“Study of the role of RAF-1 in MAPK signaling in thyroid
cancer cells”
Dissertação de Mestrado
2º Ciclo de Biotecnologia para as Ciências da Saúde
L
ISANDRA
M
ARISA
F
LORES
C
ASTRO
Trabalho realizado sob a orientação da
Professora Doutora Ana Preto
E co-orientação da
Professora Doutora Paula Soares
UNIVERSIDADE DE TRÁS-OS-MONTES E ALTO DOURO
“Study of the role of RAF-1 in MAPK signaling in thyroid
cancer cells”
Dissertação de Mestrado
2º Ciclo de Biotecnologia para as Ciências da Saúde
L
ISANDRA
M
ARISA
F
LORES
C
ASTRO
Trabalho realizado sob a orientação da
Professora Doutora Ana Preto
E co-orientação da
Professora Doutora Paula Soares
___________________________________
Orientadora:
Professora Doutora Ana A. Lopes Preto de Almeida
___________________________________
Co-Orientadora:
Professora Doutora Ana Paula Soares Dias Ferreira
___________________________________
Diretor do Curso de Mestrado:
Professor Doutor Valdemar Pedrosa Carnide
_____________________________________
Júri de Apreciação Presidente: ________________________________________________________ 1º Vogal: _________________________________________________________ 2º Vogal: __________________________________________________________ Classificação: __________________________ Data: ____ / ____ / ____
Este trabalho foi realizado no Departamento de Biologia da Universidade do Minho e no IPATIMUP sob a supervisão da Professora Doutora Ana Preto e da Professora Doutora Paula Soares.
ix
ACKNOWLEDGEMENTS
Chegada ao fim de mais uma importante etapa da minha vida, gostaria de expressar o meu sincero reconhecimento pelo apoio, disponibilidade e colaborações concedidas por diversas pessoas que de forma directa ou indirecta contribuíram para a realização deste trabalho. A elas, os meus mais sinceros agradecimentos:
À Professora Doutora Ana A. Lopes Preto de Almeida, quero agradecer desde já, por ter aceite ser minha orientadora, pelo voto de confiança, por todos os ensinamentos transmitidos quer a nível profissional quer a nível pessoal, pela revisão crítica do texto, pelo apoio, disponibilidade, atenção, preocupação e sobretudo pela paciência e tolerância que teve comigo. Foi sem dúvida um prazer realizar este trabalho e uma honra trabalhar consigo. O meu muito obrigada!
À Professora Doutora Ana Paula Soares Dias Ferreira, por ter aceite ser minha co-orientadora de tese. Desde já agradeço pela disponibilidade, pelos ensinamentos transmitidos, pela atenção, pela grande ajuda prestada na revisão crítica do texto, pela paciência e tolerância. Ficará para sempre na memória.
À Comissão de Projectos e à Coordenação do mestrado da UTAD, pela aceitação do projecto de mestrado.
Aos meus Colegas de Laboratório quer do IPATIMUP quer da Universidade do
Minho, por me terem recebido e por se terem mostrado sempre disponíveis para ajudar
em tudo. À Joana Nunes, Joana Torres, ao João Vinagre, Joana Silva, Ricardo Celestino e Sara Alves, por me terem ajudado à integração no trabalho de laboratório. Ao Cristovão e Jorge Lima, pelos ensinamentos e pelas dicas brilhantes que me deram para a realização deste trabalho. Às meninas, em especial à Suellen, à Stéphanie, à Paula, à Vera, à Cristina Carvalho, à Ana Oliveira, à Carla, à Dalila, à Juliana, à Alice, à Carolina e à Eugénia pela boa disposição, alegria e companheirismo durante todas as horas passadas no laboratório e pela ajuda sempre prestada quando necessário.
x
À Sara, pela preciosa ajuda, pela tua disponibilidade e incansável dedicação mesmo quando estavas no estrangeiro e cheia de trabalho! Um obrigada muito especial!
Aos colegas de mestrado e aos amigos de Vila Real. Obrigado pela amizade e pelo companheirismo. A vida sem amigos de fato não é nada.
À minha família em especial aos meus pais e à minha avó materna, porque sem eles nada faria sentido. A eles dedico todo o meu esforço e todo o meu trabalho.
xi
Study of the role of RAF-1 in MAPK signaling in thyroid cancer cells
ABSTRACT
Thyroid cancer is the most common endocrine malignancy, accounting to 94% of endocrine system cancers and 66% of deaths due to cancers of the endocrine origin. BRAF, RAS mutations and RET/PTC rearrangements are among the most frequent events in thyroid cancer. These mutations are associated with aberrant activation of the MAPK- ERK signaling pathway and/or the activation of the PI3K pathway.
In a previous work, our group demonstrated that inhibition of BRAF does not change the levels of pERKs in TPC-1, a papillary thyroid cancer (PTC) cell line harboring a RET/PTC1 rearrangement. It was also sugested that in TPC-1 cells RAF-1 seems to be the dominant molecule, but little is known about the role of RAF-1 in MAPK-ERK signaling regulation in those cells. Thus, we hypothesized that RAF-1 might be the molecule responsible for RET/PTC1-MAPK-ERK signaling and might be essential for the survival of those cells.
In the present study, we aimed to assess the role of RAF-1 in MAPK signaling in thyroid cancer cells harboring a RET/PTC1 rearrangement in comparison to cells with RAS and BRAF mutations. We tested the phenotypic alterations induced by RAF-1 and/or BRAF inhibition by RNAi as well as induced by MEK or PI3K pathway inhibition in TPC-1 cells harboring RET/PTC1 rearrangement, and compare them with C643 and 8505C cells harboring a HRASG13R and BRAFV600E mutations respectively. In TPC-1 cell line, inhibition of RAF-1 decreased proliferation and increased apoptosis to a greater extent, comparatively with BRAF inhibition, although no effect in the levels of ERK activation was observed. In the 8505C and C643 cell lines, RAF-1 inhibition did not affect significantly proliferation, apoptosis and ERK activation (with exception for C643) in contrast to the effect observed upon BRAF inhibition in these cells. These results show that in cell lines with BRAF and RAS mutations, BRAF is the main regulator of ERK activation. Moreover, our data support the hypothesis that thyroid cancer cells with RET/PTC1 rearrangement are more dependent on RAF-1 and less dependent on BRAF signaling pathway for proliferation and apoptosis than cells with RAS or BRAF activating mutations.
We also tested a MEK inhibitor (PD98059) and a PI3K inhibitor (LY294002) and little or no effects were observed in the levels of pERKs respectively sugesting that
xii
ERK activation in TPC-1 is activated by MEK but discretely and not regulated by PI3K. We also showed that in TPC-1 cells regulation of proliferation is more dependent on the PI3K-AKT signaling in comparison with RAF-1 and BRAF pathways. Our data sugest that in TPC-1 cells, RET/PTC1 might activate ERKs by a mechanism independent of BRAF, RAF-1, PI3K and the classical MAPK-ERK pathway.
Taken together, our results support a prime hypothesis that TPC-1 thyroid cancer cells might have an alternative mechanism of ERK activation such as MAPK-ERK pathway mutations.
Understanding the mechanisms underlying the constitutive ERK activation in thyroid cancer cells with RET/PTC1 rearrangements, might lead to find new therapeutic approaches, of prime importance for these type of cancer.
xiii
RESUMO
O cancro da tireóide é o tumor maligno mais comum do sistema endócrino, correspondendo a 94% dos cancros do sistema endócrino e a 66% das mortes de cancros de origem endócrina. Mutações nos genes BRAF e RAS e rearranjos do RET/PTC estão entre os eventos mais frequentes no cancro da tireóide. Estas mutações estão associadas com a ativação aberrante da via de sinalização MAPK-ERK e/ou a ativação da via PI3K.
Num trabalho anterior, o nosso grupo demonstrou que a inibição do BRAF não altera os níveis das pERKs na TPC-1, uma linha celular de cancro papilar da tireóide (PTC) com o rearranjo RET/PTC1. Também se sugeriu que nas células TPC-1, o RAF-1 parece ser a molécula dominante, mas pouco se conhece acerca do papel do RAF-1 na regulação da sinalização MAPK-ERK nestas células. Deste modo, colocámos a hipótese de que o RAF-1 possa ser a molécula responsável pela sinalização RET/PTC1-MAPK-ERK e possa ser essencial para a sobrevivência destas células.
