, conselhos e incentivos;
À Dr.a Silvane Murta e todo o pessoal do Centro de Pesquisa René Rachou, pela ajuda durante os experimentos em Belo Horizonte;
À minha namorada Isabela Silveira Miceli, pelo apoio, amor, compreensão e incentivo durante todo o meu mestrado;
Aos colegas do mestrado, principalmente, a Nilza Satie Hangai uma verdadeira mãe;
A Maísa Silva, grande amiga durante a graduação e mestrado;
A todos os amigos dos laboratórios do NUPEB;
Aos moradores e ex-moradores da República Nóis é Nóis, pela amizade, carinho e boas risadas;
A Cida, secretária prestativa e atenciosa, sempre pronta para ajudar;
Sumário
1- Introdução ... 1
1.1- Doença de Chagas e Trypanosoma cruzi... 2
1.2- Tratamento da doença de Chagas ... 4
1.3- Susceptibilidade e resistência a drogas... 4
1.4- Genoma do T. cruzi ... 6
1.5- Proteassoma e ubiquitinação ... 8
1.6- Proteassoma e T. cruzi... 12
2- Objetivos... 14
3- Material e Métodos... 16
3.1-Obtenção de cepas ... 17
3.2-Extração de RNA total e verificação da integridade ... 17
3.3-Transferência dos RNAs para a membrana de náilon ... 19
3.4- Extração de DNA genômico... 19
3.5- Digestão do DNA genômico com enzimas de restrição e transferência para membrana de náilon (Southern Blot)... 20
3.6- Eletroforese em Gel de Campo Alternado (PFGE) ... 21
3.7- Transferência dos cromossomos do Gel de PFGE para a membrana de náilon.. 21
3.8- Obtenção dos cDNAs ... 21
3.9- Purificação dos cDNAs ... 23
3.10- Obtenção das sondas... 23
3.11- Reação de hibridização... 24
3.12- Lavagem das membranas ... 24
3.13- Autoradiografia... 25
3.14- Obtenção do extrato bruto de proteínas totais do parasito ... 25
3.15- Preparação de fração enriquecida de proteassoma ... 26
3.16- SDS-PAGE e ensaios de Western Blot ... 26
3.17- Medida da atividade proteolítica exógena dos parasitos ... 27
4- Resultados ... 28
4.1- Localização Cromossômica dos Genes Beta-1, -2 e -5 ... 29
4.2- Determinação do número de cópias dos genes Beta-1, -2 e -5... 31
4.3- Northern Blot... 34
4.4- Detecção de conjugados proteína-ubiquitina... 36
4.5- Detecção do proteassoma 20S ... 37
4.6- Atividades enzimáticas do proteassoma 20S... 38
5- Discussão ... 41
6- Conclusões ... 52
7- Perspectivas... 54
A doença de Chagas é causada pelo protozoário Trypanosoma cruzi.
Segundo a WHO existem 16-18 milhões de pessoas infectadas com este protozoário na América Latina. Devido ao envolvimento do proteassoma na biologia celular do T. cruzi
e a baixa eficiência das drogas utilizadas no tratamento de indivíduos com doença de Chagas, entendemos que a caracterização das subunidades catalíticas do proteassoma 20S, poderá ajudar no desenvolvimento de futuros quimioterápicos contra essa doença.
Neste sentido, este trabalho mostra a caracterização molecular e bioquímica das subunidades catalíticas do proteassoma 20S em um padrão de cepas do T. cruzi com
sensibilidade e resistência induzida “in vitro” ao benzonidazol (17WTS e 17LER), sensibilidade e resistência selecionada “in vivo” ao benzonidazol (BZS e BZR) e cepas naturalmente sensíveis e resistentes a essa droga (CL e Colombiana, respectivamente).
As análises de localização cromossômica utilizando a técnica de eletroforese em gel de campo alternado, mostraram vários perfis de hibridização, sugerindo que estes genes não estão associados com o fenótipo de resistência do T. cruzi a drogas.
Além disso, utilizando a técnica de Southern blot, mostramos que estes genes estão presentes em no mínimo duas cópias por genoma haplóide do parasito.
A expressão desses genes foi avaliada através da técnica de Northern blot e os níveis do proteassoma 20S identificados utilizando Western blot e anticorpos contra subunidades alfa. Podemos constatar que enquanto os níveis de mRNA não variaram entre as cepas, uma maior quantidade de proteassoma 20S foi detectado nas cepas sensíveis (17WTS, BZS e CL) quando comparado as cepas resistentes (17LER, BZR e Colombiana). Além disso, também não detectamos acúmulo de conjugados poliubiquitinados, sugerindo que o processo de poliubiquitinação e degradação de proteínas intracelulares dependente do proteassoma 26S são fortemente regulados no padrão de cepas de T. cruzi estudadas.
A medida da atividade peptidásica utilizando substratos específicos para as subunidade Beta-1, -2 e -5, mostraram uma atividade peptidásica principal semelhante à tripsina em todas as cepas analisadas, entretanto, as cepas 17WTS e 17LER também apresentaram uma alta atividade peptidásica semelhante à quimotripsina.
Chagas disease is caused by Trypanosoma cruzi. According to WHO, in
Latin America, 16-18 million people is infected with this protozoan. Due the involvement of the proteasome in the cellular biology of the T. cruzi and the low
efficiency of the drugs used in the individuals treatment with Chagas disease, we believed that the characterization of the catalytic subunits of the 20S proteasome, would help in the development of future drugs that could be used in the therapeutics of the Chagas disease. This work shows the molecular and biochemical characterization of the catalytic subunits of the 20S proteasome in a T. cruzi population with in vitro induced
(17WTS and 17LER), in vivo (BZS and BZR) and strains naturally sensitive and
resistant to benznidazole (CL and Colombian). Chromosomal localization by using the pulse field gel electrophoresis and hybridization profiles with specific probes showed several pattern in the different strains, suggesting that these genes are not associated with the drugs phenotype resistance in T. cruzi. Southern blot analyses showed that
these genes are present in at least two copies for genome haploid of the parasite. The expression of those genes was evaluated through of Northern blot analyses and the levels of the 20S proteasome identified using Western blot and antibodies against alpha subunits. The mRNA levels did not vary among the strains, but a larger level of 20S proteasome were detected in the sensitive strains (17WTS, BZS and CL) when compared the resistant strains (17LER, BZR and Colombian). The absent of accumulation of poly-ubiquitin conjugated, suggest that the poly-ubiquitination pathway and degradation by 26S proteasome, are strongly regulated in the strains of T. cruzi
studied. The measure of the peptidase activity using specific substrate for the subunit Beta-1, -2 and -5, showed that the main proteolytic activity is tripsin-like in all strains. Moreover, the strains 17WTS and 17LER also presented a high quimotripsin-like activity. These results obtained until the moment suggest that, in the epimastigote development stage of the T. cruzi the post-translation modifications are more efficient
αP32 ATP Adenosina trifosfato raiomarcada na posição α com fósforo 32.
Bis-Acrilamida N, N’ Metileno-Bis-Acrilamida BCA Ácido bicinchoninico
BCIP 5-bromo-4-cloro-3 indolil-fosfato BSA Albumina sérica bovina
BZ Benzonidazol cDNA DNA complementar DEPC Dietilpirocarbonato gDNA DNA genômico
dNTP Deoxinucleotídeo trifosfato (N= A, C, G ou T) D.O. Densidade Óptica
EDTA Ácido etilenodiaminotetracético (sal dissódico) MOPS [Ácido 3-(N-Mofolino)Propanosulfônico] mRNA RNA mensageiro
NBT Nitrobluetetrazolium
PFGE Eletroforese em gel de campo alternado TEMED N,N,N’,N’,-Tetrametil Etilenodiamina Tris Tris-hidroximetilaminometano
1.1- Doença de Chagas e Trypanosoma cruzi
A doença de Chagas também conhecida como Tripanosomíase Americana é uma doença causada pelo protozoário parasito Trypanosoma cruzi (Família:
Tripanosomatidae, Ordem: Kinetoplastida). Segundo a WHO, existem 13 milhões de pessoas infectadas com o T. cruzi na América Latina
(www.who.int/health-topics/chagas.html). Cerca de 20% dessas pessoas desenvolvem os sintomas clínicos que caracterizam a doença e aproximadamente 14000 mortes têm sido atribuídas anualmente a esta doença (WHO, 2005). A Tripanosomíase Americana apresenta algumas características clínicas associadas com a origem geográfica da cepa do T.
cruzi, como: maior freqüência de magaesôfago e megacólon no Brasil central (Cabral et
al., 1999) e rara ocorrência ao norte do rio Amazonas. Na Argentina, Chile e no Estado do Rio Grande do Sul ocorrem diferenças na resposta ao tratamento etiológico, com altos níveis de cura. A transmissão congênita no Brasil é menor comparada com a Bolívia, Chile e algumas partes da Argentina (WHO, 2000).
