Pró-Reitoria de Pós-Graduação e Pesquisa
Stricto Sensu em Ciências Genômicas e Biotecnologia
DESENVOLVIMENTO DE MODELO MULTIDIMENSIONAL DE
PREDIÇÃO DE PEPTÍDEOS ANTIMICROBIANOS UTILIZANDO
UM SISTEMA DE INFERÊNCIA NEURO-
FUZZY
ADAPTATIVO
(ANFIS)
Brasília - DF
2012
FABIANO CAVALCANTI FERNANDES
DESENVOLVIMENTO DE MODELO MULTIDIMENSIONAL DE PREDIÇÃO DE PEPTÍDEOS ANTIMICROBIANOS UTILIZANDO UM SISTEMA DE INFERÊNCIA
NEURO-FUZZY ADAPTATIVO (ANFIS)
Tese apresentada ao Programa de Pós-Graduação Stricto Sensu em Ciências Genômicas e Biotecnologia da Universidade Católica de Brasília, como requisito parcial para a obtenção do Título de Doutor em Ciências Genômicas e Biotecnologia.
Orientador: Prof. Dr. Octavio Luiz Franco
AGRADECIMENTOS
Agradeço ao meu orientador professor Dr. Octavio Luiz Franco, pelas importantes sugestões ao trabalho, pelo incentivo, apoio e direcionamento em todos os momentos.
Agradeço ao professor Dr. Daniel John Rigden e à professora Dra. Simoni Campos Dias pelas importantes sugestões ao trabalho. Agradeço à professora Dra. Lourdes Mattos Brasil e ao professor Dr. Renato da Veiga Guadagnin pelo incentivo ao ingresso no programa.
RESUMO
FERNANDES, Fabiano Cavalcanti. Desenvolvimento de modelo multidimensional de predição de peptídeos antimicrobianos utilizando um Sistema de Inferência Neuro-Fuzzy Adaptativo (ANFIS). 2012. 169f. Tese (Doutorado em Ciências Genômicas e Biotecnologia) – Universidade Católica de Brasília, Brasília – DF, 2012.
Atualmente observa-se um aumento significativo de microrganismos patogênicos resistentes aos antibióticos comercializados, principalmente em pacientes hospitalizados. Os peptídeos antimicrobianos podem ser uma solução promissora para o crescente problema da resistência bacteriana a antibióticos, pois, fazem parte do sistema imunológico de vários organismos e formam uma linha de defesa natural contra infecções. Estudos prévios têm relatado a presença de inúmeras proteínas de defesa contra a ação de microrganismos patogênicos em mamíferos, em diversas espécies de vegetais, assim como em fungos, insetos e crustáceos. Estas proteínas têm sido frequentemente identificadas e suas sequências de aminoácidos armazenadas em bancos de dados com sua função assinalada. Além disso, inúmeras sequências putativas têm sido descobertas a partir de projetos genoma e proteoma e também criadas e sintetizadas a partir de projetos in silico. As ferramentas de predição de peptídeos antimicrobianos são imprescindíveis na descoberta de novos fármacos, pois, representam uma economia de tempo e de recursos na seleção dos melhores candidatos para serem testados em laboratório. Já foram identificados padrões de estruturas e de sequências nos peptídeos antimicrobianos e existem diversos algoritmos de predição dos mesmos, porém, até o presente momento não foi encontrado nenhum modelo específico que avalie as múltiplas dimensões (primárias, secundárias e terciárias) das proteínas putativas de forma qualitativa e que utilize um sistema de inferência neuro-fuzzy como técnica de predição. O presente estudo propõe um novo modelo de predição que utiliza como informação de entrada características multidimensionais dos peptídeos putativos e como ferramenta de reconhecimento de padrões um sistema de inferência neuro-fuzzy adaptativo (Adaptive Neuro-Fuzzy Inference System - ANFIS), obtendo uma acurácia total de 99,5% e um MCC de 0,989. O modelo proposto também utiliza um número reduzido de parâmetros de entrada necessários para a rede ANFIS, possibilitando com isso predições mais rápidas e precisas e um melhor entendimento estrutural, levando desta maneira a um aumento de eficiência no projeto e na descoberta de novos peptídeos antimicrobianos.
ABSTRACT
Antimicrobial peptides (AMPs) are defense molecules widely distributed and represent a promising alternative for solving the problem of antibiotic resistance. Nevertheless, the experimental time needed to screen putative AMPs makes computational simulations based on peptide sequence analysis and/or molecular modeling extremely attractive. Artificial intelligence methods acting as simulation and prediction tools are of great importance in helping to efficiently discover and further novel AMPs design. Hitherto were not found prediction methods using as input information qualitative data from protein primary, secondary and tertiary structures altogether. Furthermore none of the main current prediction methods employ a neuro-fuzzy system as prediction tool. In the present study, state-of-the-art published outcomes using different prediction methods and databases were compared to an Adaptive Neuro-Fuzzy Inference System (ANFIS) model that uses peptide multidimensional information such as primary sequence, physicochemical characteristics and structural characteristics. Data from our study show that ANFIS obtained a global accuracy of 99.5% and a Mathews Correlation Coefficient (MCC) of 0.989, which proved it to be an efficient model for pattern recognition in antimicrobial peptide prediction. Moreover, a lower number of input parameters were needed for the ANFIS model, improving the speed and precision of prediction. In summary, due to the fuzzy nature of AMP structural and physicochemical properties, the ANFIS approach presented here can provide an efficient solution for screening putative AMP sequences and for exploration of properties characteristic of AMPs.
LISTA DE FIGURAS
Figura 1- Ciclo básico de trabalho para análise de peptídeos antimicrobianos
putativos ... 21
Figura 2- Modelo multidimensional proposto de predição de peptídeos antimicrobianos ... 22
Figura 3 – Modelo não-linear de um neurônio (HAYKIN, 2001) ... 48
Figura 4– (a) Função de pertinência em forma de sino com k=2 e m=1. (b) Função de pertinência gama com k=2 e a=1. (c) Função de pertinência gaussiana com m=8 e k=0,1. (d) Função de pertinência trapezoidal com a=-2,5, m=0, n=2,5 e b =5. ... 53
Figura 5 – Estrutura de uma rede ANFIS (JANG, 1993) ... 55
Figura 6– Estágio de pré-processamento dos dados (Bishop, 2007) ... 57
Figura 7 – Curva ROC performance de um classificador binário através da relação entre a sensibilidade do método (taxa de verdadeiro-positivo) e 1- a especificidade (1- taxa de verdadeiro-negativo) do método ... 61
Figura 8- Matriz de contingência ... 61
Figura 9 - Modelo proposto com quatro estágios, para predição de AMPs ... 62
Figura 10 – Metodologia de construção do conjunto positivo de dados multidimensionais de AMPs ... 65
Figura 11 – Metodologia de construção do conjunto negativo de dados multidimensionais de AMPs ... 66
Figura 12 - Fluxograma de extração de características físico-químicas ... 67
Figura 13 - Fluxograma de extração de características estruturais ... 68
Figura 14 - Fluxograma de Detecção e Seleção de Dados ... 70
Figura 15 – Fluxograma de Pré-Processamento com validação cruzada (método de resistência) ... 71
Figura 16 - Fluxograma de Classificação utilizando a rede ANFIS ... 72