No presente estudo, procurou-se avaliar o papel do RAF-1 na sinalização MAPK em células do cancro da tireóide com o rearranjo RET/PTC1 em comparação com células com mutações BRAF e RAS. Testamos as alterações fenotípicas induzidas pela inibição do RAF-1 e/ou inibição do BRAF por RNAi e induzidas pela inibição das via das MEK e da PI3K em células TPC-1 com rearranjo RET/PTC1, e comparamos com células C643 e 8505C com a mutação HRASG13R e a mutação BRAFV600E, respetivamente. Na linha celular TPC-1, a inibição do RAF-1 diminui a proliferação e aumenta a apoptose em maior extensão, comparativamente com a inibição do BRAF, embora nenhum efeito foi observado nos níveis da ativação das ERKs. Nas linhas celulares 8505C e C643 a inibição do RAF-1 não afeta significativamente a apoptose, a proliferação e a activação das ERK (com exceção da C643) em contraste com o que foi observado após inibição do BRAF, nestas células. Os nossos resultados mostram que, em linhas com mutações BRAF e RAS, o BRAF é o principal regulador da ativação das ERKs. Estes resultados apoiam a hipótese de que as células do cancro da tireóide com o rearranjo RET/PTC1 são mais dependentes de RAF-1 e menos dependentes da via de sinalização BRAF para a proliferação e apoptose, do que aquelas com ativação de mutações RAS ou BRAF.
Foi também testado, um inibidor das MEKs (PD98059) e um inibidor do PI3K (LY294002) e pouco ou nenhum efeito foi verificado nos níveis de pERKs
xiv
respetivamente sugerindo que a ativação das ERKs na TPC-1 é activada discretamente pela MEK e não regulada pelo PI3K.
Mostramos também que, na linha celular TPC-1, a regulação da proliferação é mais dependente da sinalização PI3K-AKT em comparação com as vias RAF-1 e BRAF. Os nossos dados também sugerem que nas células TPC-1, o RET/PTC1 pode ativar as ERKs por um mecanismo independente do BRAF, RAF-1 e PI3K e da via MAPK-ERK clássica.
Os nossos resultados suportam a hipótese principal de que as células do cancro da tireóide, TPC-1, podem ter um mecanismo alternativo de ativação das ERK, tais como mutações na via MAPK-ERK.
A compreensão dos mecanismos subjacentes à ativação constitutiva das ERK em células de cancro da tireóide com o rearranjo RET/PTC1, pode levar a encontrar novas abordagens terapêuticas, de primordial importância para este tipo de cancro.
xv TABLE OF CONTENTS ACKNOWLEDGEMENT...ix ABSTRACT ...xi RESUMO ...xiii TABLE OF CONTENTS ...xv ABBREVIATIONS ...xix LIST OF FIGURES………..…………..…..xxiii LIST OF TABLES………..xxvii 1 INTRODUCTION ...1
1.1 Cancer epidemiology and hallmarks ...3
1.2 Thyroid cancer ...6
1.3 Important signaling pathways in thyroid cancer...11
1.3.1 MAPK-ERK pathway ...13
1.3.2 PI3K-AKT pathway………...15
1.4 The role of oncogene mutations in thyroid tumorigenesis………...16
1.4.1 BRAF mutations...17
1.4.2 RET/PTC rearrangements...18
1.4.3 RAS mutations………...20
1.5 Thyroid cancer therapies...20
1.6 Rationale and aim of the project...22
1.6.1 Rationale.………...………..22
1.6.2 General aim………..………...23
1.6.3 Specific aims………...23
xvi
2.1 Cell culture conditions ...27
2.2 RNA interference assay...27
2.2.1 Optimization of RNAi conditions...28
2.2.2 RAF-1 and BRAF silencing by RNAi...29
2.3 Drug treatment ...29
2.3.1 PD98059………..…...29
2.3.2 LY294002...30
2.4 Assessment of DNA synthesis by BrdU incorporation ...30
2.5 TUNEL assay………..…...31
2.6 SRB assay………...33
2.7 Protein extraction and western blotting analys...34
2.7.1 Preparation of total protein extracts...34
2.7.2 Protein quantification...34
2.7.3 Western blotting………...35
2.8 Statistical analysis ...37
3 RESULTS………...39
3.1 Effect of RAF-1 and BRAF inhibition in thyroid cancer cells... 41
3.1.1 Optimization of RNAi conditions ………...…...41
3.1.1.1 Determination of siRNA cellular uptake …....…….…...41
3.1.2 Effect of RAF-1 inhibition by RNAi in proliferation and apoptosis of thyroid cancer cells………...………..……..…...43
3.1.3 Effect of RAF-1 inhibition in ERK phosphorylation of thyroid cancer cells………..49
3.1.4 Effect of BRAF and RAF-1 co-inhibition in proliferation and apoptosis of thyroid cancer cells……….……..………...…...50
xvii
3.1.5 Effect of BRAF and RAF-1 co-inhibition in ERK phosphorylation of thyroid
cancer cells…….………..………...56
3.2 Effect of MEK inhibition in thyroid cancer cells.………..……...58
3.2.1 Effect of MEK inhibition in proliferation of thyroid cancer cells………..58
3.2.2 Effect of MEK inhibition in apoptosis of thyroid cancer cells…………...62
3.2.3 Effect of MEK inhibition in ERK phosphorylation………....64
3.3 Effect of PI3K inhibition in thyroid cancer cells...67
3.3.1 Effect of PI3K inhibition in proliferation of thyroid cancer cells….……...67
3.3.2 Effect of PI3K inhibition in apoptosis of thyroid cancer cells…………..…..69
3.3.3 Effect of PI3K inhibition in ERK phosphorylation ………...71
4 DISCUSSION……….……….77
5 CONCLUSIONS AND FUTURE PERSPECTIVES ...85
xix
ABBREVIATIONS AGO2 - Argonaute 2 AP1 - Activator protein 1 APS - Ammonium persulfate
ARAF - V-raf murine sarcoma 3611 viral oncogene homolog ATC - Anaplastic thyroid cancer
ATP - Adenosine triphosphate
BRAF - V-RAF murine sarcoma viral oncogene homolog B BrdU - 5-bromo-2-deoxyuridine
BSA - Bovine serum albumin CC - Coiled-coil domain
DAPI - 4',6-diamidino-2-phenylindole DMSO - Dimethylsulphoxide
DNA - Deoxyribonucleic acid
dUTP - 2`-deoxyuridine, 5`- triphosphate EDTA - Ethylenediamine tetraacetic acid EGFR - Epidermal growth factor receptor ELE1 - RET-activating gene
ERK - Extracellular-signal-regulated kinase FBS - Fetal bovine serum
FITC - Fluorescein isothiocyanate FLT-3 - Fms-like tyrosine kinase 3 FTC - Follicular thyroid cancer GF - Growth factor
GTP - Guanosine triphosphate GTPase - Guanosine triphosphatase HCl - Hydrochloric acid
HRAS - Harvey rat sarcoma viral oncogene homolog IF - Immunofluorescence
IGFR - Insulin-like growth factor receptor IgG - Immunoglobulin G
ISNT - In Situ Nick Translation JNK - c-Jun N- terminal kinase
xx
KRAS - Kirsten rat sarcoma viral oncogene homolog MAPK - Mitogen-activated protein kinase
MEK - Mitogen-activated protein kinase kinase MEN2 - Multiple endocrine neoplasia type 2 mRNA - Messenger RNA
MTC - Medullary thyroid cancer NaF - Sodium fluoride
Na3VO4 - Sodium orthovanadate
NCOA4 – Nuclear receptor coactivator 4
NF-қB - Nuclear factor kappa-light-chain-enhancer of activated B cells NP-40 - Nonidet P 40
NRAS - Neuroblastoma rat sarcoma viral oncogene homolog NTRK1 - Neurotrophic tyrosine kinase receptor type 1
PAX8-PPARγ - Paired box8 fusion with peroxisome proliferator-activated receptors
gamma
PBS - Phosphate buffer saline
PBST - Phosphate buffer saline 0.05% Tween-20 PDGFR - Platelet-derived growth factor receptor