Cerca de 100 espécies de mamíferos silvestres atuam como reservatórios da doença de Chagas. A sua transmissão natural é feita principalmente pelos insetos pertencentes à família Reduviidae subfamília Triatominae que infectam animais selvagens durante o repasto sanguíneo, mantendo o ciclo silvestre da doença. O T. cruzi
formas se multiplicam por divisão binária até se diferenciarem em formas tripomastigotas antes do rompimento da célula parasitada (WHO, 2000).
No hospedeiro vertebrado, a infecção pelo parasito ocorre quando as formas tripomastigotas metacíclicas eliminadas juntamente com as fezes dos barbeiros durante o repasto sanguíneo, atingem a camada da pele lesada pelo triatomíneo ou a mucosa. Essas formas invadem células locais dando início à multiplicação intracelular sob a forma amastigota. As células hospedeiras contendo um grande número de parasitos se rompem, liberando as formas tripomastigotas que através da circulação sanguínea seguem para outros órgãos ou tecidos do organismo onde reiniciam um novo ciclo intracelular. Ao sugar o sangue de um animal ou homem infectado com o T. cruzi, o
triatomíneo ingere as formas tripomastigotas sanguíneas que se transformam em paramastigotas que se transformam a seguir em epimastigotas no tubo digestivo do inseto, onde passam a dividir-se intensivamente. Após algumas semanas de infecção, as formas epimastigotas diferenciam-se em tripomastigotas metacíclicos que se concentram nas porções terminais do intestino, sendo eliminados com as fezes durante o próximo repasto sanguíneo do vetor, reiniciando um novo ciclo evolutivo. Os passos desse ciclo encontram-se representados na figura abaixo (Dias, 1934 e Brack, C. 1968).
Figura 1: Ciclo de vida do T. cruzi. As diferentes formas do T. cruzi ao longo de seu ciclo
1.2- Tratamento da doença de Chagas
Infelizmente, ainda não existem tratamentos quimioterápicos eficientes para indivíduos com doença de Chagas. Algumas drogas como o Nifurtimox, 5-nitrofurano [3-Metil-4-(5-nitrofurfuridileno-amino)-tetrahidro-4H-1,4-tiazina-1,1-dióxido] comer-cializado como Lampit (Bayer), e o Benzonidazol, 2-nitroimidazol-(N-benzil-2-nitro-1-imidazolacetamina), comercializado como Radamil ou Rochagan (Roche) apresentam baixa eficiência para a fase crônica da doença e são altamente tóxicas para os pacientes, causando muitos efeitos colaterais (comumente hipersensibilidade, alterações hematológicas e polineuropatia periférica) (Filardi e Brenner, 1987; Urbina e Docampo, 2003).
Essas drogas são geralmente utilizadas na fase aguda da doença e em reativações da infecção em indivíduos imunossuprimidos. Existem divergências entre autores sobre o tratamento específico durante a fase crônica devido aos baixos índices de cura encontrado, e os efeitos colaterais observados.
A procura de novas abordagens terapêuticas para o tratamento da doença de Chagas ainda é extremamente importante devido a grande prevalência e mortalidade da doença (WHO, 1991).
1.3- Susceptibilidade e resistência a drogas
As diferenças de susceptibilidade ao benzonidazol e nifurtimox entre cepas de T. cruzi (Neal e Van Bueren, 1988)podem explicar, pelo menos em parte, a eficácia
das causas da baixa eficácia de cura observada em pacientes chagásicos tratados com estas drogas (Filardi e Brener, 1987).
Vários protocolos têm sido descritos na literatura para a obtenção “in vitro” de cepas do T. cruzi resistentes ao nifurtimox (Nozaki T. et al 1996), benzonidazol
(Nirdé P. et al 1995), azole (Buckner F. et al 1998) e inibidores de enzimas cisteíno-proteases (Engel J. 2000 and Yong V. 2000). Nirdé et al. (1995) conseguiram obter “in vitro” uma população do T. cruzi, 23 vezes mais resistente ao BZ (17LER) comparado
com seu par sensível (17WTS). Os autores observaram que o fenótipo de resistência foi estável mesmo após diferenciação de epimastigotas para amastigotas. Entretanto, estudos comparativos de susceptibilidade a drogas entre cepas do T. cruzi não
detectaram correlação entre susceptibilidade a droga “in vitro” comparado com testes “in vivo” (Scott e Matthews, 1987; Neal e Van Bueren, 1988; Ribeiro- Rodrigues et. al., 1995). Diante disso, com o objetivo de obter um modelo de resistência do T. cruzi a
drogas mais próximo do que acontece no homem, Murta & Romanha (1998) fizeram a seleção “in vivo” de uma população de T. cruzi e clones resistentes para o
benzonidazol.
Murta e colaboradores (1998) realizaram um estudo com 45 cepas de T.
cruzi sensíveis e naturalmente resistentes ao benzonidazol e nifurtimox analisando 6
diferentes marcadores moleculares. Nesse estudo, o perfil heterozigoto (Zimodema B), agrupou exclusivamente cepas sensíveis e ocorreu principalmente em áreas geográficas onde o tratamento da doença de Chagas tem sido relatado como mais eficaz, demonstrando que este zimodema está associado ao fenótipo de sensibilidade a drogas pelo T. cruzi. Já os perfis isoenzimáticos Z1 e Z2 foram encontrados em populações de
T. cruzi sensíveis e naturalmente resistentes a drogas.
Em abril de 1999, durante o Simpósio Internacional Comemorativo dos 90 anos da descoberta da doença de Chagas, no Rio de Janeiro, foi adotada a subdivisão da espécie de T. cruzi em dois grupos principais. O grupo T. cruzi I está associado com o
ciclo de transmissão silvestre e infecção de marsupiais (Clark et al., 1994). O grupo T.
cruzi II apresenta cinco sub-grupos chamados: IIa, IIb, IIc, IId, e IIe (Brisse et al.,
A preocupação pela identificação de marcadores moleculares que caracterizam a susceptibilidade ou resistência do T. cruzi a drogas é importante porque
possibilita uma futura utilização no prognóstico de cura da doença de Chagas (Murta e Romanha, 1998).
1.4- Genoma do T. cruzi
Informações a respeito da organização estrutural e expressão do genoma do
T. cruzi são de grande interesse, uma vez que o ciclo de desenvolvimento do parasito e
os mecanismos de infecção são direcionados pela modulação e expressão da informação contida no genoma nuclear.
Uma das particularidades observadas no genoma do T. cruzi é a grande
variação no conteúdo de DNA entre diferentes cepas e até mesmo entre clones de uma mesma cepa uma vez que diferenças de 30-70% no conteúdo total de DNA foram observadas (Mcdaniel e Dvorak, 1993). Diante disso, um primeiro passo para o seqüênciamento genômico do T. cruzi, foi escolher uma das várias cepas. A cepa
escolhida foi a CL Brener que pertence ao subgrupo IIe. Esta escolha foi feita porque esta cepa está muito bem caracterizada em diferentes aspectos biológicos e moleculares (Zingales B. et.al., 1997), e por ser aparentemente um híbrido derivado de T. cruzi I
(Brisse et. al., 1998; Machado, C. A. et. al., 2001; Gaunt et. al., 2003; Brisse et. al., 2003).
A organização dos genes nos tripanosomas difere daquela conhecida em outros eucariotos. A maioria dos genes codificadores de proteínas estão presentes em múltiplas cópias na célula, sendo o espaçamento entre os genes de um dado arranjo bem curto (variando de 100 até algumas centenas de pares de base). Genes que codificam tubulinas, calmodulinas, ubiquitinas, antígenos de superfície de membrana e enzimas da via glicolítica existem em arranjos em “tandem” no genoma (Ullu e Nilsen, 1995; El-Sayed et al., 2005).
106 pb) possui um tamanho relativamente maior que o tamanho dos genomas de outros parasitos, como, por exemplo, Leishmania (45-65 x 106 pb) e Trypanosoma brucei (25
x 106 pb), porém, a complexidade do genoma do T. cruzi é significativamente inferior à
do genoma humano, cujo tamanho é da ordem de 3000 x 106 pb (El-Sayed et al., 2005). Mais de 50% do genoma consiste de seqüências repetitivas tais como retrotransposons e genes para grandes famílias de moléculas de superfície, como: Trans-sialidases, mucinas, gp63s e uma grande família (contendo pelo menos 1300 cópias) de genes de mucinas-associadas a proteínas de superfícies. (El-Sayed et al., 2005).
As análises dos genomas do T. cruzi, T. brucei e L. major (Tritryp)
evidenciaram diferenças em relação a outros eucariontes no reparo de DNA (estes organismos são capazes de catalisar mais vias de reparo), início da replicação (os Tritryp apresentam uma maquinaria de início de replicação diferente dos eucariontes superiores) e DNA mitocondrial (El-Sayed et al. 2005).