Figura 17 – Gráfico em radar com heurística para seleção de dados de entrada para a rede ANFIS. O par (1,5) mostra que a agregação in vitro juntamente com o comprimento do peptídeo atingiram o menor valor inicial de RMSE ... 75
LISTA DE QUADROS
LISTA DE TABELAS
LISTA DE ABREVIATURAS
AMPER - banco de dados e ferramenta de descoberta de peptídeos antimicrobianos (database and an automated discovery tool for antimicrobial peptides)
AMPs - peptídeos antimicrobianos (antimicrobial peptides)
AMSDB - banco de dados de sequências antimicrobianas (antimicrobial sequences database)
ANFIS - sistema de inferência neuro-fuzzy adaptativo (adaptive neuro-fuzzy inference system)
APD - banco de dados de peptídeos antimicrobianos (antimicrobial peptide database)
APD2 - banco de dados atualizado de peptídeos antimicrobianos (updated antimicrobial peptide database)
ATP - adenosina trifosfato (adenosine triphosphate) AuC - área sob a curva (area under curve)
CAMEL - banco de dados de polipeptídeos catiônicos sintéticos (synthetic cationic polypeptides database)
CAMP - coleção de peptídeos antimicrobianos (collection of antimicrobial peptides) DA - análise de discriminante (discriminant analysis)
ESBL –β-lactamase de espectro estendido (extended spectrum β-lactamase) HMMs - modelos ocultos de Markov (hidden Markov models)
LPS - lipopolissacarídeos
MF – função de pertinência (membership function)
MIC - concentração mínima inibitória (minimum inhibitory concentration) PDB - banco de dados de proteínas (protein data bank)
QM - matrizes quantitativas (quantitative matrices)
QSAR - correlação quantitativa estrutura-atividade (quantitative structure–activity relationship)
RF - florestas aleatórias (random forests)
RMSE - raiz quadrada do erro médio quadrático (root mean squared error) RNA - rede neural artificial
ROC - receiver-operating characteristic
SVM - máquinas de vetor de suporte (support vector machine) UNIPROT - universal protein resource
SUMÁRIO
1 INTRODUÇÃO ... 20
1.1 ESCOLHA E APRESENTAÇÃO DO TEMA ... 20
1.2 FORMULAÇÃO E DELIMITAÇÃO DO PROBLEMA ... 22
1.3 OBJETIVOS GERAIS ... 23
1.4 OBJETIVOS ESPECÍFICOS ... 23
1.5 HIPÓTESES ... 23
1.6 RELEVÂNCIA DO ESTUDO ... 23
2 REVISÃO DA LITERATURA ... 25
2.1 ANTIBIÓTICOS ... 25
2.2 INFECÇÃO NOSOCOMIAL ... 27
2.3 BACTÉRIAS PATOGÊNICAS CAUSADORAS DE INFECÇÕES NOSOCOMIAIS ... 28
2.4 RESISTÊNCIA BACTERIANA A ANTIBIÓTICOS CONVENCIONAIS ... 31
2.5 PEPTÍDEOS ANTIMICROBIANOS ... 35
2.6 RESISTÊNCIA BACTERIANA A AMPS ... 38
2.7 BANCOS DE DADOS DE PEPTÍDEOS ANTIMICROBIANOS ... 40
2.7.1 The antimicrobial peptide database (APD) e the updated antimicrobial peptide database (APD2) ... 40
2.7.2 Antimicrobial peptides database (AMSDb) ... 41
2.7.3 AMPER ... 41
2.7.4 BACTIBASE ... 41
2.7.5 Collection of antimicrobial peptides (CAMP) ... 42
2.7.6 CyBase ... 42
2.8 MÉTODOS DE PREDIÇÃO DE PEPTÍDEOS ANTIMICROBIANOS ... 42
2.9 MODELAGEM MOLECULAR ... 44
2.9.1 Modelagem molecular por homologia ... 46
2.9.2 Modelagem molecular Ab-initio ... 46
2.10 REDES NEURAIS ARTIFICIAIS (RNA) ... 47
2.10.1 Conceitos ... 47
2.10.2 Redes MLP (multilayer perceptron) ... 49
2.10.4 Aprendizagem ... 51
2.11 SISTEMAS FUZZY ... 52
2.12 O SISTEMA DE INFERÊNCIA NEURO-FUZZY ADAPTATIVO (ANFIS) ... 54
2.13 RECONHECIMENTO DE PADRÕES ... 55
2.13.1 Pré-processamento, segmentação e extração de características . 56 2.13.2 Curva polinomial ... 57
2.13.3 Reconhecimento supervisionado e não-supervisionado ... 58
2.13.4 Classificadores ... 58
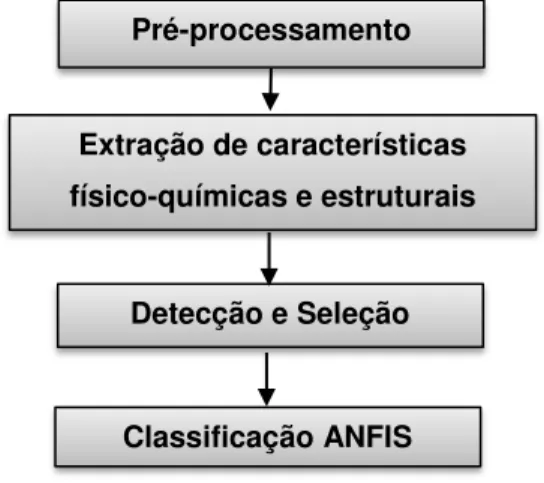
3 MATERIAL E MÉTODOS ... 62
3.1 O MODELO PROPOSTO ... 62
3.2 ESTÁGIO DE PRÉ-PROCESSAMENTO ... 62
3.2.1 Coleta e análise de dados ... 62
3.2.2 O banco de dados positivo de AMP ... 64
3.2.3 O banco de dados negativo de AMPs (não-AMPs) ... 65
3.3 EXTRAÇÃO DE CARACTERÍSTICAS FÍS.-QUÍMICAS E ESTRUTURAIS.. 66
3.4 ESTÁGIO DE DETECÇÃO E SELEÇÃO DE DADOS ... 69
3.5 ESTÁGIO DE CLASSIFICAÇÃO ... 70
4 RESULTADOS ... 73
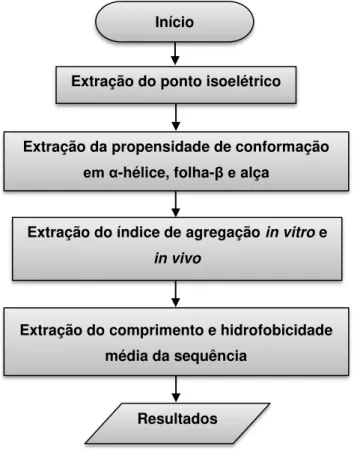
4.1 EXTRAÇÃO DE CARACTERÍSTICAS FÍSICO-QUÍMICAS ... 73
4.2 DETECÇÃO E SEL. DE CARACTERÍSTICAS FÍSICO-QUÍMICAS ... 73
4.3 EXTRAÇÃO DE CARACTERÍSTICAS ESTRUTURAIS ... 76
4.4 DETECÇÃO E SEL. DE CARACTERÍSTICAS ESTRUTURAIS ... 76
4.5 DETECÇÃO E SEL. DE CARACTERÍSTICAS MULTIDIMENSIONAIS ... 81
4.6 CLASSIFICAÇÃO ANFIS ... 83
5 DISCUSSÃO ... 97
6 CONCLUSÃO ... 109
7 TRABALHOS FUTUROS ... 110
8 REFERÊNCIAS ... 111
1 INTRODUÇÃO
O presente estudo se propõe a mapear a intersecção de três áreas do conhecimento, Reconhecimento de Padrões, Lógica Fuzzy e Bioquímica, através da construção de um sistema de predição de peptídeos antimicrobianos (antimicrobial peptides - AMPs) que pondere as diversas dimensões dos peptídeos putativos ou candidatos utilizando um sistema de inferência neuro-fuzzy adaptativo como ferramenta de reconhecimento de padrões. O desenvolvimento de um protótipo possibilita a realização de testes que demonstrem a eficiência do sistema.
1.1 ESCOLHA E APRESENTAÇÃO DO TEMA
A escolha do tema foi o resultado da identificação da necessidade de uma nova ferramenta de predição de AMPs que contemple suas dimensões primária, secundária e terciária aumentando desta forma a eficiência da predição. As ferramentas atuais de predição de AMPs abordam majoritariamente as características derivadas da sequência de aminoácidos (dimensão primária) que formam o peptídeo em estudo e algumas ferramentas utilizam informações relacionadas à sua estrutura para abordagens utilizando a técnica de relação quantitativa de atividade-estrutura (Quantitative Structure–Activity Relationship - QSAR). Com a evolução das técnicas de modelagem molecular por homologia e ab-initio e sua utilização como parte do processo de pesquisa e entendimento de novos AMPs, há a possibilidade de se utilizar novas informações para a detecção de padrões e características, auxiliando assim o projeto de novos AMPs.
Sistema de Inferência Neuro-Fuzzy Adaptativo (Adaptive Neuro-Fuzzy Inference System - ANFIS) (JANG, 1993) como ferramenta de predição.
Ao purificar e determinar a sequência polipeptídica de potenciais candidatos a peptídeos antimicrobianos, o pesquisador segue por uma análise de padrões, similaridades, alinhamentos e análise de estrutura primária. Em seguida uma análise de características físico-químicas como carga, anfipaticidade, etc. Depois são realizadas várias análises tridimensionais, através de modelagem molecular, por homologia ou ab-initio dependendo de cada caso, e predição de função, utilizando diversos algoritmos disponíveis, conforme Figura 1.
Figura 1- Ciclo básico de trabalho para análise de peptídeos antimicrobianos putativos
1- Determinação da sequencia de aminoácidos de um provável peptídeo antimicrobiano, ex: RGGRLCYCRRRFCVCVGRC
2- Busca por similaridades, padrões e alinhamento e análise de estrutura primária
3- Análise de características fisico-químicas tais como: carga, hidrofobicidade,
anfipaticidade, etc.