PDK-1 - Protein serine/treonine kinase 3´-phosphoinositide-dependent kinase-1 PH - Pleckstrin homology
PI3K - Phosphatidylinositol 3 kinase
PIK3CA - Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha PIP2 - Phosphoinositol 4,5-biphosphate
PIP3 - Phosphatidylinositol-3,4,5-triphosphate PKB or Akt - Protein kinase B
PKC - Protein kinase C
PMSF - Phenylmethylsulfonyl fluoride PTC- Papillary thyroid cancer
PTEN - Phosphatase and tensin homolog PVDF - Polyvinylidene difluoride
RAF - Mitogen-activated protein kinase kinase kinase
RAF1 or CRAF - V-raf-1 murine leukemia viral oncogene homolog RAS - Rat sarcoma viral oncogene homolog
xxi
RET - Rearranged during transfection
RET/PTC1- Rearranged during transfection in papillary thyroid cancer type 1 RISC - RNA- induced silencing complex
RKIP - Raf kinase inhibitory protein RNA - Ribonucleic acid
RNAi- RNA interference ROS - Reactive oxygen species RPM - Rotations per minute RT - Room temperature
RTK - Receptor tyrosine kinase SDS - Sodium dodecyl sulfate
SDS-PAGE - Sodium dodecyl sulfate polyacrylamide gel electrophoresis SiRNA - Small interfering RNA
SRB - Sulforhodamine B
TdT - Terminal deoxynucleotidyl transferase TEMED - Tetramethylethylenediamine TK - Tyrosine kinase domain
TM - Transmembrane domain
TPBS-B - PBS 0.5% Tween-20, 0.05% BSA TUNEL - Terminal dUTP Nick-End Labeling
VEGFR-2 - Vascular endothelial growth factor receptor 2 VEGFR-3 - Vascular endothelial growth factor receptor 3
xxiii
LIST OF FIGURES
Figure 1.1: The hallmarks of cancer: acquired capabilities of cancer cells during
carcinogenesis………5
Figure 1.2: Estimated thyroid cancer cases and deaths by sex, USA, 2012………..7 Figure 1.3: A proposed model of thyroid cancer development……….8 Figure 1.4: Schematic representation of PI3K-AKT and MAPK-ERK pathways..12 Figure 1.5: Schematic representation of involvement of the MAPK-ERK and
PI3K-AKT pathways in the follicular cell derived thyroid tumorigenesis………..13
Figure 1.6: Schematic representation of the Ras-Raf-MEK-ERK1/2 MAP kinase
pathway.………...15
Figure 1.7: Mechanisms of chromosomal rearrangements generating transforming
fusion genes……….19
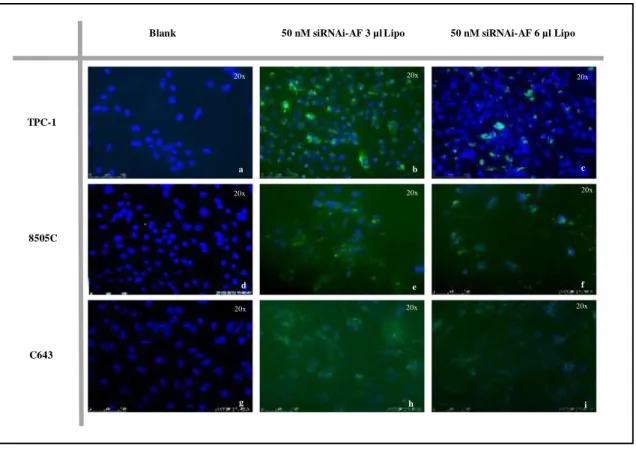
Figure 3.1: Study the uptake with Lipogen in TPC-1, 8505C and C643…………42 Figure 3.2: Representative photos of the uptake with lipogen in TPC-1, 8505C and
C643………43
Figure 3.3: A)-Western-blot analysis of RAF-1 and BRAF expression levels after
RNAi treatment. B)-Quantification of protein levels of RAF-1 (a) and BRAF (b) by Western Blotting in TPC-1, 8505C and C643 cell lines treated with siRNA HsRAF-1………...……44
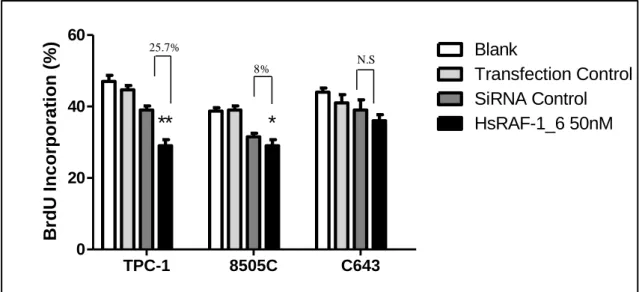
Figure 3.4: BrdU analysis after RNAi treatment in thyroid cancer cells………....45 Figure 3.5: BrdU analysis after RNAi treatment in thyroid cancer cells
(representative photos)………....46
Figure 3.6: TUNEL analysis after RNAi treatment in thyroid cancer cells…...….47 Figure 3.7: TUNEL analysis after RNAi treatment in thyroid cancer cells
(representative photos)………...…...…...48
Figure 3.8: A)-Western-blot analysis of ERK phosphorylation levels after RNAi
treatment. B)-Quantification of protein levels of pERKs………...49
Figure 3.9: A)- Western-blot analysis of RAF-1, BRAF and co-inhibition
expression levels after RNAi treatment. B)- Quantification of protein levels of RAF-1 (a) and BRAF (b)……….51
xxiv
Figure 3.11: BrdU analysis after RNAi treatment in thyroid cancer cells
(representative photos)………...….53
Figure 3.12: TUNEL analysis after RNAi treatment in thyroid cancer cells……..54 Figure 3.13: TUNEL analysis after RNAi treatment in thyroid cancer cells
(representative photos)………...….55
Figure 3.14: A)-Western-blot analysis of ERK phosphorylation levels after RNAi
treatment. B)- Quantification of protein levels of pERKs………..57
Figure 3.15: Effect of PD98059 on cell proliferation determined by SRB
assay……….59
Figure 3.16: Representative phase contrast photos of TPC-1, 8505C and C643 after
PD98059 treatment………..59
Figure 3.17: BrdU analysis after PD98059 treatment……….60 Figure 3.18: Representative photos of TPC-1 (A), 8505C (B) and C643 (C) cell
lines showing decreased incorporation of BrdU in treated cells when compared with DMSO control………...61
Figure 3.19: TUNEL analysis after PD98059 treatment……….62 Figure 3.20: Representative photos of TPC-1 (A), 8505C (B) and C643 (C) cell
lines showing increased TUNEL labelling in treated cells when compared with DMSO control………..63
Figure 3.21: A)-Western-blot analysis of ERK phosphorylation levels after
PD98059 treatment. B)- Quantification of protein levels of pERKs by Western Blotting in TPC-1, 8505C and C643 cell lines treated with PD98059 50 ìM for 1 hour and 48 hours………65
Figure 3.22: A)-Western-blot analysis of ERK phosphorylation levels after
PD98059 treatment. B)- Quantification of protein levels of pERKs by Western Blotting in TPC-1, 8505C and C643 cell lines treated with PD98059 75 and 100 μM………...66
Figure 3.23: BrdU analysis after LY294002 treatment………...67 Figure 3.24: Representative photos of TPC-1 (A), 8505C (B) and C643 (C) cell
lines showing decreased incorporation of BrdU in treated cells when compared with DMSO control……….68
Figure 3.25: TUNEL analysis after LY294002 treatment……….……….69 Figure 3.26: Representative photos of TPC-1 (A), 8505C (B) and C643 (C) cell
lines showing increased TUNEL labeling in treated cells when compared with DMSO control………..70
xxv
Figure 3.27: Western-blot analysis of ERK and AKT phosphorylation levels after
LY294002 treatment………72
Figure 3.28: Western-blot analysis of ERK and AKT phosphorylation levels after
LY294002 treatment………..74,75,76
Figure 4.1: General outline of the project and the hypotheses presented based in
xxvii
LIST OF TABLES
3
1.1 Cancer epidemiology and hallmarks
In the past decades cancer has emerged as a major health issue being responsible for more than 10% of deaths worldwide. Despite consistent developments in medical research, the global burden of cancer has more than doubled during the last 30 years, being today a major cause of death, becoming a disease with a great impact in the world (Accardi, Adebamowo et al. 2008).
In the year of 2008 cancer was responsible for 7.6 million deaths (about 13% of all deaths worldwide), with 63% of the cancer deaths occurring in the less developed regions of the world (Accardi, Adebamowo et al. 2008). The most commonly diagnosed cancers worldwide are lung, breast and colorectal cancers (1.23 million, 9.7% of the total). In USA about 1.638.910 new cancer cases are expected to be diagnosed in 2012 (Society 2012). It could be expected that, by the year of 2030, 27 million new cases of cancer and 17 million cancer related deaths will have occurred (Accardi, Adebamowo et al. 2008).