Uma das características mais marcantes da família trypanosomatidae é a presença do cinetoplasto, uma região especializada na única mitocôndria da célula que contém o DNA mitocondrial ou do cinetoplasto, também conhecido como kDNA. O kDNA é uma estrutura complexa, organizada como uma rede compacta constituída por dois tipos diferentes de moléculas de DNA: os maxicírculos e os minicírculos (Shapiro e Englund, 1995).
Embora os Tritryp não apresentem muitas classes de moléculas sinalizadoras como por exemplo: receptores tipo serpentina, proteínas G heterotriméricas, muitas classes de receptores catalíticos, domínios de interação SH2 e SH3 e fatores de transcrição regulatórios, seus quinomas mostraram uma grande diversidade de proteínas quinases e fosfatases, o que sugere um papel importante da fosforilação na biologia celular destes parasitos (El-Sayed et. al., 2005).
Através da utilização da técnica de eletroforese de campo pulsátil (PFGE), foi possível verificar que o genoma do T. cruzi está organizado em pelo menos 20-25
al., 1995; Henriksson et al., 1995). Com essa técnica foi possível observar a existência de um grande polimorfismo no genoma de diferentes cepas do T. cruzi no que se refere
ao número de bandas cromossômicas e a localização de determinados marcadores moleculares, entretanto, apesar desta variabilidade no cariótipo, a organização cromossômica em T. cruzi parece conservada em algum aspecto (Aymerich e
Goldenberg, 1989 e Henriksson et al., 1995). A localização de vários marcadores genéticos em uma mesma banda cromossômica permitiu a identificação de oito grupos de ligações conservadas. Além disso, Henriksson e colaboradores (1993) observaram uma correlação entre a variabilidade do cariótipo com a classificação de zimodemas.
É bom ressaltar que Porcele et al. (2003) promoveram um refinamento do cariótipo molecular do clone CL Brenner (cepa referência do projeto genoma do T.
cruzi). Utilizando 210 marcadores genéticos para bandas cromossômicas separadas por
PFGE, esse grupo identificou 61 marcadores específicos de cromossomos e dois cromossomos com tamanhos polimórficos. Além disso, esse grupo construiu o mapa integrado da banda cromossômica XX. O isolamento de marcadores cromossômicos específicos e o mapa físico das bandas cromossomais contribuíram muito para o seqüênciamento do genoma nuclear do parasito.
Os esforços para o seqüênciamento genômico e localização cromossômica do T. cruzi, auxiliarão no entendimento da biologia do parasito, o que certamente
permitirá o desenvolvimento de futuros alvos terapêuticos.
1.5- Proteassoma e ubiquitinação
O proteassoma 20S é um complexo proteolítico com peso molecular de 750 kDa, presente em archaeobactérias (Seemuller et al., 1995), leveduras (Hilt & Wolf, 1995), Entamoeba histolytica (Scholze et al., 1996), Leishmania mexicana (Robertson,
1999), Trypanosoma brucei brucei (Lomo, et al., 1997), T. cruzi (Gonzales et al., 1996)
diferenciação celular, respostas a estresse, adaptação metabólica e resposta imune celular.
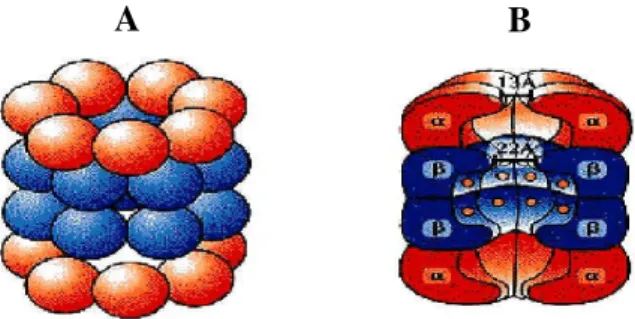
O proteassoma de mamíferos apresenta 14 tipos diferentes de subunidades polipeptídicas, com peso molecular variando entre 22-34 kDa e pontos isoelétricos entre 4,5 a 8,7. Essas subunidades são agrupadas em duas famílias distintas: alfa e beta (Hendil et al., 1995), sendo as alfas estruturais e as betas catalíticas (Coux et al., 1996). Estudos utilizando microscopia eletrônica demonstraram a existência de uma estrutura cilíndrica constituída de quatro anéis superpostos, sendo os dois anéis externos, constituídos de sete subunidades alfa, e os anéis internos, constituídos de sete subunidades betas. Esse arranjo alfa1-7beta1-7beta 1-7alfa1-7 formam um “barril” com um canal central constituído de três cavidades, por onde passam as proteínas desenoveladas para serem degradadas. As duas cavidades externas estão localizadas nas interfaces entre os anéis alfa e beta. A terceira cavidade está localizada no centro do complexo e é formada pelos anéis beta. O complexo 20S apresenta uma atividade hidrolítica semelhante à caspase, tripsina e quimotripsina localizadas, respectivamente, nas subunidades beta-1,-2 e -5 (Tanaka, 1998). Cada uma dessas subunidades apresenta um resíduo de treonina na porção amino-terminal, o que caracteriza o proteassoma como membro da subfamília T1A de peptidases. Essa característica das subunidades catalíticas do proteassoma (único membro dessa subfamília) é que permitiu o desenvolvimento de inibidores específicos dessa protease. A estrutura do proteassoma 20S encontra-se ilustrada na figura 2.
Figura 2: Estrutura do proteassoma 20S. Em A, encontra-se representado de vermelho os
dois anéis heptaméricos constituídos pelas subunidades α, e de azul os anéis heptaméricos internos contituídos de subunidades β. Em B, destacam-se as três cavidades formadas pelas subunidades α e β, sendo o sítio catalítico representado de azul e formado pelas subunidades β. Figura adaptada de Tanaka et al., 1998.
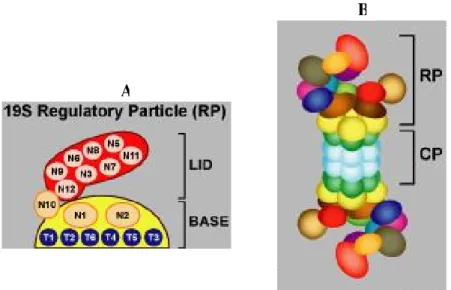
O proteassoma 20S pode se associar a outros complexos como: complexo regulatório 19S (também conhecido por Proteasome Activation 700) e Complexo 11S (também conhecido por Proteasome Activation 28). Quando o complexo 20S se associa ao complexo regulatório 19S, ocorre à formação do proteassoma 26S, que está envolvido no processo de degradação de proteínas poliubiquitinadas. O PA700 é constituído por subunidades diferentes, com peso molecular variando entre 25-110 kDa. Essas subunidades heterólogas são classificadas em dois subgrupos: subunidades com funções ATPásicas e não ATPásicas. O papel das ATPases está relacionado ao desenovelamento das proteínas e fornecimento de energia necessária a degradação seletiva. As subunidades não ATPásicas, como a RPN10 e RPN11, apresentam funções de reconhecimento de proteínas poliubiquitinadas e ação desubiquitinadora, respectivamente (Tanaka, 1998, Baumesteir et al., 1998). Diversas evidências experimentais sugerem que o proteassoma 20S, ligado ao ativador PA28 ou 11S, está envolvido no processo de apresentação de antígenos (Zhang et al., 1998). A figura 3 ilustra a estrutura do complexo 19S e o proteassoma 26S.
Figura 3: Estrutura do complexo 19S e formação do complexo 26S. Em A, encontra-se
representado o complexo regulatório 19S formado por 6 subunidades ATPásicas representadas por azul e 11 subunidades não ATPásicas representadas por círculos claros. O conjunto dessas subunidades formam uma base e uma tampa. Em B, encontra-se representado o complexo 26S formado pela união dos complexos 20S e 19S. Figura adaptada de Hicke L. 1999. Trends Cell Biol. 9: 107-112.
A ubiquitina é um polipeptídeo constituído por 76 aminoácidos, presente em todas as células eucarióticas, sendo encontrada no citosol, núcleo e superfície celular.
A
Em eucariontes superiores, um dos papéis da ubiquitina é marcar proteínas que serão degradadas via proteassoma. Este processo requer a participação de três famílias de enzimas: E1 (enzima ativadora da ubiquitina), E2 (enzima conjugadora de ubiquitina) e E3 (enzima ligante de ubiquitina), todas envolvidas concomitantemente no processo de conjugação da ubiquitina às proteínas (Jentsch, 1995; Finley, 1991; Wilkinson, 1995).