4- Análise conformacional, alinhamento 3D, Z-score, cobertura, etc.
Foi desenvolvido neste trabalho um modelo multidimensional de predição de peptídeos antimicrobianos, segundo Figura 2. Essa ferramenta disponibilizou uma camada fuzzy para tratar as informações de seqüências de aminoácidos, suas propriedades físico-químicas e também informações da sua estrutura tridimensional e submeteu a uma rede neural artificial (RNA), estas duas etapas compõem a rede ANFIS, que funcionou como ferramenta de reconhecimento de padrões.
Figura 2- Modelo multidimensional proposto de predição de peptídeos antimicrobianos
1.2 FORMULAÇÃO E DELIMITAÇÃO DO PROBLEMA
O problema definido no presente trabalho consiste em melhorar a eficiência da predição antimicrobiana de um peptídeo candidato através da adição de características estruturais da molécula, além das características primárias já utilizadas, em um sistema de reconhecimento de padrões.
A avaliação de um sistema neuro-fuzzy adaptativo como ferramenta de reconhecimento de padrões e sua comparação com ferramentas atuais na predição de peptídeos antimicrobianos também faz parte do problema a ser solucionado, trazendo uma maior eficiência no processo de predição de peptídeos candidatos e consequentemente uma maior economia de tempo e de recursos no desenvolvimento de novos antibióticos para a população.
Sequência primária do peptídeo candidato
ANFIS
Modelo tridimensional do peptídeo candidato
1.3 OBJETIVOS GERAIS
O objetivo geral deste trabalho consiste na construção de um modelo multidimensional neuro-fuzzy adaptativo de predição de peptídeos antimicrobianos para auxiliar no projeto e na pesquisa de novos fármacos.
1.4 OBJETIVOS ESPECÍFICOS
1. Identificar a intersecção entre as áreas citadas e sua aplicação na predição de peptídeos antimicrobianos;
2. Realizar simulações para identificar a melhor solução para o problema de reconhecimento de padrões em peptídeos antimicrobianos;
3. Desenvolver um modelo e um protótipo de um sistema que possibilite uma predição efetiva de peptídeos antimicrobianos e testar o mesmo em novas sequências putativas de aminoácidos;
1.5 HIPÓTESES
Esta pesquisa pretende testar a seguinte hipótese: Um modelo multidimensional neuro-fuzzy adaptativo de predição de peptídeos antimicrobianos pode aumentar a eficiência da análise de peptídeos putativos.
1.6 RELEVÂNCIA DO ESTUDO
2 REVISÃO DA LITERATURA
2.1 ANTIBIÓTICOS
Os antibióticos foram originalmente definidos como substâncias produzidas por micro-organismos, que em pequenas concentrações inibem o crescimento de outros micro-organismos conforme Waksman (1944 apud DENYER et al., 2011). Esta definição atualmente ultrapassada implica em uma ocorrência natural dos antibióticos, excluindo as substâncias resultantes de síntese química. O uso do termo antibiótico vem mudando com o tempo, pois, muitas substâncias sintéticas análogas foram desenvolvidas com as mesmas características dos antibióticos tradicionais tais como potência, baixa toxicidade e ação sistêmica (DENYER et al., 2011). A definição mais aceita hoje é a de uma substância produzida por micro-organismos ou por uma substância semelhante, produzida integralmente ou parcialmente por síntese química, que em baixas concentrações inibe o crescimento ou mata outros micro-organismos. Com base nesta definição mais ampla podem-se incluir as sulfonamidas que foram utilizadas em terapias no início do século XX e sua descoberta precedeu a penicilina (FLEMING, 1929) em cerca de dez anos. O período da segunda guerra mundial serviu como incentivo à indústria farmacêutica americana para a produção em larga escala de antibióticos e sua posterior popularização e utilização ampla (CHRETIEN et al., 2007).
(ALANIS, 2005). Além disso, a descoberta das sulfonamidas, antibióticos de amplo espectro de ação, resultou em modificações na forma de tratamento dessas infecções. Neste sentido, ocorreu a transição da terapia antimicrobiana patógeno-específica para a terapia não patógeno-específica, conhecida também como terapia empírica (CASADEVALL, 1996). Esta terapia, amplamente difundida até os dias atuais (POGUE et al., 2011), permite o tratamento imediato e efetivo de uma infecção bacteriana sem a necessidade da identificação do patógeno envolvido (CASADEVALL, 1996).
A resistência bacteriana a antibióticos é um fenômeno natural e anterior à pressão seletiva atual causada pelo uso clínico de antibióticos, porém o seu uso inadequado tem criado uma força motriz que influencia a seleção de genes de resistência previamente existentes. (D'COSTA et al., 2011; SCHMIEDER; EDWARDS, 2012). Como consequência adicional do uso inadequado da penicilina, em apenas um ano de uso observou-se um número significante de cepas de S. aureus resistentes a este medicamento e, em 1948, 50% das cepas hospitalares isoladas já se mostravam resistentes a inúmeros tipos de antibióticos, atingindo níveis mundiais de 80% entre 1955-1957 (ALANIS, 2005; SYKES, 2010). À medida que outros agentes como a eritromicina, cloranfenicol e tetraciclinas tornaram-se medicamentos de escolha no tratamento de S. aureus já resistente à penicilina, a resistência bacteriana a esses agentes também cresceu, limitando seu uso clínico por volta de 1950. O mesmo ocorreu com a meticilina, derivado semissintético do núcleo ativo da penicilina (ácido 6-aminopenicilânico), que assumiu importante papel no controle da infecção de S. aureus. Bactérias resistentes a meticilina causaram inúmeros casos de infecção hospitalar na Europa em 1960 (KOLLEF; FRASER, 2001). A resistência de S. aureus a meticilina atingiu cerca de 60% em 2011, cujas cepas são clones dos isolados endêmicos brasileiros (ROSSI, 2011).
espalhando na comunidade e provocando doenças severas em pacientes previamente saudáveis (ALANIS, 2005). Dessa forma, os agentes antimicrobianos são essenciais para a saúde e bem-estar humano e animal, e a resistência a esses agentes é uma preocupação de saúde pública mundial (WHO, 2005).
2.2 INFECÇÃO NOSOCOMIAL
De acordo com a Organização Mundial de Saúde (World Health Organization - WHO), a infecção hospitalar ou nosocomial pode ser definida como “[...] infecção adquirida durante o período de internação hospitalar que não estava presente ou incubada na admissão [...]” (WHO, 2002). Infecções que ocorrem mais de 48 h após a admissão são geralmente consideradas nosocomiais. A maior susceptibilidade pode ocorrer em pacientes imunodeprimidos, mas estas também podem ser adquiridas por visitantes e funcionários do próprio hospital (WHO, 2002). Atualmente, este tipo de infecção representa um grave problema da saúde pública mundial. Em países desenvolvidos a prevalência de infecções nosocomiais varia de 4,5% a 10,5%, sendo nos Estados Unidos a menor taxa e no Canadá a maior. Enquanto que em países em desenvolvimento a menor prevalência é registrada na Letônia (5,7%) e a maior na Albânia (19,1%). No Brasil, a taxa média de infecção nosocomial atual registrada é de 14%, enquanto que a taxa geral em unidades de terapia intensiva neonatal é de 38,6% (ALLEGRANZI, 2010). Nos Estados Unidos, a infecção nosocomial afeta 1,7 milhões de pacientes anualmente, dos quais 99.000 morrem devido a estas infecções (KLEVENS et al., 2007).
a séria limitação quanto à escolha dos antibióticos adequados para o tratamento, especialmente em pacientes em estado de saúde crítico. Assim, a resistência a medicamentos pode ser relacionada como o maior sinal de debilidade no tratamento de doenças infecciosas severas (ALANIS, 2005; FISCHBACH; WALSH, 2009), consistindo em um grande desafio que requer uma ação multidisciplinar e um esforço comum envolvendo todas as unidades de saúde públicas e privadas (WHO, 2005).