In Europe, the incidence and mortality increased in the period between 2004 and 2006, with an estimated 3.191.600 new cases diagnosed and 1 703 000 deaths. In the year of 2008, in Europe alone, cancer was responsible for 1.72 million deaths, 56% of them in men and 44% in women. In the same year, about 3.2 million new cases, 53% of them in men and 47% in women have been diagnosed. The most common cancers were colorectal (13.6% of the total), followed by breast (13.1%), lung (12.2%) and prostate cancer (11.9%). The major cause of cancer related deaths in Europe was lung cancer accounting for one fifth of the total number of cancer related deaths. The second major cause of cancer death is colorectal cancer (12.3%), followed by breast (7.5%) and stomach cancer (6.8%) (Ferlay, Parkin et al. 2010). In the more recent work performed in Portugal focusing on the incidence of cancer, carried out by Pinheiro and colleagues (2003), the authors predicted for the year 2000 a total of 19 880 new cancer cases among men, and 17 000 new cancer cases in women in Portugal (Pinheiro, Tyczynski et al. 2003)
The 5-year relative survival rate for all cancers diagnosed between 2001 and 2007 is 67% (it was 49% in 1975-1977). The improvement in survival reflects both progress in diagnosing certain cancers at an earlier stage and improvements in treatment (Society 2012).
4
Due to the high incidence and worldwide mortality, cancer is one of the areas of research where enormous efforts are being developed in order to better understand this disease, and consequently, to discover more efficient treatments and/or diagnostic tests.
Cancer is difficult to define with precision but it could be described as a result of a stepwise process, designated carcinogenesis, that consists in the accumulation of genetic mutations and epigenetic changes (Pelengaris and Khan 2006). These changes include mutations, translocations and other chromosomal alterations and/or abnormal expression of oncogenes or tumor suppressor genes, (Hanahan and Weinberg 2000; Argyle and Blacking 2008). Most proto-oncogenes are key genes involved in the control of cell proliferation and growth, namely growth factors, growth factor receptors, protein kinases, signal transducers, nuclear proteins and transcription factors (Hanahan and Weinberg 2000; Hanahan and Weinberg 2011).
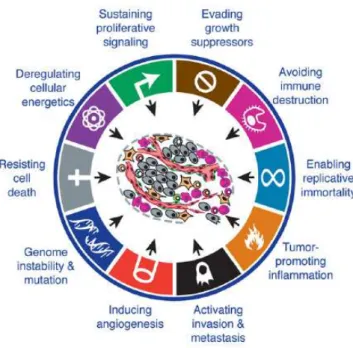
Changes caused by several genetic or epigenetic alterations impair the control of cell proliferation, survival, differentiation, migration and interaction with neighbor cells (Pelengaris and Khan 2006). The hallmarks responsible for the malignant transformation of the cells, are acquired during carcinogenesis and are the follow: sustained proliferative signals, evading growth suppressors, enabling replicative immortality, activating invasion and metastasis, inducing angiogenesis, resisting cell death, deregulating cellular energetics, avoiding immune destruction, tumor-promoting inflammation and genome instability and mutation (Figure 1.1) (Mantovani 2009; Hanahan and Weinberg 2011).
5
Figure 1.1: The hallmarks of cancer: acquired capabilities of cancer cells during carcinogenesis.
Despite being considered a complex disease, cancer can be defined by these acquired alterations (Adapted from (Hanahan and Weinberg 2011)).
In a simple way, cancer can be considered as a breakdown in cellular homeostasis, which can lead to an uncontrolled cell division and ultimately to a disease stage. The cancer formation is the phenotypic outcome of a series of changes that require a long period of time to develop (Argyle and Blacking 2008).
Genetic alterations can occur as germline mutations, resulting in inherited cancer predisposition or, more commonly, occur in somatic cells (somatic mutations), where their accumulation in association with several environmental factors give rise to sporadic tumors (Souglakos 2007). The presence of inherited mutations can initiate cancer, but other modifications such as somatic mutations and epigenetic alterations are still needed for cancer to develop (Pelengaris and Khan 2006). Numerous studies report that the formation of cancer is thought to be a combination of multiple genetic and environmental risk factors (Pharoah, Dunning et al. 2004). It is important to take into account that the common genetic alterations may not have, by themselves, substantial impact but when associated with exposure to environmental carcinogens could lead to tumor development (Bartsch and Hietanen 1996).
Epidemiological studies have estimated that up to 80 to 90% of all cancers are related to environmental factors, tobacco smoke, and diet (Doll and Peto 1981).
6
Tobacco smoking is unquestionably a major human carcinogen factor, as about 30% of all cancer cases worldwide are caused by tobacco, particularly lung cancer. Modifiable risk factors for cancer have been identified, including alcohol consumption, excessive exposure to sunlight, lack of physical activity, overweight and obesity, dietary factors, occupational exposures and chronic infection (Bartsch and Hietanen 1996; Accardi, Adebamowo et al. 2008). The knowledge about the involvement of these risk factors in various types of tumors is still limited. Thus, it is extremely important to decode and understand the mechanisms involved in the development of such tumors. It is therefore important to develop preventive measures, new diagnostic tools and more effective therapeutic approaches.
1.2 Thyroid cancer
Thyroid cancer is the most common endocrine malignancy, accounting to 94% of endocrine cancers and 66% of deaths due to cancers with endocrine origin (Society 2007). The incidence of thyroid cancer has increased 50% since 1973, and it is the type of cancer that shows the most rapidly increasing incidence among women and the 2nd among men (Hundahl, Fleming et al. 1998). Some investigators have attributed the increase to environmental radiation (Mangano 1996) while others didn’t found reasons for such increase (Haselkorn, Bernstein et al. 2000).
The current incidence of thyroid cancer in Europe is 40/1 000 000 inhabitants per year, with a nearly three times higher incidence in women (Hay and Klee 1993). As shown in Figure 1.2, an estimated 56.460 new cases of thyroid cancer are expected to be diagnosed in 2012 in the USA, with 3 in 4 cases occurring in women. The incidence rate of thyroid cancer has been increasing sharply since the mid-1990s, and it is the fastest-increasing cancer in both men and women. Since 2004, incidence rates have been increasing by 5.5% per year in men and 6.6% per year in women (Society 2012). An estimated 1.780 deaths from thyroid cancer are expected in 2012 in the USA. From 2004 to 2008, the death rate for thyroid cancer increased slightly from 0.47 (per 100,000) to 0.50 in men, and from 0.47 to 0.52 in women (Figure 1.2) (Society 2012).
7
Figure 1.2: Estimated thyroid cancer cases and deaths by sex, USA, 2012. (Adapted from (Society
2012)).
There are many risk factors associated with thyroid cancer, such as age, gender, exposure to radioactive substances and predisposing genetic alterations (certain rare genetics syndromes also increase risk) (Suzuki, Willingham et al. 2002). More risks include having a record of goiter (enlarge thyroid) thyroid nodules or a family history of thyroid cancer. Patients that are positive for an abnormal gene that causes a hereditary form of thyroid cancer can decrease the chance of developing the disease by surgical removal of the thyroid gland. Exposure to radiation as atomic weapons tests and nuclear accidents, such as Chernobyl, has also been linked to increased risk of thyroid cancer, especially in children. Unlike other adult cancers, for which older age increases risk, 80% of newly diagnosed thyroid cancer patients are under 65 years of age (Society 2012).
Although numerous studies have been conducted to unravel the etiology of thyroid cancer, the understanding of the development of thyroid cancer is still largely incomplete (Suzuki, Willingham et al. 2002). The thyroid gland is part of the endocrine system, which regulates hormones in the body. The thyroid gland absorbs iodine from the bloodstream to produce thyroid hormones, which regulate metabolism. It contains two types of cells: follicular cells, which are responsible for the production of thyroid hormone (thyroxine), and C cells or parafollicular cells, which make calcitonin, a hormone that participates in calcium metabolism. The follicular cells are of endodermal origin and C cells are of neural crest origin (Mansouri, Chowdhury et al. 1998). Thyroid cancer can be developed from both follicular cells or C cells (Figure 1.3). Those cells give rise to the four major types of thyroid cancers: papillary thyroid cancer (PTC), follicular thyroid cancer (FTC), anaplastic thyroid cancer (ATC) and medullary thyroid
8
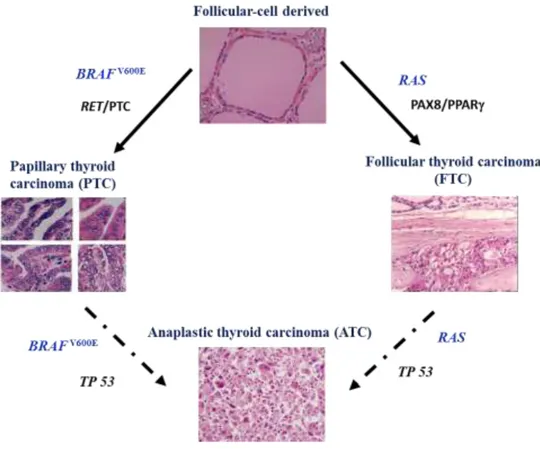
cancer (MTC). The first three (PTC, FTC and ATC) derived from follicular cells and MTC derived from C cells (Figure 1.3) (Maciel, Kimura et al. 2005).