Para a ubiquitinação de uma proteína alvo, inicialmente ocorre à formação de uma ligação tio-éster entre a carboxila terminal da ubiquitina e o grupo sulfidrila da enzima E1, sendo o ATP necessário para esta reação. Posteriormente, a ubiquitina ativada é transferida para uma sulfidrila da enzima E2 e, finalmente, a enzima E3 catalisa a transferência da ubiquitina de E2 para a proteína alvo (Wilkinson et al., 1995). Após sucessivos ciclos, as proteínas alvos vão tornar-se poliubiquitinadas, sendo que em alguns casos requer a participação da enzima E4 para esse processo (revisado por Hoppe T. 2005). Os passos da ubiquitinação encontram-se ilustrados na figura 4.
Figura 4: Poliubiquitinação de proteínas alvos. Esse processo de poliubiquitinação requer a
É importante ressaltar que esse processo de ubiquitinação é reversível. Existem enzimas desubiquitinadoras (Dubs) que podem promover a desubiquitinação de proteínas ligadas a ubiquitinas.
1.6- Proteassoma e T. cruzi
González e colaboradores (1996) foram os primeiros grupos a mostrar que os inibidores MG132 e lactocistina preveniam a transformação de tripomastigota em amastigota em meio axênico. No T. cruzi, o envolvimento do proteassoma na
transformação estágio-específico de tripomastigota para amastigota tem recebido uma atenção especial por parte dos pesquisadores. O “turnover” de proteínas em tripomastigotas é dependente da via proteassoma-ubiquitina, sendo acentuada durante a transformação em amastigotas. Quando a atividade do proteassoma é inibida, profundas mudanças morfológicas ocorrem durante a transformação, demonstrando que as proteínas do citoesqueleto associadas ao flagelo são alvos da via dependente de ATP, ubiquitina e proteassoma (Gonzáles et al., 1996 e De Diego, J. et al.,2001).
O proteassoma 26S da forma epimastigota do T. cruzi foi identificado como
tendo um peso molecular de 1400 kDa, apresentando uma atividade quimotripsina-símile dependente de ATP ao substrato Suc-LLVY-Amc (De Diego, J.L. et al.,2001). Mais de 30 proteínas abrangendo 25-110 kDa foram detectadas no purificado de proteassoma 26S de T. cruzi (De Diego, J. et al., 2001).
Zou C. B. e colaboradores (2000) clonaram e caracterizaram pela primeira vez os genes RPN1-1 (Regulatory-Particle Non-ATPase subunit 1) e RPN1-2 do
proteassoma de T. cruzi. Além disso, esse grupo localizou a posição desses genes no
DNA cromossomal (2300 e 1900 kb, respectivamente). Os produtos gênicos, Rpn1-1 e Rpn1-2, atuam como a Nas1, uma subunidade homologa a Rpn1 em Saccharomyces
cerevisiae.
os inibidores lactocistina e MG132 bloquearam o crescimento do parasito e previniram a encistação (Makioka, A. et al. 2000 e Robertson, C.D 1999); no Plasmodium berghei,
a lactocistina bloqueou a replicação, porém, não impediu a entrada do parasito nas hemácias (Gantt, S.M. et al. 1998); no Plasmodium falciparum, a lactocistina bloqueou
a replicação de linhagens sensíveis e resistentes a cloroquinona (Certad, G. et al. 1999); no T. brucei, a lactocistina bloqueou a progressão do ciclo celular, contudo, não inibiu a
diferenciação de formas sangüíneas em formas procíclicas (Nkengu-Njinkeng, J. et al. 2002); finalmente, no T. cruzi, o MG132 e a lactocistina inibiram a transformação da
forma tripomastigota em amastigota, no entanto, não bloquearam a invasão celular (De Diego, J. et al. 2001).
Diferenças entre proteassomas dos protozoários e dos mamíferos envolvem imunoreatividade (Trypanossoma e Toxoplasma) e atividades de peptidases, nos mamíferos a atividade principal é quimotripsina-símile, enquanto que no T. cruzi a
atividade principal é tripsina-símile (revisado por Paugam A. et al., 2003).
Essas diferenças reforçam a possibilidade do proteassoma servir como um futuro alvo terapêutico. Entretanto, a limitada informação sobre a biologia do proteassoma de T. cruzi e a natureza das diferenças observadas com os proteassomas de
mamíferos requer estudos mais aprofundados tanto na parte estrutural quanto funcional para que se torne concreto o uso de drogas específicas que bloqueiam o proteassoma do
Considerando que uma das características marcantes do T. cruzi é a grande
heterogeneidade tanto genômica como fenotípica, entre as cepas e também clones de uma mesma cepa, entendemos que este parasito constitui não só um bom modelo para avaliar a relevância biológica da via ubiquitina-proteassoma como também um bom exemplo de um organismo onde alterações na proteólise mediada pelo proteassoma poderiam influenciar comportamentos biológicos como taxa de crescimento, patogenicidade e susceptibilidade às drogas.
Diante destas considerações, este trabalho teve como objetivo geral estudar aspectos moleculares e bioquímicos do proteassoma 20S em cepas com o fenótipo de resistência e susceptibilidade a drogas. De modo a estabelecer estas correlações, utilizamos as cepas do T. cruzi: 17LER, com resistência induzida “in vitro” ao
benzonidazol (BZ) e seu par sensível 17WTS; BZR com resistência selecionada “in vivo” ao BZ e seu par sensível BZS; CL (sensível) e Colombiana (naturalmente resistente ao BZ) e os seguintes objetivos específicos:
1- Localizar os genes beta-1, -2 e -5 nos cromossomos do parasito; 2- Verificar o tamanho desses transcritos por Northern Blot;
3- Determinar o número de cópias, utilizando a técnica de Southern blot; 4- Investigar a presença de conjugados proteína-ubiquitina;
5- Analisar os níveis de proteassoma 20S;
3.1-Obtenção de cepas
Todas as cepas utilizadas neste trabalho foram gentilmente cedidas pelo Laboratório de Parasitologia Celular (Centro de Pesquisa René-Rachou, Fiocruz) e encontram-se representadas na tabela 1. Os blocos dos parasitos utilizados na técnica de PFGE foram também cedidos pelo Laboratório de Parasitologia Celular (Centro de Pesquisa René-Rachou Fiocruz). Além das cepas listadas na tabela abaixo, foram utilizadas as cepas Yuyu, Noel, Berenice, Ja e Romano na técnica de PFGE.
Tabela 1- Característica das cepas utilizadas neste trabalho.
Cepas Origem Hospedeiro Susceptibilidade* Zimodema
17WTS México Triatomíneo Susceptível 1
17LER México Triatomíneo Resistente 1
BZS São Paulo Caso Agudo
Humano Susceptível 2
BZR São Paulo Caso Agudo Humano Resistente 2
Cl RGS** Triatomíneo Susceptível B
Colombiana Colômbia Caso Crônico Humano Resistente 1 * Susceptibilidade ao Benzonidazol (Murta e Romanha, 1998; Nirdé et al, 1995; Filardi & Brener, 1987).
**RGS= Rio Grande do Sul
3.2-Extração de RNA total e verificação da integridade
Nos experimentos de extração de RNA, a água utilizada foi inicialmente tratada com dietilpirocarbonato (DEPC-Sigma) na proporção de 1/1000, deixada em repouso por 12 horas e a seguir esterilizada com auxílio de uma autoclave, 1 atm 120°C, para tornar-se isenta de RNases. Todos os materiais, tais como: ponteiras, tubos eppendorfs, e vidrarias foram lavados com água DEPC e autoclavados por 40 minutos.
clorofórmio ao homogenado, e estes agitados vigorosamente por 1 minuto em vórtex. A mistura resultante foi incubada por 20 minutos à temperatura ambiente e a seguir centrifugada por 10 minutos a 10.000 xg. A fase aquosa foi então transferida para um
eppendorf de 1,5 mL e adicionado 500 µL de isopropanol (Sigma). A mistura foi homogenizada por inversão e incubada por 30 minutos a -20ºC. Após este período os tubos foram centrifugados por 10 minutos a 10.000 xg e o sobrenadante descartado. O
precipitado foi lavado com etanol 70% e centrifugado por 5 minutos a 10.000 xg, descartando novamente o sobrenadante.
Posteriormente, o RNA total foi mantido por 20 minutos à temperatura ambiente para a evaporação total do etanol. A seguir, foi ressuspenso em 25 a 50 µL de água DEPC. Para quantificar o RNA, foram diluídos 4 µL da solução em 1000 µL de água. A leitura foi processada em 260 e 280 nm, para estimar o grau de contaminação com proteínas. Segundo SAMBROOK e Cols., (1989), 1 unidade de absorção na DO a 260 nm corresponde a 40 µg de RNA/mL.