2.3 BACTÉRIAS PATOGÊNICAS CAUSADORAS DE INFECÇÕES NOSOCOMIAIS
Diferentes microrganismos podem ser patogênicos ao homem, muitos dos quais são frequentes em casos de infecção nosocomial. As bactérias têm sido relatadas como as principais responsáveis por este tipo de infecção (LEVY, 2004). Entretanto, os índices reais e atuais sobre infecções nosocomiais são difíceis de calcular, principalmente por falta de informações oriundas dos sistemas de saúde de todo o mundo (FISCHBACH; WALSH, 2009; ALLEGRANZI, 2010). Em uma estimativa europeia, realizada em unidades pediátricas no ano 2000 (RAYMOND; AUJARD, 2000), os fungos foram associados a aproximadamente 10% dos casos de infecções hospitalares, as viroses a cerca de 22% dos casos, e as bactérias a 68% dos casos. Os patógenos que lideram o ranking das infecções hospitalares estão descritos no Quadro 1. A Enterobacteriaceae é considerada a maior e mais heterogênea família de bactérias Gram-negativas e está diretamente implicada nestas infecções, sendo identificadas em 95% das amostras de casos clínicos de infecções nosocomiais bacterianas (LEVY, 2004).
Quadro 1- Agentes infecciosos mais comuns de infecções nosocomiais
Patógeno Regiões comuns de isolamento do patógeno Bactérias Gram-negativas
E. coli Trato urinário, feridas cirúrgicas, sangue Pseudomonas sp. Trato urinário, trato respiratório, queimaduras
Patógeno Regiões comuns de isolamento do patógeno Proteus sp. Trato urinário, feridas cirúrgicas
Klebsiella sp. Trato urinário, trato respiratório, feridas cirúrgicas Enterobacter sp. Trato urinário, trato respiratório, feridas cirúrgicas Serratia sp. Trato urinário, trato respiratório, feridas cirúrgicas Bactérias Gram-positivas
Streptococcussp. Trato urinário, trato respiratório, feridas cirúrgicas S. aureus Pele, feridas cirúrgicas, sangue
Staphyloccus epidermitis Pele, feridas cirúrgicas, sangue Fungos
Cândida albicans Trato urinário, sangue
Outros Trato urinário, sangue, trato respiratório Fonte: Adaptada de (LEVY, 2004; MARIA-NETO, 2011)
suficiente para alcançar o trato respiratório ou a presença de um microrganismo altamente virulento (LEVY, 2004).
2.4 RESISTÊNCIA BACTERIANA A ANTIBIÓTICOS CONVENCIONAIS
Em geral, tem sido visto que a resistência bacteriana tem fundamentação em nível genético, indicando que mudanças na composição genética das bactérias previamente suscetíveis ocorrem tanto por mutação quanto por introdução de um novo material genético (JOLIVET-GOUGEON et al., 2011). Dessa forma, a expressão gênica modificada resulta em modificações em um ou mais mecanismos biológicos da bactéria, determinando o tipo de resistência que irá desenvolver (ALANIS, 2005; SYKES, 2010). Esses genes de resistência podem ser adquiridos de outras espécies e transmitidos por replicação (MACLEAN et al., 2010). Resumidamente, para o desenvolvimento de resistência, normalmente tem sido necessário que dois elementos chave combinem-se: a presença de um antibiótico bactericida ou bacteriostático e uma colônia bacteriana heterogênea onde pelo menos uma célula bacteriana carregue o determinante genético capaz de expressar resistência ao antibiótico (HUJER et al., 2006; DAVIES; DAVIES, 2010). A resistência a antibióticos pode ser natural, também conhecida como intrínseca, ou pode ser adquirida. O tipo mais frequente de resistência é a adquirida e ela é geralmente transmitida horizontalmente através de plasmídeos. A forma de resistência natural herdável pode ser causada por mutação gênica, principalmente através de falhas e deficiências no sistema de reparo de bases mal emparelhadas (MMR - Methyl-directed Mismatch Repair) que podem resultar em um estímulo a recombinação de ADN (ALANIS, 2005; JOLIVET-GOUGEON et al., 2011).
antimicrobianos durante a criação de animais para consumo como alimento humano nas décadas de 1980 e 1990. Entre 1988 e 1990, devido ao uso excessivo de ceftazidima, Enterobacter cloacae mostrou resistência a cefalosporina. Recentemente, com a propagação de bactérias resistentes fora do ambiente hospitalar, determinadas cepas de microrganismos multirresistentes vêm sendo disseminados. Entre estes, podem ser citadas Streptococcus sp. resistente a macrolídeos; Streptococcus pneumoniae resistente a diferentes classes de antibióticos, incluindo penicilina; cepas mais virulentas de S. aureus resistentes a meticilina, devido à expressão de determinadas toxinas; assim como enterococos que se tornaram resistentes a vancomicina (ALANIS, 2005; DAVIES; DAVIES, 2010; SYKES, 2010).
enquanto um quarto das Enterobacteriaceas isoladas são resistentes a combinação de amoxicilina com o inibidor de TEM-1 clavulanato. Já para os antibióticos não β -lactâmicos, a taxa de isolados de E. coli resistentes a ciprofloxacina é 12,5 %, 24,4 % a trimetropim/sulfametoxazol, e 5,8% a gentamicina.
Dessa forma, o preditor principal de mortalidade causada por E. coli resistentes, como as produtora de ESBLs, consiste na terapia antimicrobiana inicial inadequada (definida como o início do tratamento com agentes antimicrobianos até 72 h após o primeiro teste de cultura sanguínea positivo) (TUMBARELLO et al., 2007). Tratamentos alternativos de severas infecções por E. coli produtora de ESBLs, incluem carbapenamicos, amikacinas, tigeciclinas e combinações de inibidor de β-lactamases/β-lactâmicos, porém há poucas evidências clínicas comparativas sobre a sua eficácia (OTEO et al., 2010). Ademais, carbapenamicos, além de medicamentos dispendiosos e de administração intravenosa, são reservados para o tratamento de pacientes com infecções severas e imunocomprometidos. De modo que este recurso especial será perdido caso as carbapenamases se tornem o medicamento de primeira escolha no tratamento de infecções por E. coli (ERB et al., 2007). Por sua facilidade em adquirir resistência, isolados de E. coli com resistência generalizada a antibióticos têm sido identificados em fontes humanas, animais e ambientais, resultando em uma maior difusão dos elementos de resistência graças ao compartilhamento de material genético entre microrganismos de diferentes fontes (AJIBOYE et al., 2009).
existentes, com mecanismos de ação semelhantes. Dessa forma, o desafio atual não consiste simplesmente na busca de novos agentes antibióticos, mas sim na busca de antibióticos com mecanismos de ação inovadores (COATES et al., 2011).
Quadro 2- Principais classes de antibióticos, seus mecanismos de ação, resistência e doenças bacterianas relacionadas
Mecanismo de ação Família de antibióticos Bactéria resistente Doença Inibição da síntese de
parede celular cefalosporinas, -lactâmicos (penicilinas, carbapenêmicos)
S. pneumoniae Pneumonia N. gonorrhoeae Gonorreia
tifoide Glicopeptídeos S. aureus Nosocomiais Lipopeptídeos cíclicos
(daptomicina) S. aureus Nosocomiais Oxazolidononas Enterococcus spp. Nosocomiais
Streptograminas S. aureus Nosocomiais Cetolídeos Enterococcus spp. Nosocomiais Macrolídeos Enterococcus spp. Nosocomiais
Lincosamidas Enterococcus spp. Nosocomiais Inibição da síntese de
proteínas Tetraciclinas N. gonorrhoeae Gonorreia tifoide Aminoglicosídeos
Inibição da síntese de
ADN Fluoroquinolonas P. aeruginosa Nosocomiais S. pneumoniae Pneumonia Inibição da síntese de
ARN Rifampina
K. pneumoniae Pneumonia Inibição da síntese do
ácido fólico Sulfonamidas Mycoplasma pneumoniae
Pneumonia Trimetoprim S. aureus Nosocomiais
Agentes
desorganizadores de membrana
Polimixinas (Polimixina B,
Colistina) S. aureus Nosocomiais Outros mecanismos Metronidazol Clostridium
perfringens
Gastrenterites
2.5 PEPTÍDEOS ANTIMICROBIANOS
Todos os organismos vivos, abrangendo desde microrganismos até plantas e animais, evoluíram de forma a desenvolver mecanismos para defender-se ativamente contra o ataque de patógenos. O mais sofisticado desses mecanismos consiste no desenvolvimento de anticorpos após um primeiro reconhecimento de antígenos, os quais permitem eliminar invasores específicos (THOMMA et al., 2002; FRECER et al., 2004). Porém, essa ação de resposta imune adquirida no reconhecimento de organismos estranhos, tem sido apenas elaborada por um pequeno subtipo de organismos, denominados vertebrados superiores. A imunidade natural consiste no mecanismo mais comum de defesa, valendo-se da produção de compostos fenólicos, substâncias secundárias e compostos proteicos antimicrobianos como resposta a patógenos invasores (CEDERLUND et al., 2011).