Figure 1.3: A proposed model of thyroid cancer development. Both papillary and follicular thyroid
cancers are originated from thyroid follicular cells. Anaplastic thyroid cancer is developed from either papillary thyroid cancer or follicular thyroid cancer. Medullary thyroid cancer is originated from calcitonin secreting C-cells (Adapted from (Maciel, Kimura et al. 2005)).
Both papillary and follicular thyroid cancers are mostly well-differentiated and slow growing. PTC and FTC account for ~95% of total thyroid cancer cases (Nikiforova and Nikiforov 2008). Metastasis to distant organs is rarely observed in PTC and FTC patients. The 5-year survival rate for well-differentiated thyroid cancer is more than 95% (Riesco-Eizaguirre and Santisteban 2007).
9
Papillary thyroid cancer is the most frequent type of thyroid malignancy and includes about 80% of thyroid cancer cases (Nikiforova and Nikiforov 2008). In most cases, PTC has a very good prognosis, particularly in patients younger than 45 years at the time of diagnosis (Bilimoria, Bentrem et al. 2007). PTC is usually an indolent cancer and curable with surgical thyroidectomy, often followed by radioiodine treatment (Hay, Bergstralh et al. 1993). This type of thyroid cancer is sporadic in about 95% of the cases and familiar in the remaining (Soares, Trovisco et al. 2003). PTC is generally considered curable, with about 97% survival rate at 5-year (Riesco-Eizaguirre and Santisteban 2007). PTCs are associated with nonoverlapping mutations of genes coding for mitogen-activated protein kinase signaling effectors, including mutations in BRAF or RAS, and rearrangements of the RET or NTRK1 (Sobrinho-Simoes, Preto et al. 2005; Xing 2005; Trovisco, Soares et al. 2007). BRAF mutation is the most common genetic alteration in PTC and is found in approximately 45% of PTCs patients (Soares, Trovisco et al. 2003; Soares, Trovisco et al. 2004; Sobrinho-Simoes, Preto et al. 2005; Trovisco, Soares et al. 2005; Xing 2005; Adeniran, Zhu et al. 2006; Trovisco, Soares et al. 2007; Nikiforova and Nikiforov 2008). Somatic rearrangements of the RET proto-oncogene have been detected in 3-60% of sporadic PTC (Nikiforov 2002).
These mutations are mutually exclusive, providing genetic evidence that mutations in MAPK signaling pathway are required for transformation into PTC. Thus, inappropriate activation of any of these effectors is sufficient to change the phenotype (Soares, Trovisco et al. 2003; Mesa, Mirza et al. 2006; Preto, Goncalves et al. 2009; Soares, Lima et al. 2011).
Follicular thyroid cancer is the second most frequent thyroid cancer and represents about 15% of total thyroid cancer cases (Nikiforova and Nikiforov 2008). It arises also from thyroxine-secreting follicular cells and is well-differentiated and slow growing (Riesco-Eizaguirre and Santisteban 2007). It usually occurs after the age of 40 and is more common in women than in men. Although risk factors for follicular cell thyroid cancer include radiation exposure and a family history of thyroid cancer, it is important to note that the majority of patients have no risk factors. It is assumed that FTC can develop from thyroid follicular adenoma, a precancerous stage not seen in PTC (Figure 1.3). There are three types of genetic alterations that are frequently found in FTC: RAS point mutation (Vasko, Ferrand et al. 2003), PAX8-PPARγ rearrangement (Kroll, Sarraf
10
et al. 2000) and alterations in the PI3K signaling pathway. Point mutations of RAS occur with a frequency ranging from 40% to 50% (Nikiforova and Nikiforov 2008) and it has been associated to more advanced and malignant characteristics, such as dedifferentiation and metastasis to distant organs (Karga, Lee et al. 1991; Manenti, Pilotti et al. 1994). The PAX8-PPARγ fusion occurs in ~35% of FTC tumors (Nikiforova, Biddinger et al. 2002) and it results from the chromosomal rearrangement between t(2;3)(q13;p25) (Kroll, Sarraf et al. 2000; Sobrinho-Simoes, Preto et al. 2005; Castro, Rebocho et al. 2006; Trovisco, Soares et al. 2007). This rearrangement results in fusion of the DNA binding domains of the thyroid transcription factor PAX8 todomains A to F of the peroxisome proliferator-activated receptorPPARγ1 (Kroll, Sarraf et al. 2000). PAX8-PPARγ fusion is more common in younger patients and often associated with small tumor size (French, Alexander et al. 2003; Nikiforova, Lynch et al. 2003). This mutation also occurs in up to 13% of follicular adenomas (Marques, Espadinha et al. 2002).
Medullary thyroid cancer (MTC) makes up about 3% of thyroid cancer cases (Nikiforova and Nikiforov 2008). MTC is originated from calcitonin-secreting thyroid C cells and is also present in the genetic syndrome called multiple endocrine neoplasia type 2 (MEN2) (Hofstra, Landsvater et al. 1994). MTC has a more guarded prognosis than well-differentiated thyroid cancer (PTC and FTC). Almost 12-100% of the MTCs are caused by a mutation on the RET gene, depending on the reported series (Marx 2005). Since RET mutation can be inherited, it is considered as a tumor-initiation event in MTC (de Groot, Links et al. 2006). Recently Moura et al, 2011 showed that RAS mutations were present in the RET-negative MTC and in the RET-positive MTC, suggesting that activation of the proto-oncogenes RAS and RET represents alternative genetic events in sporadic MTC tumorigenesis (Moura, Cavaco et al. 2011).
Although only representing 1% to 2% of total thyroid cancer incidences, ATC contributes to approximately 40% of thyroid cancer deaths (Lin, Gao et al. 2008). The mean survival time for ATC patients after diagnosis is merely 6 months (Lin, Gao et al. 2008). ATC is one of the most rapidly growing and invasive type of human malignancy and probably the most destructive cancers encountered in humans. It is not known what causes ATC, but often well-differentiated thyroid cancers (such as PTC and FTC) can degenerate and turn into anaplastic thyroid cancer (Lin, Gao et al. 2008). The most frequent genetic alteration in ATC is p53 mutation, which is found in more than 70% of samples (Nikiforova and Nikiforov 2008; Sobrinho-Simoes, Eloy et al. 2010; Soares,
11
Lima et al. 2011). Mutations in p53 inhibit the DNA binding ability of p53, inactivating its tumor suppressor effect (Fagin, Tang et al. 1996). Other mutations, such as PI3K, β-catenin BRAF, RAS and PTEN, have also been found in ATC (Nikiforova and Nikiforov 2008).
To address the critical need for novel and real therapeutic strategies for aggressive and/or disseminated thyroid cancers, it is important to obtain a comprehensive insight into the signaling cascades which trigger the uncontrolled proliferation and/or resistance of thyroid cancer cells to pro-apoptotic stimuli (Mitsiades, Negri et al. 2007).
1.3 Important signaling pathways in thyroid cancer
Carcinogenesis involves an accumulation of genetic alterations that in turn lead to alterations in important biological pathways of cell proliferation and cell death, such as phosphotidylinositol-3 kinase (PI3K)/protein kinase B (PKB/AKT) and mitogen-activated protein kinases (MAPKs) [extracellular-signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38].
Many of the genetic alterations implicated in thyroid carcinogenesis involve the aberrant activation of MAPK-ERK pathway (Nikiforov 2002) and/or the activation of the PI3K-AKT pathway (Figure 1.4 and 1.5) (Soares, Trovisco et al. 2003; Trovisco, Soares et al. 2007; Wang, Hou et al. 2007; Saji and Ringel 2010; Brzezianska and Pastuszak-Lewandoska 2011).