A integridade da preparação foi verificada em gel de agarose a 1,2% em MOPS 1X diluído com água DEPC (Mops 21g, Acetato de sódio diidratado 3,4g, EDTA tetrassódico 1,9g, água DEPC 500mL, o pH 7 e autoclavado) e encontra-se representado na figura 5. Antes de aplicar as amostras no gel, 10 µg de RNA foram adicionados a 15µl de tampão de amostra (formamida 187,5 µL, formaldeído 27,5 µL, Mops 10X 37,5 µL, azul de bromofenol [100mg/mL] 1 µL, água DEPC 42 µL e brometo de etídeo 0,5 µL). Essa mistura foi desnaturada por 15 minutos a 65°C, seguida de banho de gelo por 3 minutos. As amostras foram aplicadas no gel e adotou-se uma voltagem de 45 a 50 volts adotou-sendo o tampão de corrida MOPS 1X. O padrão de RNA (0,24 a 9,5 Kb Gibco) foi tratado nas condições descritas acima.
Figura 5: Análise em gel de agarose/formaldeído do RNA total de T. cruzi. Cerca de 5 µg
de RNA total obtido como descrito no item 3.2 foram analisados.
2,1
1,7
1,3
17
W
T
S
17
L
E
R
B
Z
S
B
Z
R
C
3.3-Transferência dos RNAs para a membrana de náilon
Após a corrida eletroforética, o formaldeído foi retirado do gel de agarose por meio de duas lavagens de 30 minutos cada, com solução de SSC 10X (solução estoque de SSC 20X= cloreto de sódio 175,3g, citrato trissódico 88,2g e água q.s.p 1000 mL em pH=7,0). O sistema de transferência foi montado de acordo com a seqüência: travessa de vidro Pirex (25 x 16 x 5 cm) contendo1000 mL de SSC 10X e
uma espuma com densidade 23, quatro papéis Whatman 3 mm de 16x16 cm, gel de agarose invertido com tamanho 15x15cm, uma membrana de náilon (Genescreen plus) 15x15 cm previamente molhada em água (rapidamente) e incubada durante 10 minutos em SSC 10X, quatro papéis Whatman 3 mm 15x15 cm molhados em SSC 10X (neste momento uma pipeta estéril foi rolada por cima dos itens mencionados para retirar as bolhas) e, finalmente, uma pilha de papel toalha (25 cm), uma placa de vidro e um peso de 0,5 Kg foram colocados em cima de todo o conjunto. A transferência dos RNAs do gel para a membrana, ocorreu por 12 horas à temperatura ambiente.
Após a transferência a membrana foi exposta a luz ultra violeta durante 5 minutos para fixar o RNA na membrana. Em seguida a membrana foi lavada por 10 minutos em SSC 2 X e acondicionada em papel Whatman 3mm até o uso.
3.4- Extração de DNA genômico
água milli-q e armazenados a 4ºC. A integridade do DNA genômico, foi analisada em gel de agarose a 0,7% corado com brometo de etídeo.
3.5- Digestão do DNA genômico com enzimas de restrição e transferência para membrana de náilon (Southern Blot)
Cerca de 5 µg de DNA genômico (DNAg) foram digeridos com uma quantidade apropriada de diversas enzimas de restrição, durante 16 horas, segundo recomendações do fabricante. As enzimas de restrição utilizadas para o gene beta 1 (855pb) foram: BamHI que corta o gene uma vez na posição 762 e XbaI que não corta o gene. Para o gene beta 2 (906pb) foram utilizadas: XhoI que corta o gene uma vez na posição 449 e XbaI que não corta o gene. Finalmente, para o gene beta 5 (933pb) foram utilizados: BamHI que corta uma vez na posição 148 e EcoRI que não corta o gene. Os fragmentos de DNA gerados foram submetidos a uma corrida em gel de agarose 0,7% sob uma voltagem de 40 V. Posteriormente, o gel foi corado com brometo de etídeo (0,5 µg/mL) durante 30 minutos, e lavado por 5 minutos para documentação.
3.6- Eletroforese em Gel de Campo Alternado (PFGE)
A metodologia PFGE foi utilizada para localizar os genes (β1, β2 e β5) nos
cromossomos do T. cruzi. A eletroforese de pulso alternado foi realizada utilizando o
aparelho “gene navigatorTM system” (Pharmacia). Os blocos contendo as diferentes cepas do T. cruzi foram cedidos pelo Laboratório de Parasitologia Celular
(LPCM/Fiocruz-BH). Os blocos foram colocados nos poços do gel de agarose à 1% em tampão TBE 1X. A corrida eletroforética foi realizada com uma voltagem constante de 180Volts (10-12 mA) a 9oC. Conforme as condições padronizadas no laboratório LPCM foram aplicados pulsos homogêneos (N/S, L/O) de 70 seg por 9h, 90 seg por 24h, 200 seg por 17h e 400 seg por 22h, sem interpolação. O gel foi corado com brometo de etídio (1µg/ml) e documentado pelo sistema Eagle Eye Stratagene II.
3.7- Transferência dos cromossomos do Gel de PFGE para a membrana de náilon
O DNA foi desnaturado incubando o gel em uma solução de HCl 0,25 M por 2 vezes de 20 minutos. A seguir o gel foi neutralizado em uma solução NaOH 0,5M e NaCl 1,5M e submetida a incubação por 2 vezes de 15 minutos. Finalmente, a solução foi descartada e o gel incubado por 2 vezes de 20 minutos em uma solução de NaCl 1,5M e Tris-HCl 0,5M pH 7,4. Posteriormente, os cromossomos foram transferidos para a membrana de náilon conforme descrito no item 3.5. Após a transferência, os cromossomos foram fixados na membrana pela exposição por 5 minutos em luz ultravioleta e a seguir lavada por 10 minutos em uma solução de SSC 2X.
3.8- Obtenção dos cDNAs
Os oligonucleotídeos utilizados para a obtenção dos cDNAs correspondentes aos genes das subunidades beta-1, -2 e -5 do proteassoma 20S, foram desenhados com base nas seqüências de nucleotídeos (EST) de T. cruzi, depositadas no
brucei, Leishmania sp e Plasmodium sp. Todas estas seqüências encontram-se
disponíveis em www.genedb.org/genedb/tcruzi. A Tabela 2 apresenta o número de acesso dos genes, o tamanho dos genes amplificados, o desenho dos iniciadores e a temperatura de anelamento adotada.
Tabela 2- Número de acesso, seqüência dos iniciadores, tamanho esperado e temperatura de anelamento para os genes Beta-1, -2 e -5.
Gene Número de acesso Seqüência dos iniciadores Tamanho T. M
Beta-1 Tc00.1047053507603.40 Tc00.1047053509429.110
F:5’GGAGCATTGCTTCATGAG3’ R:5’TCGCAGAGCTGCTGAATG3’
798pb 50°C
Beta-2
Tc00.1047053508461.430 Tc00.1047053510287.30 Tc00.1047053503891.100
F: 5’TGGTTATTATGGCATGTG3’
R: 5’TGTTTCGGGTACTTACGC3’ 809pb 51°C
Beta-5 Tc00.1047053507639.40 Tc00.1047053503781.70
F:5’CAACGCTTAGCTCGTATG3’
R:5’CTACAACACGTAGCGATC3’ 805pb 49°C
TM = Temperatura de Anelamento.
A primeira fita do cDNA foi obtida utilizando 5 µg de RNA total e o kit thermoscript RT-PCR (GIBCO-BRL). As condições para esse experimento foram exatamente iguais às fornecidas pelo fabricante.
Para a obtenção do cDNA, utilizamos um programa de amplificação com 35 ciclos, cada um composto por: uma etapa de desnaturação 95°C por 1 minuto, 1 minuto de anelamento (conforme indicado na Tabela 2) e 1 minuto e 30 segundos de extensão a 72°C. A seguir uma alíquota dessa reação foi analisada em gel de agarose a 1,2%, e os cDNAs visualizados pela coloração com brometo de etídeo. A identidade do produto obtido foi confirmada por seqüênciamento.
de gDNA obtido conforme descrito no item 3.4. Esses resultados encontram-se representados na figura 6.
Figura 6: Gel de agarose 1,2% corado com brometo de Etídeo. Amplificação dos genes
beta-1, -2 e -5, com diferentes temperaturas de anelamento (50, 51 e 49ºC, respectivamente).
3.9- Purificação dos cDNAs
Foram adicionados 15 µL de acetato de sódio 3M, pH 7, e 375 µL de etanol absoluto em 150 µL dos cDNAs obtidos como descrito no item 3.8. Essa mistura foi incubada a -20°C por 30 minutos, seguida de uma centrifugação por 10 minutos a 14.000 xg sendo o sobrenadante descartado. A seguir, 500 µL de etanol 70% foram adicionados ao precipitado seguido de uma centrifugação a 14.000 xg por 5 minutos. O sobrenadante foi novamente descartado e o precipitado ressuspendido em 10 µL de água estéril. Após a purificação uma alíquota foi analisada em gel de agarose.