AMPs são particularmente potentes e apresentam um amplo espectro de ação. Normalmente sua atividade não se restringe a um único tipo de patógeno como bactérias Gram-positivas ou Gram-negativas, fungos ou protozoários. Recentemente, tem sido evidenciado que AMPs exercem funções na modulação da imunidade, as quais têm impacto em infecções e inflamações (BROWN; HANCOCK, 2006; RAHNAMAEIAN, 2011). Entretanto, alguns AMPs helicoidais também tendem a ser hemolíticos, podendo causar efeitos colaterais (HANCOCK et al., 2006). Diversos modelos têm sido propostos para explicar o mecanismo de ação dos AMPs (Quadro 3), mas basicamente os peptídeos catiônicos inicialmente se ligam a carga negativa do LPS e a fosfolipídios na face externa da membrana bacteriana, formando acumulados e agregados na superfície da membrana e, finalmente, permeabilizando/desintegrando a membrana bacteriana através de vários mecanismos possíveis de formação de poros (FRECER et al., 2004; RAHNAMAEIAN, 2011).
Quadro 3- Modelos de ação propostos para peptídeos antimicrobianos
Alvos Principais Alvo Específico/Modo de Ação Exemplos de Peptídeos Referências Proteínas
externas Ativação da autolisina pep5, nisina (ZHAO; KINNUNEN, 2003; BROGDEN, 2005) Ativação da fosfolipase A2 magainina II, indolicidina (BROGDEN, 2005) Lipídeos de
superfície externa Permeabilização de LPS (Gram-negativa) cecropina P1 (STRAUSS et al., 2010) Lipídeo II (precursor de
peptideoglicano)
defensinas e nisina (HSU et al., 2004; DE LEEUW et al., 2010; SASS et al., 2010)
Proteínas de membrana externa (Gram-negativa)
Proteína de membrana externa I (OprI)
SMAP-29, CAP-18 (LIN et al., 2010)
Inibição da proteína D associada a LPS (LptD)
Análogos
peptidomiméticos protegrina I
(SRINIVAS et al., 2010)
Proteína de membrana
externa F (OmpF) HP (2–20) análogo (APETREI et al., 2010) Membrana interna Formação de poro
barrel-stave
alameticina (BROGDEN, 2005) Micelização por
detergente dermaseptina, cecropina (BROGDEN, 2005) Poro toroidal magainina II, melitina,
protegrina I (BROGDEN, 2005; TANG; HONG, 2009)
Alvos Principais Alvo Específico/Modo de Ação Exemplos de Peptídeos Referências Poro toroidal desordenado Análogo de magainina,
melitina (SENGUPTA et al., 2008) Espessamento/afinamento
de membrana
PGLa, LL-37 (LOHNER, 2009) Aglomeração de lipídeos
carregados Análogos de magainina, peptídeos ricos em arginina
(EPAND; EPAND, 2011)
Sem formação de bicamada intermediária
gramicidina S (HANEY et al., 2010)
Fosfolipídios oxidados temporina L, indolicidina (MATTILA et al., 2008)
Carregador aniônico indolicidina (ROKITSKAYA et al., 2011)
Despolarização não lítica
da membrana lactoferricina bovina, daptomicina (JEU; FUNG, 2004; GIFFORD et al., 2005)
Eletroporação NK-lisina (JEU; FUNG, 2004) Proteínas integrais
de membrana Proteínas relacionadas a translocação de prótons clavanina A (VAN KAN et al., 2002) Ácidos nucleicos ADN (geral) buforina 2, tachiplesina,
indolicidina
(BROGDEN, 2005) ADN (interações
covalentes) indolicidina (MARCHAND et al., 2006) ADN ramificado WRWYCR (SU, L. Y. et al.,
2010)
ARN (geral) buforina 2 (BROGDEN, 2005) Proteínas
intracelulares
Inibição de ADNK, chaperonina GroEL
pirrocoricina, drosocina, apidaecina
(BROGDEN, 2005) Proteassomo 20S,
proteínas contendo SH3 PR-39 (ZANETTI, 2004; ANBANANDAM et al., 2008)
Fonte: Adaptada de (MARIA-NETO, 2011; NGUYEN et al., 2011)
proteicas de AMPs de sementes comumente observadas, podem ser incluídas as proteínas inibidoras de ribossomo (NG; PARKASH, 2002), proteínas transferidoras de lipídeos (MOLINA et al., 1993; CRUZ et al., 2010), proteínas de ligação à quitina (YOKOYAMA et al., 2009), defensinas (FRANCO et al., 2006; DOS SANTOS et al., 2010), proteínas semelhantes à taumatina (CHU; NG, 2003) e proteínas semelhantes à heveína (KOO et al., 1998; SHUKUROV et al., 2012). Ainda, podem ser citadas as proteínas de armazenamento, como as proteínas ricas em glicina e as albuminas 2S, ricas em metionina (PELEGRINI et al., 2006; PELEGRINI et al., 2008; ODINTSOVA et al., 2010; MARIA-NETO et al., 2011).
Já no reino animal, os AMPs exercem um papel fundamental no sistema imune, atuando como barreira natural inata frente a infecções microbianas e processos inflamatórios (STROMINGER, 2009). Dessa forma, tem sido relatado o isolamento de AMPs em vertebrados (GANZ, 2004) e invertebrados (OTERO-GONZALEZ et al., 2010). Dentre estes se incluem, desde humanos (WIESNER; VILCINSKAS, 2010), insetos (BOULANGER et al., 2006), aracnídeos (GAO et al., 2005), até anfíbios (KONIG; BININDA-EMONDS, 2011). De fato, a pele de anfíbios consiste em uma das fontes mais ricas em substâncias com propriedades farmacológicas, como citotoxicidade, atividade analgésica, anti-inflamatória, antiviral e antimicrobiana (CALKOSINSKI et al., 2009).
2.6 RESISTÊNCIA BACTERIANA A AMPS
Supõe-se que a resistência a AMPs pode surgir naturalmente a partir de mutações espontâneas ou de genes de resistência previamente existentes em uma população heterogênea, resultado do curto tempo de geração bacteriano e da pressão seletiva exercida quando for constante a exposição bacteriana aos AMPs (D'COSTA et al., 2011; SCHMIEDER; EDWARDS, 2012). Desta forma, entender os mecanismos de resistência a AMPs é uma etapa fundamental para o planejamento racional da geração subsequente de antibióticos (FJELL et al., 2011; NGUYEN et al., 2011).
bactérias Gram-negativas a polimixina B tem sido associada a modificações nos lipopolissacarídeos (LPS), um dos principais componentes de membrana externa (FERNANDEZ et al., 2010). Conforme demonstrado em P. aeruginosa, por genética molecular, este mecanismo de resistência parece ser desencadeado pela indução de um sistema regulatório de dois componentes como o sistema PhoP-PhoQ (MACFARLANE et al., 2000; KWON; LU, 2006) e o sistema PmrA-PmrB (MCPHEE et al., 2003). O sistema PhoP-PhoQ ativa o sistema PmrA-PmrB, que regula a expressão gênica de proteínas periplasmáticas e integrais de membrana, bem como alguns produtos citoplasmáticos, resultando em modificações pontuais no lipídeo A. Ademais, tem sido relatado que bactérias Gram-negativas resistentes a polimixina empregam o açúcar 4-amino-4-desoxi-L-arabinose (L-Ara4N) carregado positivamente para modificar o grupamento fosfato do lipídeo A da estrutura do LPS (BURTNICK; WOODS, 1999). Publicações recentes utilizando bactérias nocaute e técnicas cromatográficas, têm reportado o operon pmrCAB como mediador das resistências a polimixina (ARROYO et al., 2011) e a colistina (BECEIRO et al., 2011) em A. baumannii, através de modificações na fosfoetanolamina do lipídeo A. A redução na carga negativa da membrana como resultado destas modificações poderia reduzir a afinidade entre os compostos proteicos antimicrobianos catiônicos e a membrana celular e, portanto, reduzir a eficácia de sua ligação .
TZENG et al., 2005), um complexo de transporte das exoproteínas pelo espaço periplasmático. Já em V. cholerae, a partir de experimentos de mutação sítio-dirigida, o gene msbB tem sido indicado como essencial para a resistência de cepas El Tor, causando a perda do grupamento acil do lipídeo A (MATSON et al., 2010). Modificações na composição dos fosfolipídios aniônicos de membrana e consequente redução da afinidade por compostos proteicos antimicrobianos catiônicos poderia ser alcançado pela enzima MprF, que usa L-Lys e L-Ala para introduzir cargas positivas à superfície da membrana (ERNST; PESCHEL, 2011). Experimentos de espectrometria de massa encontraram nas bactérias patogênicas ao homem P. aeruginosa, E. faecalis, Proteus mirabilis e Streptococcus pyogenes, proteinases capazes de degradar e inativar o peptídeo de defesa LL-37 (SCHMIDTCHEN et al., 2002). Em síntese, os mecanismos de resistência bacteriana a AMPs mostram alta complexidade e não aparentam seguir um padrão único até então registrado. Estas observações sugerem que AMPs não são meramente moléculas que causam o rompimento de membranas (MARIA-NETO, 2011).