12
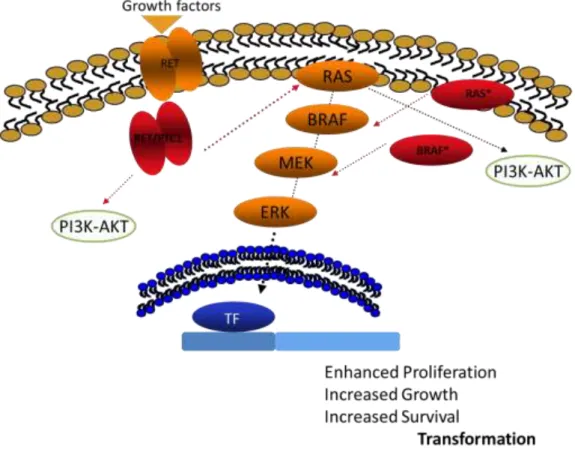
Figure 1.4: Schematic representation of PI3K-AKT and MAPK-ERK pathways. The small GTPase
protein RAS when activated by, for example, growth factors and receptor tyrosine kinases (RTK), changes from the inactive RAS-GDP to the active RAS-GTP form, being able to activate the class I phosphatidylinositol-3 kinases (PI3K) and the protein RAF. RAF phosphorylates MEK1/2 and consequently activates ERK1/2 pathway. These pathways are involved on various cellular functions, apoptosis, cell cycle regulation, cell growth and metabolism. The protein MEK1/2 could be inhibited by PD98059 and the PI3K pathway could be suppressed by several drugs such as, LY294002 (used in this work). Adapted from (Soares, Trovisco et al. 2003).
13
Figure 1.5: Schematic representation of involvement of the MAPK-ERK and PI3K-AKT pathways in the follicular cell derived thyroid tumorigenesis. The two pathways are involved in the different types thyroid cancer. The PI3K-AKT pathway is more involved in FTC and ATC and MAPK pathway is more involved in PTC (Adapted from (Soares, Trovisco et al. 2003; Trovisco, Soares et al. 2007)).
1.3.1 MAPK-ERK pathway
The extracellular signal‐regulated kinase (ERK) signaling pathway is a classical conserved intracellular signaling pathway that is often altered in human cancer. MAPK proteins phosphorylate a number of substrates involved in several cellular events, such as: proliferation, differentiation, apoptosis and survival (Torii, Nakayama et al. 2004; MacCorkle and Tan 2005; Cuevas, Abell et al. 2007). It is known that when ERK signaling pathway is aberrantly activated may induce tumorigenesis (Sebolt-Leopold and Herrera 2004; Kohno and Pouyssegur 2006).
In mammalian, RAF proteins are serine/threonine kinases that are located downstream of RAS in the signaling pathway. Humans have three RAF genes: ARAF, CRAF-1, and BRAF (Fukushima, Suzuki et al. 2003).
These proteins are recruited to the membrane and bind to RAS being subjected to phosphorylation/dephosphorylation on different domains and to dissociation with the
14
RAF kinase inhibitory protein (RKIP) promoting RAF activation (McCubrey, Steelman et al. 2007). When activated, the protein RAF (MAPKKK) can phosphorylate two MAPKK proteins, MEK1 and MEK2, which consequently activates the MAP kinases ERK1 and ERK2 (McCubrey, Steelman et al. 2007). Subsequently, the activated ERK phosphorylate regulatory protein molecules in the nucleus and in the cytosol (Xing 2007) (Figure 1.6).
In the cytosol, ERK can activate NF-κB by phosphorylating and activating the inhibitor κB kinase, as well as phosphorylate caspase 9 on residue Thr125 leading to its inactivation. In the nucleus, ERK1/2 promotes the phosphorylation of many transcription factors, including c-Jun, c-Myc and c-Ets-1 that are required for activator protein 1 (AP-1) expression (McCubrey, Steelman et al. 2007).
Activation of MAPK-ERK pathway has been demonstrated to be involved in PTC progression (Nikiforov 2002). Mutations upstream of this pathway are frequent in PTC, including RET/PTC (Soares, Trovisco et al. 2003), NTRK (Frattini, Ferrario et al. 2004), RAS (Vasko, Ferrand et al. 2003) or BRAF (Kimura, Nikiforova et al. 2003) and are found in two thirds of cases being mutually exclusive (Kimura, Nikiforova et al. 2003). This data provides genetic evidences, that constitutive signaling along RET/PTC-RAS-BRAF-MEK-ERK is a key in the development of PTCs (Mitsutake, Miyagishi et al. 2006). In other words, one activating mutation along the RAS–MAPK pathway is sufficient for thyroid cell malignant transformation (Melillo, Castellone et al. 2005; Xing, Westra et al. 2005).
There are several compounds found to inhibit the enzyme activity of MAP kinases (Malemud 2007), one of them, PD98059, showed to inhibit MEK1 and MEK2 and partially MEK5 with an impact on ERK signaling (McCubrey, Steelman et al. 2007).
15
Figure 1.6: Schematic representation of the Ras-Raf-MEK-ERK1/2 MAP kinase pathway. MAPK
pathway is physiologically active by binding of growth factors to receptor tyrosine kinases (RTKs). RAS activates RAF that in turn phosphorylates MEK1/2 and consequently activates ERK1/2 pathway. ERK pathway is involved in various biological responses, such as cell proliferation, survival, cell migration and differentiations. GF - growth factor; RTK - receptor tyrosine kinase. (From (Frémin and Meloche 2010)).
1.3.2 PI3K-AKT Pathway
The phosphatidylinositol-3-kinase (PI3K) signaling pathway is crucial to many aspects of cell growth and survival (Hennessy, Smith et al. 2005). The PI3Ks are a family of lipid kinases that are classified according to sequence homology and subtract specificity into class I, class II and class III. Class I PI3K is the major class and is involved in cell growth, proliferation and apoptosis (Zhao and Vogt 2008). This class of PI3K can be activated by receptor tyrosine kinases (RTK), such as PDGFR, EGFR or IGFR through interaction with phosphotyrosine residues, and by G protein-coupled receptors. RAS is a classical player in the MAPK pathway, but it also plays an important role in the signaling of the PI3K-AKT pathway (Xing 2010).
16
The activation of PI3K leads to the conversion of the lipid substrate phosphoinositol 4,5-bisphosphate (PIP2) to the second messenger, the phosphatidylinositol-3,4,5- triphosphate (PIP3) at the inner side of the plasma membrane. PIP3 binds to target proteins that contain pleckstrin homology (PH) domains, such as PKA, PKC and AKT/PKB, the latter being the primary downstream mediator of PI3K effects (Fresno Vara, Casado et al. 2004; Michl and Downward 2005; Zhao and Vogt 2008).
Activating mutations in the genes encoding the Class I PI3K catalytic subunit (PIK3CA) have been described in cancers (Samuels, Wang et al. 2004). In thyroid cancers, PIK3CA mutations have been described by several groups (Hou, Ji et al. 2008; Ricarte-Filho, Ryder et al. 2009), occurring more frequently in FTC and ATC, in comparison to PTC and benign thyroid tumors. Amplification of PIK3CA has been described to be a common event in thyroid tumors. In several recent studies, approximately 13% of follicular adenomas, 16% of PTCs, 30% of FTC and 50% of ATC exhibited amplification, sugesting an increased frequency of PIK3CA amplification in more aggressive histological subtypes (Hou, Ji et al. 2008; Santarpia, El-Naggar et al. 2008).
Several studies have sugested that RET/PTC is involved in the signaling of the PI3K-AKT pathway and that RET/PTC is an activator of the PI3K-AKT pathway in thyroid tumors (Miyagi, Braga-Basaria et al. 2004; Castellone, De Falco et al. 2009; Xing 2010). Therefore, RET/PTC is an important player in the altered signaling of the PI3K-AKT pathway in thyroid tumorigenesis (Xing 2010).
Overactivation of PI3K signaling plays an important role in the development and progression of thyroid cancer (Saji and Ringel 2010). The best known pharmacological inhibitors of PI3K are wortmannin and LY294002, which target the p110 catalytic subunit of class I PI3K with antitumor activity (Hennessy, Smith et al. 2005; LoPiccolo, Blumenthal et al. 2008).
1.4 The role of oncogene mutations in thyroid tumorigenesis
PTCs harbor point mutations of the BRAF, RET/PTC rearrangements and RAS genes, all of which are able to activate the mitogen-activated protein kinase (MAPK) pathway. These mutually exclusive mutations are found in more than 70% of PTCs (Kimura, Nikiforova et al. 2003; Soares, Trovisco et al. 2003; Frattini, Ferrario et al.
17
2004; Adeniran, Zhu et al. 2006). In FTC point mutation of RAS are more frequent and occurs in 40% to 50% (Nikiforova and Nikiforov 2008). In ATC this mutations occurs in 68% of the RET-negative and in only 2.5% of the RET-positive sporadic MTC (Moura, Cavaco et al. 2011).