3.10- Obtenção das sondas
Para marcação do DNA com P32, foram utilizados 5 µL dos cDNAs purificados e 33 µL de água. As amostras foram incubadas a 96ºC por cinco minutos, seguido de banho de gelo por 2 minutos. Após a desnaturacao, foi adicionado à mistura 5 µL de mix dNTP menos dCTP, 5 µL do fragmento Klenow da DNA polimerase I (AMBOS DO KIT nicktranslation – INVITROGEN) e 2 µL de dCTP marcado com 32P na posição α, este último manipulado dentro da câmara de acrílico. A solução foi então
incubada a 16ºC por uma hora, sendo a reação interrompida pela adição de tampão de parada, fornecido pelo kit. Para a remoção dos nucleotídeos não incorporados, as sondas foram purificadas em uma coluna de Sephadex G-50, previamente equilibrada com o tampão NT (Tris-HCL 10 mM, NaCl 50 mM, EDTA 0,1 mM pH=8,0).
Βeta-1 Βeta-2 Βeta-5
Para a obtenção das sondas não radioativas, utilizamos como molde 250 ng dos cDNAs correspondentes aos genes beta-1, -2 e -5. As sondas marcadas com fosfatase alcalina foram obtidas utilizando o kit Gene Images AlkaPhos direct Labeling
and Detection System (Amersham Biosciences), conforme instruções do fabricante.
3.11- Reação de hibridização
Primeiramente, as membranas de náilon foram pré-hibridizadas, em cilindros, com a solução de pré-hibridização (Tampão Fosfato de Sódio 0,5 M pH 6,8, EDTA 0,5 M, água destilada, BSA 5%, SDS 7%). Foi utilizada a temperatura de 56°C para hibridizar as membranas de Northern e Southern e 60°C para hibridizar as
membranas de PFGE. As membranas foram incubadas em forno de hibridização durante 1 hora.
Depois desse período, foi adicionada a sonda radioativa, previamente desnaturada por 5 minutos a 100°C, com atividade específica de 2x108 µCi/µL. A reação de hibridização foi realizada durante 16 horas, sob agitação leve.
As membranas a serem marcadas não radioativamente foram acondicionadas em sacos plásticos com cerca de 20 mL de tampão de hibridação (0,25 mL/cm2). Esse conjunto foi pré-hibridado em banho Maria a 55°C por 30 minutos.
Após este intervalo, foram adicionados 32 µL da sonda específica marcada com fosfatase alcalina, e a reação foi incubada por 18 horas em banho Maria a 55°C.
3.12- Lavagem das membranas
As membranas marcadas com sondas não radioativas foram transferidas para novos sacos plásticos e lavadas duas vezes de 10 minutos a 55°C com tampão primário (uréia 2M, SDS 0,1%, fosfato de sódio 0,5M pH 7, NaCl 150mM, MgCl2
1mM, reagente de bloqueio 0,2%). As membranas foram transferidas para recipientes plásticos e lavadas por 2 vezes de 5 minutos a temperatura ambiente sob agitação com tampão secundário (solução estoque 20 vezes: Tris-Base 1M, NaCl 2M, pH 10). Após este período elas foram secadas em papel de filtro.
3.13- Autoradiografia
Para verificar as regiões de homologia, as membranas marcadas com radioativo foram embrulhadas em sacos plásticos e acondicionadas em cassetes contendo intensificadores e filmes Kodak (X-OMAT-XAR-5). Os cassetes foram acondicionados a -70ºC por 1 semana. As revelações foram feitas com as soluções da Kodak, de acordo com as instruções do fabricante (Revelador e Reforçador GBX Kodac e Fixador e Revelador GBX Kodac).
As membranas marcadas não radioativamente foram acondicionadas em cassetes juntamente com 2 mL de reagente de detecção (CDP-StarTM, Amersham
Bioscience). Esse conjunto foi deixado por 20 horas. As revelações foram feitas de acordo com as instruções do fabricante.
3.14- Obtenção do extrato bruto de proteínas totais do parasito
Inicialmente, a 2x108 epimastigotas foram adicionados 1 mL do tampão de homogeneização (Tris-HCl 5mM pH 8,0, glicerol 1%, EDTA 1mM, leupeptina 50µM e
β-mercaptoetanol 1mM). A seguir, a mistura foi estocada em banho de gelo e sonicada
em sonicador adotando-se 4 pulsos de 45 segundos com intervalos de 1 minuto.
3.15- Preparação de fração enriquecida de proteassoma
Inicialmente 4 x 108 formas epimastigotas do T. cruzi foram
homogeneizadas em 1mL de tampão 20S (Tris-HCl 25 mM pH=7,5; DTT 1 mM, Glicerol 10%, EDTA 1mM, Leupeptina 1 mM, NEN 10 mM). O homogenado foi transferido para um eppendorff e submetido a uma sonicação por quatro vezes de 45 segundos a 60 watts com intervalo de 30 segundos em banho de gelo. A seguir, as amostras foram centrifugadas por 20 minutos a 10.000 g e o sobrenadante descartado. Feito isso, o material resultante foi ultracentrifugado por 1 hora a 100.000 g. Posteriormente, o sobrenadante foi transferido para um novo eppendorf e centrifugado por 6 horas nas mesmas condições. O precipitado foi ressuspendido em 100 µL do tampão 20S. A determinação da concentração protéica foi realizada segundo o método de Lowry (1951), usando soro albumina bovina para a construção da curva padrão.
3.16- SDS-PAGE e ensaios de Western Blot
Cerca de 20 µg de extrato bruto ou 5 µg da fração enriquecida de proteassoma foram fracionados em gel de SDS-PAGE 10% como descrito por Laemmli (1970). Foi utilizado como padrão de peso molecular o MultiMark Multi-Colored Standart (Invitrogen). A voltagem adotada foi de 120 Volts. Após a corrida, um dos géis foi corado com comassie blue. O outro gel foi preparado para a transferência de acordo com o método descrito por TOWBIN e Cols., 1979, com modificações no tampão de transferência (Tris-HCl 25 mM pH 8,3, glicina 192 mM, etanol 18% e SDS 0,02%). A voltagem aplicada no sistema foi de 25 Volts a uma temperatura de 4ºC por 16 horas. Após o término da transferência, a membrana foi corada com Ponceau (0,25% em ácido acético 1%) por 5 minutos e descorada com água para visualização das proteínas.
proteassoma 20S e anti-ubiquitina para detectar proteínas ubiquitinadas) numa diluição 1:1000 em TBS-T por 3 horas à temperatura ambiente. O anticorpo foi removido e a membrana lavada três vezes rapidamente com TBS-T. Em seguida a membrana foi incubada por 1 hora, à temperatura ambiente, com o anticorpo secundário (anti-IgG de rato conjugado com fosfatase alcalina para a detecção das subunidades α e anti-IgG de
coelho conjugado com fosfatase alcalina para a detecção de proteínas ubiquitinadas) na diluição 1:1000. Após este período, a membrana foi lavada por três vezes durante 5 minutos em tampão de revelação (Tris pH=9,5, NaCl 5M, MgCl2 1M e H2O q.s.p 100ml). Finalmente, foram adicionados 5 mL de tampão de revelação colorimétrica, 66 µL de NBT e 33 µL de BCIP agitando-se o conteúdo por 30 minutos.
3.17- Medida da atividade proteolítica exógena dos parasitos
Nos ensaios da atividade proteolítica exógena foram utilizados diferentes substratos fluorogênicos como: Ac-Tyr-Val-Ala-Asp-7-amido-4-metilcumarina, Cbz- Gly-Gly-Arg-7-amido-4-metilcumarina e Suc-Leu-Leu-Val-Tyr-7- amido-4-metilcumarina para a determinação das atividades semelhantes a caspase, tripsina e quimotrisina, respectivamente, do proteassoma na fração citosólica das formas epimastigotas de T. cruzi.
Foram utilizados 50µg de proteínas totais que foram obtidas após centrifugação a 100000xg por 2 horas e 13µM dos substratos fluorogênicos. O tampão
utilizado foi Tris-HCl 50mM pH 8,0 e MgCl2 10mM, na presença ou ausência do
4.1- Localização Cromossômica dos genes beta-1, -2 e -5
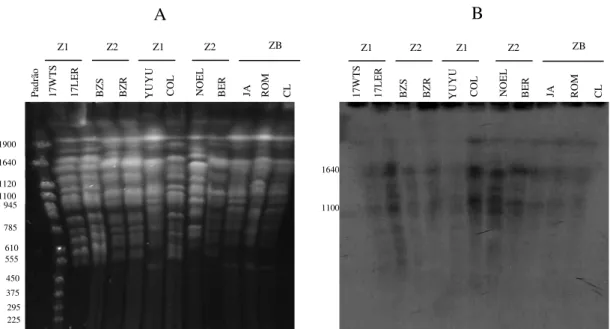
Para a localização cromossômica dos genes Beta-1, -2 e -5 utilizamos a técnica de eletroforese em gel de campo alternado (PFGE) como descrito em Material e Métodos. As figuras 7A, 8A e 9A mostram o perfil cromossômico das amostras de T.
cruzi coradas com brometo de etídio. Observamos um perfil heterogêneo de bandas
cromossômicas entre as diferentes cepas do parasito, variando de 800 a 3000 Kb. Os cromossomas foram transferidos do gel de agarose para a membrana de náilon, conforme descrito em Material e Métodos. A hibridização dos cromossomas com as sondas dos genes Beta-1, -2 e -5 (figuras 7B, 8B e 9B) mostrou heterogeneidade de tamanho e número dos cromossomas entre as diferentes populações do T. cruzi que
contém estes genes.