2.7 BANCOS DE DADOS DE PEPTÍDEOS ANTIMICROBIANOS
2.7.1 The antimicrobial peptide database (APD) e the updated antimicrobial
peptide database (APD2)
os aminoácidos mais comuns (>10%) em AMPs são Alanina, Glicina, Cisteína, Lisina e Leucina. O APD2 tem conseguido através de suas ferramentas de projeto de novos AMPs, obter novos peptídeos com maior atividade antimicrobiana. O banco de dados pode ser acessado em: http://aps.unmc.edu/AP/main.php (WANG; WANG, 2004; WANG et al., 2009).
2.7.2 Antimicrobial peptides database (AMSDb)
Este banco de dados contém sequências de peptídeos e de peptídeos antimicrobianos codificados por genes existentes. O banco também inclui, quando disponível, as sequências precursoras e os AMPs putativos, deduzidos da sequência de ADN. O AMSDb mantém apenas sequências oriundas de animais e de plantas. O banco de dados foi criado e tem sido mantido pela Universidade de Trieste e pode ser acessado em http://www.bbcm.units.it/~tossi/.
2.7.3 AMPER
Em 2007 foi lançado o AMPER, um banco de dados aberto com 890 sequências derivadas do banco AMSDb, abrangendo as mais importantes classes de AMPs. Destas, apenas 767 sequências apresentaram correspondências no UniProt (http://www.uniprot.org/) (CONSORTIUM, 2011) como peptídeos maduros e regiões propeptídicas. Em seguida, através de buscas iterativas no Swiss-Prot (CONSORTIUM, 2011) por AMPs desconhecidos, obteve-se um conjunto final de 1045 peptídeos maduros e 253 propeptídeos (FJELL et al., 2007).
2.7.4 BACTIBASE
múltiplas sequências, modelos ocultos de Markov, modelagem molecular entre outras. O BACTIBASE pode ser acessado em http://bactibase.pfba-lab-tun.org (HAMMAMI et al., 2007; HAMMAMI et al., 2010).
2.7.5 Collection of antimicrobial peptides (CAMP)
O CAMP consiste em um banco de dados de acesso livre que possui 2867 sequências antimicrobianas validadas experimentalmente e 1153 sequências antimicrobianas preditas. O banco oferece informações relacionadas à sequência, definição da proteína, números de acesso, atividade, taxonomia do organismo fonte, organismos alvo com valores de MIC, atividade hemolítica e links para bancos de dados externos tais como UniProt (http://www.uniprot.org/) e Protein Data Bank (PDB - http://www.rcsb.org/). O CAMP pode ser acessado em http://www.bicnirrh.res.in/antimicrobial/ (THOMAS et al., 2010).
2.7.6 CyBase
O CyBase foi originalmente desenvolvido em 2006 como um banco de dados de proteínas de backbone cíclico, uma promissora classe de core estrutural para o desenvolvimento de novas drogas, oferecendo ferramentas de busca, visualização e análise de função. No lançamento da sua segunda versão em 2007, ele possuía 251 sequências de proteínas, 49 sequências de ácidos nucleicos, 39 estruturas e 91 entradas relacionadas à atividade de cinco classes de proteínas circulares. O CyBase também possui dados de análogos sintéticos que auxiliam o projeto de desenvolvimento de novas drogas (MULVENNA et al., 2006; WANG et al., 2008).
2.8 MÉTODOS DE PREDIÇÃO DE PEPTÍDEOS ANTIMICROBIANOS
O número de possíveis combinações de seqüências de aminoácidos torna difícil a descoberta de novos AMPs exclusivamente através de testes experimentais em laboratório. Para um peptídeo de comprimento de amino ácidos, existem sequências possíveis a serem testadas, variando tipicamente de a ,
grupos de pesquisa estão, portanto, buscando ferramentas que realizem predição in silico da atividade antimicrobiana, para fornecer conjuntos específicos de candidatos mais prováveis para a avaliação clínica (HADLEY; HANCOCK, 2010).
O surgimento dos bancos de dados de peptídeos antimicrobianos tornou mais fácil o entendimento e projeto racional de AMPs por ser uma fonte de informações disponível para comparações, análises e testes com AMPs e AMPs putativos através dos mais diversos métodos e algoritmos.
Em 2004, um conjunto de 101 polipeptídeos catiônicos sintéticos, chamado de CAMEL-s foi utilizado como base de dados para cálculos de Relação Estrutura-Atividade Quantitativa (QSAR) e Redes Neurais Artificiais (RNA) para predizer função antimicrobiana (CHERKASOV; JANKOVIC, 2004). Em 2007, o banco de dados AMPER utilizou modelos ocultos de Markov (HMM) para predizer classes individuais de AMPs (FJELL et al., 2007). No mesmo ano, o Antibp (LATA et al., 2007), um método que utilizou um subconjunto de 486 sequências provenientes do banco de dados APD2, empregou RNA, Matrizes Quantitativas (QM) e Support Vector Machine (SVM) para predizer atividade antimicrobiana, utilizando proteínas não secretadas validadas pelo algoritmo Mitpred (KUMAR et al., 2006) como conjunto de dados negativos. Em 2009, o banco de dados atualizado de peptídeos antimicrobianos (ADP2) (WANG; WANG, 2004; WANG et al., 2009) demonstrou que em AMPs alguns resíduos específicos de aminoácidos são mais comuns do que outros. Sua ferramenta de previsão pode ser baseada em similaridade de seqüências e de certos princípios conhecidos de AMPs. No mesmo ano, o banco de dados chamado RANDOM (FJELL et al., 2009) pesquisou AMPs em duas bibliotecas aleatórias com 1.400 peptídeos, utilizando 3D QSAR e RNAs como ferramentas de predição e um conjunto de testes de 100.000 peptídeos virtuais.
uma ferramenta de predição baseada em SVM. O AMSDb (http://www.bbcm.univ.trieste.it/~Tossi/amsdb.html) abrange as sequências peptídeos antimicrobianos codificados por genes e seqüências de proteínas de animais e vegetais e não fornece ferramentas de predição de atividade. Além disso, outros bancos de dados (BRAHMACHARY et al., 2004; JURETIC et al., 2009; HAMMAMI et al., 2010) e metodologias, tais como matrizes quantitativas (QM) (LATA et al., 2010) e transdutores ponderados de estado finito (WFSTs) (WHELAN et al., 2010), permitiram a análise computacional de AMPs, com o objetivo de acelerar e racionalizar o processo de descoberta e projeto de drogas (HAMMAMI; FLISS, 2010; WANG et al., 2011). No entanto, nenhum dos métodos acima mencionados e bases de dados têm empregado métodos híbridos que combinam as redes neurais artificiais e sistemas de inferência fuzzy, tais como o Sistema de Inferência Neuro-Fuzzy Adaptativo (ANFIS) (JANG, 1993).
2.9 MODELAGEM MOLECULAR
especialmente no Brasil, a bioinformática, juntamente com a química computacional, vem se tornando, a cada dia, indispensável no planejamento racional de novos fármacos, já com inúmeros casos de sucesso alcançados (MARSHALL, 2005; SILVA; SILVA, 2007).
Ainda não existem algoritmos de predição de função antimicrobiana que utilizem como vetor de características de entrada, informações advindas de modelagem molecular, de forma qualitativa. A modelagem molecular resulta em modelos de predição de estruturas tridimensionais de proteínas, construídos a partir das respectivas cadeias de aminoácidos, fundamentais na determinação da função e atividade da molécula e, portanto podem contribuir com informações relevantes aos classificadores e sistemas de reconhecimento de padrões (FJELL et al., 2011; MELO et al., 2012).