1.4.1 BRAF mutations
The most common BRAF mutation, found in about 80% of the mutated cases, is a thymidine to adenosine transversion at nucleotide 1796 (T1796A) that converts valine 600 to glutamate (V600E) (Davies, Bignell et al. 2002; Garnett and Marais 2004). This mutation induces proliferation and transformation (Ikenoue, Hikiba et al. 2004), showing a 138-fold transforming and oncogenic activity over wild-type BRAF (Davies, Bignell et al. 2002).
In PTCs BRAFV600E mutation is the most common genetic alteration and is found in approximately 45% of patients of PTC (Xing, Westra et al. 2005; Adeniran, Zhu et al. 2006; Nikiforova and Nikiforov 2008) and in 30% in patients with ATC (Soares, Trovisco et al. 2004). This mutation does not occur in FTC or other type of thyroid tumors (Xing, Westra et al. 2005). The hydrophobic interactions between the activation loop and the ATP binding site, maintains dephosphorylated wild-type BRAF protein in an inactive conformation. The V600E substitution disrupts these interactions and allows the formation of new interactions that keep the protein in a catalytically competent conformation, resulting in continuous phosphorylation of MEK (Wan, Garnett et al. 2004). PTCs with BRAFV600E mutation exhibit a more advanced clinical stage (Namba, Nakashima et al. 2003; Nikiforova, Lynch et al. 2003; Adeniran, Zhu et al. 2006) and show high risk of recurrence (Xing, Westra et al. 2005; Lupi, Giannini et al. 2007). This mutation has been associated with more aggressive PTCs with characteristics such as: advanced tumor stage, tumor recurrence and distant metastases (Namba, Nakashima et al. 2003; Xing, Westra et al. 2005). This invasive phenotype of PTC with the BRAFV600E mutation is probably due to secondary genetic events linked to an increase in genome instability (Mitsutake, Knauf et al. 2005). In addition, since BRAF is the downstream effector of RET and RAS signaling, inhibiting BRAF alone may have inhibitory effects on RET and RAS signaling as well.
18
1.4.2 RET/PTC rearrangements
The RET proto-oncogene is located on chromosome 10q11.2. It was isolated in 1985 and shown to be activated by a DNA rearrangement (rearranged during transfection) (Takahashi, Ritz et al. 1985). This oncogene codes for a cell membrane receptor tyrosine kinase and it is essential for the development of the sympathetic, parasympathetic, and enteric nervous systems, the kidney and the testis (Airaksinen and Saarma 2002). RET can be activated by chromosomal rearrangement known as RET/PTC rearrangement. It is formed by the fusion between the 3` portion of the RET receptor tyrosine kinase gene and the 5`portion of various unrelated genes (Nikiforov 2011). Many different genes have been found to be rearranged with RET in individual PTC patients, 15 types of RET/PTC have been reported to date (Mishra, Agrawal et al. 2009). The two most common rearrangement types RET/PTC1 and RET/PTC3, account for the vast majority of all rearrangements found in PTCs (Tallini and Asa 2001; Nikiforov 2002). There are intrachromosomal inversions as both RET and its respective fusion partner genes, H4 and NCOA4 (also known as ELE1), and are located on chromosome 10 (Figure 1.7) (Bongarzone, Butti et al. 1994; Santoro, Dathan et al. 1994). RET/PTC2 and nine more recently identified types of RET/PTC are all interchromosomal translocations (Figure 1.7) (Ciampi, Giordano et al. 2007). All rearrangement types contain the intact TK domain of the RET receptor and enables the RET/PTC oncoprotein to bind SHC and activate the RAS-RAF-MAPK cascade, initiating thyroid tumorigenesis (Knauf, Kuroda et al. 2003).
RET/PTC rearrangements are found on average in 20% of the sporadic PTCs (Nikiforova and Nikiforov 2008). The incidence of RET rearrangements for child and young-adult patients, who have been exposed to radiation, is 50%-80% and 40%-70%, respectively (Nikiforova and Nikiforov 2008).
Cancers with a RET/PTC rearrangement rarely correspond to aggressive or undifferentiated cancers (Soares, Fonseca et al. 1998; Tallini, Santoro et al. 1998; Adeniran, Zhu et al. 2006) and a clearly link to radiation exposure is evident in many studies, such as those following the well-known Chernobyl accident (Williams 2002). RET/PTC rearrangements are associated with younger patients and patients younger than 45 years, these mutations have been the most common type of the mutation in primary PTC (Nikiforov, Rowland et al. 1997; Fenton, Lukes et al. 2000).
19
In tumors arising after radiation exposure, RET/PTC1 was found to be associated with classic papillary histology, whereas RET/PTC3 type was more common in the solid variants (Nikiforov, Rowland et al. 1997). RET/PTC rearrangements and consequent constitutive PI3K and MAPK signaling are early events in radiation-related thyroid tumorigenesis (Saji and Ringel 2010).
Unlike in PTC and in MTC, RET genetic alterations are point-mutations. Almost 12-100% of MTC is caused by germline-transmitted mutation on the RET gene. Nonetheless, the effects of RET genetic alterations are virtually the same, with the constitutively activated kinase domain and downstream effectors including the oncogenic MAPK signaling pathway (Marx 2005; de Groot, Links et al. 2006).
.
Figure 1.7: Mechanisms of chromosomal rearrangements generating transforming fusion genes.
RET/PTC oncogenes, which are found in papillary thyroid cancers (PTC), are chimeric genes resulting from chromosomal rearrangements that lead to the fusion of the RET tyrosine kinase domain to different heterologous genes. The rearrangements involving H4, RIa, and RFG/ELE1 partner genes are shown. TM- transmembrane domain; TK- tyrosine kinase domain; CC- coiled-coil domain (From (Arighi, Borrello et al. 2005)).
20
1.4.3 RAS mutations
The small GTP-binding protein RAS is a common upstream molecule of several signaling pathways, including PI3K-AKT and MAPK-ERK. Three human RAS genes-
NRAS, HRAS, and KRAS - have been identified differing in the ability to activate those
different pathways (McCubrey, Steelman et al. 2007).
The upregulation of PI3K-AKT and RAF-MAP-ERK pathways by the oncogene RAS cooperates to regulate various cellular responses and generate resistance to therapy (Barault, Veyrie et al. 2008). Moreover, it was found that RAS could downregulate PTEN expression through the RAF-MAPK-ERK pathway promoting activation of the PI3K-AKT pathway, which strengthens the connection between these two pathways (Vasudevan, Burikhanov et al. 2007).
The RAS mutations are present in different cancers and give rise to constitutively active RAS proteins. Point mutations in the N-RAS, H-RAS or K-RAS genes have been identified in thyroid cancer. In FTC, point mutations of RAS are the most common genetic alteration with a frequency ranging from 40% to 50% and occur in up to 15% cases of PTC (Nikiforova and Nikiforov 2008). This mutation also occurs in ATC and in sporadic MTC (Moura, Cavaco et al. 2011).
1.5 Thyroid cancer therapies
Well-differentiated papillary and follicular cancers typically have indolent behavior and can be effectively treated by surgery followed by radioiodine therapy. However, tumors that lose differentiation and consequently the ability to trap radioiodine and that do not respond to radioiodine treatment usually have a much less favorable prognosis. These tumors are clear candidates for alternative therapeutic approaches such as molecularly targeted therapy (Nikiforov 2008).
Numerous small-molecule tyrosine kinase inhibitors along the MAPK kinase and PI3K signaling pathway are available and have shown substantial therapeutic effects in thyroid cancer. Several signaling inhibitor drugs are in preclinical evaluation or in clinical trial showing synergistic effect with different types of therapies and the ability to overcome therapy resistances (Hennessy, Smith et al. 2005; LoPiccolo, Blumenthal et al. 2008).
21
Several TK inhibitors have been shown to block oncogenic RET/PTC signaling. An anilinoquinazoline, ZD6474, is a potent inhibitor of the vascular endothelial growth factor receptor 2 (VEGFR-2) and effectively blocks phosphorylation and signaling of RET/PTC3. This inhibitor induce growth arrest of human papillary cancer cell lines carrying RET/PTC1 (Carlomagno, Anaganti et al. 2006; Santoro and Carlomagno 2006). A multikinase inhibitor SU12248 (Sunitinib) has been shown to effectively inhibit signaling from RET/PTC kinase in differentiated thyroid cancer (PTC and FTC) and MTC (Kim, Jo et al. 2006). Two other small-molecule tyrosine kinase inhibitors, the pyrazolopyrimidine compounds PP1 and PP2, have been tested to blocking RET/PTC (Carlomagno, Vitagliano et al. 2002; Carlomagno, Vitagliano et al. 2003).