A hibridização dos cromossomas com a sonda do gene Beta-1 (Fig. 7B), mostrou que este gene está localizado em bandas cromossomais entre 1640 e 1100 Kb, uma vez que observamos uma forte intensidade de hibridização da sonda nesta região. Algumas cepas apresentaram fraco perfil de hibridização (17LER, Yuyu e Colombiana), mas mesmo assim podemos visualizar bandas nesta região. Na amostra 17WTS observamos claramente a presença do gene Beta-1 nas bandas cromossomais de 1640, 1120 e 1100 Kb.
O perfil de hibridização com a sonda Beta-2 (Fig. 8B) não esta muito bem definido, pois várias bandas cromossomais foram reconhecidas pela sonda, porém com baixa intensidade. Este experimento está sendo repetido com uma seqüência mais específica, diferente daquela inicialmente usada como sonda. Em relacão ao gene Beta-5 (Fig. 9B), observamos que este gene está localizado principalmente em duas bandas cromossomais de 1640 e 1100 Kb em todas amostras do T. cruzi analisadas, com maior
Figura 7: Localização cromossomal do gene beta-1 em cepas de T. cruzi com diferentes
zimodemas e fenótipos de resistência a drogas. (A) Bandas cromossomais das cepas de T.
cruzi foram separadas por PFGE e coradas com brometo de etídeo. (B) Southern blots dos
cromossomos com uma sonda específica para o gene β1 marcado com 32P. O marcador de peso molecular foi obtido dos cromossomas de Saccharomyces cerevisae.
Figura 8: Localização cromossomal do gene beta-2 em cepas de T. cruzi com diferentes
zimodemas e fenótipos de resistência a drogas. Em A, bandas cromossomais das cepas de T.
cruzi foram separadas por PFGE e coradas com brometo de etídeo. Em B, Southern blots dos
cromossomos com uma sonda específica para o gene β2 marcado com 32P. O marcador de peso molecular foi obtido dos cromossomas de Saccharomyces cerevisae.
17 W T S 17 L E R B Z S B Z R Y U Y U C O L N O E L B E R
JA RO
M
C
L
Z1 Z2 Z1 Z2 ZB
17 W T S 17 L E R B Z S B Z R Y U Y U C O L N O E L B E R
JA RO
M
C
L
Z1 Z2 Z1 Z2 ZB
Pa dr ão 1900 1640 1120 1100 945 915 815 785 610 790 820
A B
1640 1120 1110 1640 a 1100 Kb 17 W T S 17 L E R B Z S B Z R Y U Y U C O L N O E L B E R
JA RO
M
C
L
Z1 Z2 Z1 Z2 ZB
17 W T S 17 L E R B Z S B Z R Y U Y U C O L N O E L B E R
JA RO
M
C
L
Z1 Z2 Z1 Z2 ZB
Pa dr ão 1900 1640 1120 1100 945 450 375 785 610 555 610
A B
Figura 9: Localização cromossomal do gene beta-5 em cepas de T. cruzi com diferentes
zimodemas e fenótipos de resistência a drogas. Em A, bandas cromossomais das cepas de T.
cruzi foram separadas por PFGE e coradas com brometo de etídeo. Em B, Southern blots dos
cromossomos com uma sonda específica para o gene β5 marcado com 32P. O marcador de peso molecular foi obtido dos cromossomas de Saccharomyces cerevisae.
4.2- Determinação do número de cópias dos genes beta-1, -2 e -5.
A técnica Southern blot foi utilizada para estimar o número de cópias dos
genes beta-1, -2 e -5 no genoma do T. cruzi. Para isso, cerca de 5 µg de DNA genômico foram digeridos com enzimas de restrição e submetidos à eletroforese em gel de agarose 0,8%. As enzimas de restrição utilizadas nas digestões foram selecionadas com base no mapa de restrição teórico dos genes, utilizando o programa Gene Runner e/ou a base de dados contida no site da Baylor College of Medicine (BCM search launcher).
Para o gene beta-1, utilizamos a enzima BamHI por apresentar sítio interno
na seqüência e XbaI, que não apresenta sítio. O resultado de digestão do DNA com a
enzima BamHI (Fig. 10B), mostrou que todas as cepas do T. cruzi analisadas
apresentam um mesmo padrão de bandas, ou seja, a sonda reconhece bandas de 1.48, 1.61, 2.47, 2.7 e 3.6 Kb. Para a outra enzima analisada, XbaI, a sonda reconheceu três
bandas de 1,47, 1,6 e 3,4 Kb, em todas as cepas em estudo. Desses resultados podemos concluir que há pelo menos 2 cópias do gene beta-1 por genoma haplóide do parasita.
17 W T S 17 L E R B Z S B Z R Y U Y U C O L N O E L B E R
JA RO
M
C
L
Z1 Z2 Z1 Z2 ZB
17 W T S 17 L E R B Z S B Z R Y U Y U C O L N O E L B E R
JA RO
M
C
L
Z1 Z2 Z1 Z2 ZB
Figura 10- Análises de Southern Blot do gene beta-1 (855 pb) nas cepas do T. cruzi
sensiveis e resistentes ao BZ. Em A, o DNAg dos parasitos digeridos com as enzimas BamHI
(corta uma vez o gene na posição 766pb) e XBaI (que não corta o gene) foram separados em gel de agarose a 0,7% em seguida foram transferidos para membrana de náilon. Em B, os genes beta-1 da membrana de náilon foram hibridizadas com sondas não radioativas e exposta a filme X-OMAT-XAR-5 (Kodak) representado na figura.
Para o gene beta-2, utilizamos a enzima XhoI por apresentar sítio interno na
seqüência e XbaI, que não apresenta sitio.
A Figura 11B mostra que todas as cepas analisadas apresentam um mesmo padrão de bandas, ou seja, a sonda reconhece bandas com: 1,46, 1,68, 2,86 e 3,75Kb devido à presença de um sítio interno para a enzima XhoI. Para a outra enzima
analisada, XbaI houve a presença de três bandas com 1,41, 1,55 e 3,19 Kb
respectivamente, em todas as cepas em estudo. Semelhante ao gene 1, o gene beta-2 também apresentou pelo menos beta-2 cópias por genoma haplóide do parasito.
17 W T S 17 L E R B Z S B Z R C L C ol 17 W T S 17 L E R B Z S B Z R C L C ol 17 W T S 17 L E R B Z S B Z R C L C ol 17 W T S 17 L E R B Z S B Z R C L C ol
BamHI XbaI BamHI XbaI
A B
Figura 11- Análises de Southern Blot do gene beta-2 (906 pb) nas cepas do T. cruzi
sensíveis e resistentes ao BZ. Em A, o DNAg dos parasitos digeridos com as enzimas XhoI
(corta uma vez o gene na posição 229pb) e XBaI (que não corta o gene) foram separados em gel de agarose a 0,7% em seguida foram transferidos para membrana de náilon. Em B, os genes beta-2 da membrana de náilon foram hibridizadas com sondas não radioativas e exposta a filme X-OMAT-XAR-5 (Kodak) representado na figura.
Para o gene beta-5, utilizamos a enzima BamHI por apresentar sítio interno
na seqüência e EcoRI, que não apresenta sitio.
Na Figura 12B podemos notar que a sonda Beta-5 reconhece bandas com: 0,33, 0,73, 1,07, 1,43, 1,59 e 3,03 Kb devido à presença de um sítio interno para a enzima BamHI. Para a enzima EcoRI houve a presença de três bandas com 1,55, 1,67 e
3,07 Kb respectivamente, em todas as cepas em estudo. Desses resultados podemos concluir que também há pelo menos 2 cópias do gene Beta-5 por genoma haplóide do parasito.