2.9.1 Modelagem molecular por homologia
No processo de modelagem molecular por homologia estrutural são utilizadas estruturas de proteínas determinadas experimentalmente para se predizer a conformação de outra proteína que apresenta uma sequência de aminoácidos similar. A qualidade de modelos estruturais de proteínas gerados por modelagem comparativa e a sua aplicabilidade no processo de desenvolvimento de novos fármacos dependem, predominantemente, do grau de similaridade seqüencial entre a proteína com estrutura resolvida (proteína-molde) e a proteína a qual se deseja modelar (proteína-alvo). Pesquisas recentes mostram que essa abordagem pode ser utilizada na identificação e validação de alvos terapêuticos, bem como na identificação e otimização de protótipos (SILVA; SILVA, 2007). A modelagem molecular por homologia obtém bons resultados em predições de estruturas, mas está limitada ao uso de informações estruturais de proteínas cuja estrutura já seja conhecida (proteína-molde).
2.9.2 Modelagem molecular Ab-initio
2.10 REDES NEURAIS ARTIFICIAIS (RNA)
2.10.1 Conceitos
A construção de RNA tem inspiração nos neurônios biológicos e nos sistemas nervosos. Entretanto, é importante compreender que, atualmente, as RNA estão muito distantes das RNN (Redes Neurais Naturais) e, as semelhanças são mínimas (AZEVEDO et al., 2000).
Uma RNA pode ser composta de um determinado número de nós, ou unidades, conectados por links. Cada link tem um peso numérico associado a ele. Os pesos são formas primárias de armazenamento de longo prazo em RNA, e o aprendizado acontece através da atualização desses pesos. Algumas unidades são conectadas ao ambiente externo e podem ser designadas como unidades de entrada ou de saída. Os pesos são modificados de acordo com as tentativas de tornar o comportamento de entrada ou de saída da rede mais de acordo com as entradas fornecidas pelo ambiente (RUSSELL; NORVIG, 2009).
Cada unidade pode ser composta por um conjunto de links de entrada vindos de outras unidades, um conjunto de links de saída para outras unidades, um nível de ativação momentâneo e formas de realizar um tratamento computacional no nível de ativação para o próximo passo em um tempo determinado, dadas determinadas entradas e pesos. Cada unidade executa uma computação local baseada nas informações de entradas vindas dos vizinhos, porém sem a necessidade de qualquer controle global sobre o conjunto de unidades como um todo. Na prática, a maioria das implementações de RNA deve ser feita através de software e é utilizado um controle síncrono para atualizar todas as unidades seguindo uma seqüência pré-fixada (RUSSELL; NORVIG, 2009).
A Figura 3 mostra uma unidade típica, que recebe sinais de entrada e computa um novo nível de ativação, que é enviado para o link de saída yk. A
computação do nível de ativação pode ser baseada nos valores de cada sinal de entrada recebido de um nó vizinho e dos pesos de cada link de entrada. A computação é dividida em dois componentes, isto é, o primeiro é um componente linear chamado de junção aditiva, , que computa a soma ponderada dos valores de entrada das unidades. O segundo é um componente não linear chamado de função de ativação, , que transforma a soma ponderada no valor final, e que serve como o valor de ativação da unidade, . A Equação 1 mostra que a entrada ponderada total é a soma das ativações de entrada vezes os seus respectivos pesos (RUSSELL; NORVIG, 2009). Modelos diferentes são obtidos pelo uso de diferentes funções matemáticas para . As três escolhas mais comuns são as funções sigmoidal, sinal e passo (AZEVEDO et al., 2000; HAYKIN, 2001).
∑ (1)
Para caracterizar uma RNA, é importante especificar os seguintes pontos (AZEVEDO et al., 2000):
Os componentes da rede: os neurônios;
A resposta de cada neurônio;
O estado global de ativação da rede;
A conectividade da rede dada pelos valores de conexões sinápticas;
Como se propaga a atividade da rede;
Como se estabelece a conectividade da rede;
O ambiente externo a rede;
Como o conhecimento é representado na rede.
2.10.2 Redes MLP (multilayer perceptron)
Uma rede MLP com uma camada intermediária de neurônios é suficiente para aproximar qualquer função contínua e uma rede MLP com duas camadas intermediárias é suficiente para aproximar quaisquer funções matemáticas, contínuas ou não (AZEVEDO et al., 2000).
Em uma rede MLP, o número de camadas intermediárias é determinado pela natureza do problema a ser aproximado. Em geral este número é definido de maneira empírica, dependendo da distribuição dos dados a serem utilizados e de validação subseqüente. Entretanto, Eberhart e Dobbins (1990 apud AZEVEDO et al., 2000) apresentam uma heurística onde Qni (número de neurônios da camada
intermediária) é igual à raiz quadrada de Qne (quantidade de neurônios da camada
de entrada), somada com Qns (número de neurônios da camada de saída),
conforme Equação 2.
√ (2)
ou retropropagação. O algoritmo ajusta repetidamente os pesos das conexões entre os neurônios de maneira a minimizar as diferenças entre as saídas reais e as observadas. O método é baseado em um gradiente descendente, onde a função de ativação precisa ser contínua, derivável e de preferência não decrescente, para que o gradiente possa ser calculado e o ajuste dos pesos seja direcionado. O algoritmo de retropropagação é composto de duas fases: a forward, que define a saída em função de um padrão de entrada e a backward, que a partir da saída desejada e dos valores obtidos pela rede, busca atualizar os pesos das conexões sinápticas.
Os algoritmos para treinamento de uma rede MLP não são eficientes e não garantem a convergência para um mínimo global. A utilização de uma taxa de aprendizado e de um momento , otimizam o processo de aprendizado nas épocas de treinamento e facilitam a convergência para um mínimo global (RUSSELL; NORVIG, 2009).
2.10.3 Validação cruzada (método de resistência)
A essência do aprendizado em backpropagation é codificar um mapeamento de entrada para a saída, representado por um conjunto de exemplos, em pesos sinápticos e limiares de ativação de uma rede MLP. O objetivo é que a rede esteja bem treinada e que seu processo de aprendizagem possa ser suficiente para uma futura generalização (HAYKIN, 2001). Após um ciclo de treinamento, uma rede MLP pode piorar sua taxa de acertos para entradas diferentes daquelas utilizadas para a aprendizagem, esse fenômeno é chamado de overfitting ou ajuste demasiado aos dados. Para reduzir a sua ocorrência, uma das alternativas é a utilização do método de validação cruzada, onde o conjunto de dados é dividido aleatoriamente em um conjunto de treinamento e em um conjunto de testes. O conjunto de treinamento é então dividido em dois outros subconjuntos disjuntos: (i) estimação, para seleção do modelo e (ii) validação, para testar ou validar o modelo. Segundo Kearns (1996 apud HAYKIN, 2001) 80% do conjunto de treinamento devem ser atribuído ao conjunto de estimação e 20% deve ser atribuído ao conjunto de validação (HAYKIN, 2001).
desempenho possa tornar a MLP direcionada para o conjunto de validação, o desempenho de generalização é realizada no conjunto de testes, que é diferente do conjunto de validação (HAYKIN, 2001).
2.10.4 Aprendizagem
Aprendizagem consiste em um processo pelo qual os parâmetros de uma RNA são ajustados, de forma continuada, pelo estímulo do ambiente no qual a rede está operando. O tipo de aprendizagem é definido pela maneira como ocorrem os ajustes realizados nos parâmetros, ou seja, como são alteradas as intensidades das conexões entre os neurônios (RUSSELL; NORVIG, 2009).
A aprendizagem supervisionada busca extrair de um professor ou supervisor, o conhecimento de que o mesmo dispõe sobre o ambiente, permitindo um mapeamento entrada-saída. Durante a sessão de treinamento de uma RNA, pares de entradas e saídas são apresentadas a ela. A rede toma cada entrada e produz uma resposta na saída. Esta resposta é comparada com o sinal de saída desejado. Se a resposta real difere da resposta desejada, a RNA gera um sinal de erro, o qual é, então, usado para calcular o ajuste que deve ser feito para os pesos sinápticos da rede. Assim a saída real se aproxima da saída desejada e o erro é reduzido. O processo de minimização de erro requer um circuito especial conhecido como professor ou supervisor (AZEVEDO et al., 2000). O desempenho do sistema pode ser medido através do erro médio quadrático ou da soma de erros quadrados sobre a amostra de treinamento, definida como uma função dos parâmetros livres do sistema. Esta função pode ser visualizada como uma superfície multidimensional de desempenho de erro, obtida pela média de todos os exemplos possíveis de entrada-saída (HAYKIN, 2001).