A number of RAF inhibitors have been developed and some of them have entered clinical trials (Halilovic and Solit 2008). Various BRAF inhibitors have been identified and tested as potential therapeutic agents. Moreover, since in the signaling cascade, BRAF is downstream of RET and RAS, BRAF inhibitors may be potentially effective in tumors with other mutations affecting this signaling pathway. One BRAF inhibitor in clinical trial is sorafenib (BAY 43-9006), a multikinase inhibitor with potent activity against RAF, VEGFR-2, VEGFR-3, PDGFRb, FLT-3, and c-KIT kinases (Wilhelm, Carter et al. 2004). BAY 43-9006 inhibits the BRAF signaling and growth of all thyroid cells carrying the BRAFV600E mutation (Salvatore, De Falco et al. 2006; Preto, Goncalves et al. 2009). Recently the therapeutic effect of BAY 43-9006 had also been found on cells carrying the RET/PTC rearrangement (Carlomagno, Anaganti et al. 2006; Preto, Goncalves et al. 2009). Other inhibitors of RAF kinases such as: AAL-881 and LBT-613 have also been explored in thyroid cancer and the results showed that both compounds were found to block MAPK signaling and growth of rat thyroid cells and human thyroid tumor cell lines harboring the V600E BRAF and RET/PTC1. (Ouyang, Knauf et al. 2006).
Several compounds have been found to inhibit the enzyme activity of MAP kinases (Malemud 2007). One of them, PD98059, showed to inhibit MEK1 and MEK2 and partially MEK5 with an impact on ERK signaling (McCubrey, Steelman et al. 2007). Other MEK inhibitor such as: CI-1040, a potent small-molecule MEK-selective inhibitor, which inhibits both MEK1 and MEK2, was demonstrated to inhibit the growth and proliferation of thyroid cancer cells harboring mutant BRAF (Liu, Liu et al. 2007).
22
The strategy to target PI3K pathway consists in the use of inhibitors for individual components of this pathway, such as PI3K, AKT, PDK-1 and mTOR, (Cheng, Lindsley et al. 2005; Granville, Memmott et al. 2006). The best known pharmacological inhibitors of PI3K are wortmannin and LY294002 (Hennessy, Smith et al. 2005; LoPiccolo, Blumenthal et al. 2008). Wortmannin, a natural fungal metabolite, although it was shown its ability to inhibit PI3K at low concentrations (Cheng, Lindsley et al. 2005). LY294002 is a derivate of the flavonoid quercetin, which is effective in inhibiting PI3K. LY294002 not only inhibits the ATP binding site of PI3K, but also other downstream components of the PI3K pathway (Cheng, Lindsley et al. 2005; Granville, Memmott et al. 2006). These commercially available PI3K inhibitors in combination with chemotherapeutic agents have been shown effective results in cell lines, demonstrating the advantage of their use in combination therapies. In thyroid cancer these inhibitors have been used to inhibit proliferation and used a possible therapeutic strategy to treat patients with MTC (Kunnimalaiyaan, Ndiaye et al. 2006).These compounds are not clinically useful but could provide powerful tools to study the cellular effects of PI3K inhibition and to develop other PI3K inhibitors (LoPiccolo, Blumenthal et al. 2008).
1.6 Rationale and aim of the project
1.6.1 Rationale
A major event in the neoplastic transformation of thyroid follicular cells is the constitutive activation of oncogenes: RET/PTC or RAS or BRAF genes (Soares, Trovisco et al. 2004). The intracellular pathways driven by these oncogenes are known to activate the MAPK-ERK signaling, required for maintaining cellular activities such as cell growth, proliferation, differentiation and apoptosis (Melillo, Castellone et al. 2005). Many genetic alterations have been implicated in PTCs and mostly involve the aberrant activation of MAPK pathway (Nikiforov 2002). Aberrant activation of the ERK pathway may induce tumor transformation. (Melillo, Castellone et al. 2005). One activating mutation along the RAS-MAPK pathway is sufficient for thyroid cell malignant transformation (Melillo, Castellone et al. 2005; Xing 2005).
23
Many events involved in the MAPK pathway have been studied in PTC. In a previous work (Preto, Goncalves et al. 2009) our group demonstrated that inhibition of BRAF by RNA interference (RNAi) do not change the levels of pERKs in TPC-1 (harboring a RET/PTC1 background). In other study Rocha et al, 2008, sugested that RAF-1 seems to be the dominant molecule in TPC-1 (Rocha, Paternot et al. 2008). It is important to understand the role of RAF-1 in the regulation of MAPK pathway in cells with RET/PTC1 background. Moreover, study the relevance of RAF-1 in the survival of these cells may provide new therapeutic strategies in thyroid cancer. Thus, we hypothesize that RAF-1 and not BRAF might be the molecule responsible for RET/PTC1-MAPK signaling in cells with RET/PTC1 rearrangement and might be essential in the survival of these cells.
1.6.2 General aim
The project proposal aimed to assess the role of RAF-1 in MAPK signaling in thyroid cancer cells harboring a RET/PTC1 rearrangement in comparison to cells with HRASG13R and BRAFV600E mutations. We also aimed to test the role of PI3K-AKT pathway in the regulation of ERK activation and proliferation of thyroid cancer cells with RET/PTC1 rearrangement.
1.6.3 Specific aim
To achieve our goals we will use three cell lines with various genetic profiles found in thyroid tumors in vivo: TPC-1 (harboring a RET/PTC1 rearrangement), 8505C (harboring a homozygous BRAFV600E mutation), and C643 (harboring a RASG13R mutation).
24 Specifically in the project we aimed at:
1- Study the phenotypic changes (proliferation and apoptosis) and ERK activation after inhibition of RAF-1, BRAF and co-inhibition by RNAi in TPC-1 cells and compare with the others cell lines (8505C and C643).
2- Study the phenotypic changes (proliferation and apoptosis) and ERK activation upon inhibition of MEK and PI3K-AKT pathways by PD98059 and LY294002 respectively, in TPC-1 cells in comparison with other cell lines.
II- MATERIAL AND
METHODS
27
2.1 Cell culture conditions
To meet the objectives of the present work, three human thyroid cancers derived cells lines with different genetic background were used: TPC-1, 8505C and C643.
TPC-1 was established from an papillary thyroid cancer of a female patient and harbors a RET/PTC1 rearrangement (Tanaka, Ogura et al. 1987). 8505C was established from an anaplastic thyroid cancer of a 78 year old female patient and harbors a homozygous BRAFV600E mutation (Ito, Seyama et al. 1992; Ito, Seyama et al. 1994; Meireles, Preto et al. 2007). C643 cell line was established from a fine-needle biopsy of an anaplastic thyroid cancer of a 76-year-old man and harbors a HRASG13R mutation (Mark, Ekedahl et al. 1987; Meireles, Preto et al. 2007).
TPC-1 was obtained from Dr. M. Mareel and Dr. JE. Dumont (University of Ghent, Ghent, Belgium), 8505C and C643 were obtained from Dr. M. Mareel (Meireles, Preto et al. 2007). All cell lines were obtained from the Cell Lines Bank from IPATIMUP.
Cells were grown in RPMI 1640 medium (GIBCO, Invitrogen) with L-glutamine and HEPES, supplemented with 10% heat inactivated fetal bovine serum (GIBCO, Invitrogen), 100 U/ml of penicillin and 100 μg/ml of streptomycin (GIBCO, Invitrogen). All cells were plated in 25 cm2 or 75 cm2 tissue culture flasks and maintained in a humidified incubator supplied with 95% air and 5% CO2, at 37ºC, fed
ever 3 to 4 days, and sub-cultured when 80% of confluence is reached, by treatment with a 0.05% trypsin/EDTA solution (GIBCO, Invitrogen). For the experiments cells were seeded onto six (1.5 ml) and twenty-four (500 µl) well plates at a density of 5x104 (TPC-1), 2x105 (8505C) and 2x105 (C643) cells/well.
2.2 RNA interference assay
The RNA interference (RNAi) is a mechanism for silencing gene expression by targeted degradation of a specific mRNA. Small interfering RNA (siRNA) could be used to induce post-transcriptional gene silencing. Within the cell this siRNA is then incorporated into the RNA- induced silencing complex (RISC), that leads to the cleavage of the sense strand of RNA by argonaute 2 (AGO2), resulting in the formation of an active complex RISC-siRNA. Finally, this complex recognizes, binds to and