A B
XhoI XbaI XhoI
Figura 12- Análises de Southern Blot do gene beta-5 (933 pb) nas cepas do T. cruzi
sensiveis e resistentes ao BZ. Em A, o DNAg dos parasitos digeridos com as enzimas BamHI
(corta uma vez o gene na posição 148pb) e EcoRI (que não corta o gene) foram separados em gel de agarose a 0,7% em seguida foram transferidos para membrana de náilon. Em B, os genes beta-5 da membrana de náilon foram hibridizadas com sondas não radioativas e exposta a filme X-OMAT-XAR-5 (Kodak) representado na figura.
4.3- Northern Blot
A expressão dos genes que codificam para as subunidades Beta-1, -2 e -5 foram verificados utilizando a técnica de Northern blot. Com esses experimentos
tivemos por objetivo estimar o tamanho dos transcritos codificados pelos genes, além de analisar se havia diferenças de expressão entre cepas sensíveis e resistentes ao BZ. Para isso, 15 µg de RNA total foram submetidos à eletroforese em gel de agarose formaldeído 1,2%, e posteriormente, transferidos por capilaridade em membrana de náilon. As membranas foram hibridizadas com sonda radioativa (32P) e autoradiografadas em filmes Kodak (X-OMAT_XAR-5), conforme descrito em Material e Métodos.
17 W T S 17 L E R B Z S B Z R C L C ol 17 W T S 17 L E R B Z S B Z R C L C ol 17 W T S 17 L E R B Z S B Z R C L C ol 17 W T S 17 L E R B Z S B Z R C L C ol
BamHI BamHI
EcoRI EcoRI
A B
O resultado apresentado na Figura 13A mostra que para o gene beta-1, além da mensagem com 1,09 Kb (esperado) também detectamos duas outras mensagens, fracas com 1,96 e 1,35 Kb, respectivamente. Na Figura 13B, mostramos a normalização da membrana utilizando como sonda o gene do rRNA 24S que reconhece uma banda de 1,84 Kb. Podemos notar que todas as amostras analisadas apresentam quantidades equivalentes de RNA. A razão entre o gene beta-1 e o rRNA 24S, considerado constitutivo em T. cruzi, sugere que não há aumento real de transcrição entre as
diferentes cepas deste parasito, utilizadas neste trabalho.
Para o gene beta-2, identificamos dois transcritos com 1,15 e 2,06 Kb , como poder ser observado na Figura 14A. Também podemos notar a presença de quantidades equivalentes de RNA entre as amostras estudadas e níveis equivalentes de RNA para o gene beta-2 nas cepas em estudo (figura 14B e 14C, respectivamente).
A figura 15A mostra a presença de duas mensagens para o gene beta-5, com 1.17 e 2,03 Kb, respectivamente. Como mostram as figuras 15B e 15C, não observamos para o gene beta-5 variações nos níveis de RNA entre as diferentes cepas estudadas.
Esses resultados sugerem que as bandas maiores e mais fracas, detectadas em todos os genes estudados podem representar transcritos policistrônicos, uma vez que apresentam o dobro do tamanho esperado e que o transcrito completo para os genes beta-1, -2 e -5 apresentam 1,09, 1,15 e 1,17 Kb, respectivamente.
Figura 13- Níveis de mRNA do gene beta-1 em cepas de T. cruzi. 15 µg de RNA total foram
separados em gel de agarose/formaldeído 1,2% e transferidos para membrana de náilon. Em A, encontram-se representados os transcritos marcados com sondas específicas para o gene beta-1. Em B, temos a normalização utilizando genes constitutivos rRNA 24S. Em C, temos um gráfico que representa a razão da intensidade entre o transcrito principal e o rRNA 24S.
B A 1,09 1,35 1,96 B A 1,09 1,84 17 W T S 17 L E R B Z S B Z R C L C ol
0 0,2 0,4 0,6 0,8 1 1,2
Razão transcrito/rRNA 24S
Figura 14- Nível de mRNA do gene beta-2 em cepas de T. cruzi. 15 µg de RNA total foram
separados em gel de agarose/formaldeído 1,2% e transferidos para membrana de náilon. Em A, encontram-se representados os transcritos marcados com sondas específicas para o gene beta-2. Em B, temos a normalização utilizando genes constitutivos rRNA 24S. Em C, temos um gráfico que representa a razão da intensidade entre o transcrito principal e o rRNA 24S.
Figura 15- Nível de mRNA do gene beta-5 em cepas de T. cruzi. 15 µg de RNA total foram
separados em gel de agarose/formaldeído 1,2% e transferidos para membrana de náilon. Em A, encontram-se representados os transcritos marcados com sondas específicas para o gene beta-5. Em B, temos a normalização utilizando genes constitutivos rRNA 24S. Em C, temos um gráfico que representa a razão da intensidade entre o transcrito principal e o rRNA 24S.
4.4- Detecção de conjugados proteína-ubiquitina.
Em eucariontes superiores uma das funções da ubiquitina é a de modificações de substratos protéicos, formando conjugados proteína-ubiquitina. Nesta reação, diversas moléculas de ubiquitina são adicionadas a um substrato protéico num A 1,17 B 17 W T S 17 L E R B Z S B Z R C L C ol 2,03 1,84 A 1,15 B 17 W T S 17 L E R B Z S B Z R C L C ol 2,06
1,84 0 0,2 0,4 0,6 0,8 1
Razão transcrito/rRNA 24S
17WTS 17LER BZS BZR CL COL C e p a s Beta-2 C
0 0,2 0,4 0,6 0,8 1
Razão transcrito/rRNA 24S
processo denominado ubiquitinação. Desta forma, também foi de interesse neste trabalho verificar o perfil de proteínas poliubiquitinadas em diferentes cepas de T. cruzi.
Para isso extratos totais de cepas de T. cruzi na forma epimastigota obtidos
como descrito em Material e Métodos (item 3.14), foram separados por eletroforese em gel de poliacrilamida (SDS-PAGE) a 10%, transferidos para membranas de PVDF e incubados com anticorpos policlonal anti-ubiquitina de coelho seguido de anticorpo secundário anti-IgG de coelho conjugado com fosfatase alcalina conforme descrito em Material e Métodos.
Os resultados apresentados na Figura 16B mostram o mesmo perfil de ubiquitinação em todas as cepas analisadas, além disso, nenhum acúmulo de bandas protéicas conjugadas a ubiquitina foi observado.
Figura 16- Detecção de conjugados proteína-ubiquitina. Em A, as bandas protéicas (10 µg
de proteínas) após eletroforese em gel de acrilamida a 10% foram coradas com Azul de Comassie. PM- padrão de peso molecular (Invitrogen). Em B, as bandas protéicas foram transferidas para membrana de nitrocelulose e a seguir incubadas com anticorpos anti-ubiquitina de coelho e posteriormente revelados como descrito em Materiais e Métodos.
4.5- Detecção do proteassoma 20S
Como mencionado na introdução, o proteassoma 20S apresenta dois tipos de subunidades: subunidades α e subunidades β. Essas subunidades formam quatro anéis
heptaméricos (α1-7, β1-7, β1-7, α1-7) superpostos formando o complexo proteolítico
20S. Neste trabalho detectamos os níveis de subunidades alfa e, conseqüentemente, do
17WTS 17LER BZS BZR CL COL 17WTS 17LER BZS BZR CL COL
PM
250 148
60
42
30
22
proteassoma 20S no padrão de cepas de T. cruzi estudados. Para a detecção das
subunidades estruturais, utilizamos à técnica de Western Blot e amostras de proteínas
totais extraídas das formas epimastigotas do T. cruzi estudadas.
A figura 17B mostra o padrão de expressão do proteassoma 20S. Podemos observar que todas as cepas sensíveis ao benzonidazol (17WTS, BZS e CL) apresentam uma maior expressão do proteassoma 20S em relação às cepas resistentes.
Figura 17- Detecção das subunidades α do proteassoma 20S em um padrão de cepas
sensíveis e resistentes ao benzonidazol. Em A, cerca de 10 µg de proteínas totais de cepas de
T. cruzi foram coradas com Azul de comassie. PM- padrão de peso molecular (Invitrogen). Em
B, as bandas protéicas foram transferidas para membrana de nitrocelulose e a seguir incubada com anticorpos anti-subunidades α de ratos e, posteriormente, revelada como descrito em Material e Métodos.
4.6- Atividades enzimáticas do proteassoma 20S
O proteassoma 20S apresenta três subunidades beta com funções peptidásicas: Beta-1 (atividade peptidásica semelhante à caspase), Beta-2 (atividade peptidásica semelhante à tripsina) e Beta-5 (atividade peptidásica semelhante à quimotripsina).
Para a medida da atividade enzimática semelhante à caspase utilizamos o substrato fluorogênico: Ac-Tyr-Val-Ala-Asp-7-amido-4-metilcumarina. As medidas de proteólise foram realizadas na presença e na ausência do inibidor aldeído peptídeo
17WTS 17LER BZS BZR CL COL PM
250 148
60
42
30
22
17WTS 17LER BZS BZR CL COL