2.11 SISTEMAS FUZZY
A dificuldade de se obter todas as informações e de equacionar a realidade imprecisa do mundo levou alguns cientistas a propor lógicas alternativas que seriam mais propícias à representação daquele mundo particular. A lógica fuzzy envolve a captura, representação e trabalho com noções lingüísticas, que se relacionam a objetos que possuem fronteiras indefinidas ou imprecisas (ZADEH, 1965). Um conjunto fuzzy é definido como uma coleção de objetos com valores de pertinência entre 0 (exclusão completa) e 1 (pertinência completa). Os valores de pertinência são expressos em graus de compatibilidade entre os objetos e as propriedades ou características da coleção, (PEDRYCZ; GOMIDE, 1998). O valor de pertinência A(x) quantifica a compatibilidade de x com o conceito da função A, como também expressa a incerteza da informação, onde A(x) representa a probabilidade de x ocorrer dado à ocorrência de A. Basicamente, qualquer função da forma A: X → [0,1] descreve uma função de pertinência (membership function – MF) associada com o conjunto fuzzy A. As funções de pertinência podem ser determinadas pelo contexto da aplicação, sendo mais ou menos adaptadas às mesmas.
A função de pertinência em forma de sino é definida conforme Equação 3 e um exemplo de seu gráfico para k=2 e m=1 é mostrado na Figura 4a.
(3)
A função de pertinência gama é definida conforme Equação 4 e seu gráfico ilustrado na Figura 4b.
{
(4)
A função de pertinência gaussiana é definida conforme Equação 5 e um exemplo de seu gráfico para e , é mostrado na Figura 4c.
A função de pertinência trapezoidal é definida conforme Equação 6 e seu gráfico ilustrado na Figura 4d.
[ ] (6)
Outras funções de pertinência são utilizadas em diversos domínios de aplicação tais como a função triangular, e a função polinomial, entre outras.
Figura 4– (a) Função de pertinência em forma de sino com k=2 e m=1. (b) Função de pertinência gama com k=2 e a=1. (c) Função de pertinência gaussiana com m=8 e k=0,1. (d) Função de pertinência trapezoidal com a=-2,5, m=0, n=2,5 e b =5.
Como característica de um conjunto fuzzy, a intensificação de contraste diminui os valores de pertinência menores do que ½, enquanto que os graus de pertinência maiores do que este limiar são aumentados, reduzindo a difusão do conjunto. A operação de intensificação de contraste é definida pela Equação 7.
2.12 O SISTEMA DE INFERÊNCIA NEURO-FUZZY ADAPTATIVO (ANFIS)
Um sistema de inferência neuro-fuzzy adaptativo (Adaptive Neuro-Fuzzy Inference System – ANFIS) é a combinação em uma arquitetura homogênea de uma rede neural artificial e um sistema fuzzy de forma que os algoritmos de aprendizagem da rede neural artificial são utilizados para determinar os parâmetros do sistema fuzzy (NAUCK et al., 1997). Um dos primeiros sistemas híbridos neuro-fuzzy para aproximação de funções foi o modelo ANFIS. Ele representa um sistema fuzzy do tipo Sugeno de ordem zero em uma arquitetura de RNA feedforward especial de cinco camadas (JANG, 1993). O modelo ANFIS implementa regras conforme Equação 8.
Rr: Se x1 é Aj1(1) ^ ... ^ xn é Ajn(n)Então y = α0(r)+ α1(r)x1+ ... + αn(r)xn (8)
A base de regras deve ser conhecida anteriormente. O modelo ANFIS ajusta apenas as funções de pertinência dos parâmetros antecedentes e conseqüentes. O seu treinamento pode ser feito em backpropagation ou híbrido, que é uma mistura do método backpropagation com o método de mínimos quadráticos.
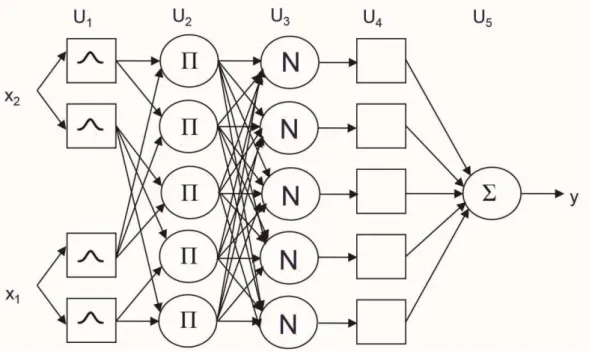
A estrutura da rede ANFIS, mostrada na Figura 5, contém η unidades de entrada na camada U0 (não computada, segundo Jang (1993)). As outras camadas
(denotadas como U1, ..., U5) têm as seguintes funcionalidades (NAUCK et al., 1997):
Camada 1: Cada unidade em U1 armazena três parâmetros para definir uma
função de pertinência em forma de sino que representa um termo lingüístico. Cada unidade é conectada a exatamente uma unidade de entrada e calcula o grau de pertinência do valor de entrada obtido.
Camada 2: Cada regra é representada por uma unidade em U2. Cada unidade
é conectada àquelas unidades na camada anterior que pertencem aos antecedentes da regra.
Camada 3: Nesta camada U3, para cada regra Rr existe uma unidade que
calcula seu grau relativo de preenchimento.
Camada 5: A unidade de saída calcula a saída final através da somatória de todas as saídas de U4.
Figura 5 – Estrutura de uma rede ANFIS (JANG, 1993)
2.13 RECONHECIMENTO DE PADRÕES
Reconhecimento de Padrões (RP) consiste em uma disciplina científica cujo objetivo relaciona-se a classificação de objetos em um número de categorias ou classes. Abrange uma grande gama de problemas de processamento de informação com grande significado prático, desde o reconhecimento de voz até o reconhecimento de escrita, de detecção de falhas em máquinas até o diagnóstico médico. Alguns desses problemas podem ser resolvidos por humanos de forma eficiente e rápida, porém são atividades muito complexas para o uso de computadores (THEODORIDIS; KOUTROUMBAS, 2009).
variáveis de entrada xi, ... , xd, para uma variável de saída y representando o nome
ou rótulo da classe mapeada. Em problemas mais complexos podem existir várias variáveis de saída, yk onde k = 1 . . .n. De modo geral não é possível determinar uma
forma apropriada para um mapeamento exceto com a ajuda de um conjunto de dados de exemplo. O mapeamento é, portanto modelado em termos de alguma função matemática que contenha um número de parâmetros ajustáveis cujos valores são determinados com ajuda dos dados (BISHOP, 2007). Essas funções podem ser escritas conforme Equação 9.
Yk = yk (x;w) (9)
Onde w denota o vetor de parâmetros. Um modelo de Rede Neural Artificial (RNA) pode ser considerado como uma escolha particular de um conjunto de funções yk (x;w). Neste caso, os parâmetros relativos à w são chamados de pesos.
2.13.1 Pré-processamento, segmentação e extração de características
De acordo com Duda (2001), a segmentação pode ser um dos problemas mais profundos do RP, pois na prática os objetos estão sobrepostos e o sistema deve determinar onde termina o objeto em questão e onde começa o próximo objeto. A fronteira conceitual entre a extração de características e a classificação é arbitrária, e o extrator de características deve caracterizar um objeto para que o mesmo seja reconhecido por medidas de objetos similares na mesma categoria.
Ainda segundo Bishop (2007), ao invés de representar uma transformação total de um conjunto de entrada xi, . . .xd para um conjunto de variáveis de saída y1, .
. .yc através de uma única função de uma RNA existe um grande benefício, de
quebrar esse mapeamento em um estágio inicial de pré-processamento seguido por uma parametrização do modelo de rede neural. Para se mapear um espaço d-dimensional x1. . .xd para uma variável de saída y deve-se dividir o espaço de
entrada em um determinado número de células M e especificar o valor de y para cada célula, resultando em um crescimento exponencial do número total de células Md, reduzindo a desempenho do sistema, fenômeno conhecido como impacto da
é uma forma ineficiente de se representar uma função não linear multivariada. As RNA feedforward são muito menos susceptíveis ao impacto da dimensionalidade. Uma das principais tarefas do estágio de pré-processamento é de reduzir o impacto da dimensionalidade dos dados antes de utilizá-los para treinar uma RNA ou outro sistema de reconhecimento de padrões. A Figura 6 mostra o estágio de pré-processamento dos dados.
Figura 6– Estágio de pré-processamento dos dados (Bishop, 2007)
2.13.2 Curva polinomial
Muitos dos aspectos importantes com relação à aplicação de RNA podem ser resumidos pelo contexto de ajuste da curva polinomial, onde se deve encaixar um polinômio a um conjunto de pontos de dados N pela técnica de minimização de função de erro. Considerando o polinômio de ordem M da Equação 10.
∑
(10)
O polinômio pode ser considerado como um mapeamento não linear que toma x como entrada e produz y como saída. A forma precisa da função y(x) é determinada pelos valores dos parâmetros w0 , . . ., wM, análogos aos pesos de uma
RNA. É conveniente denotar o conjunto de parâmetros (w0, . . . wM) pelo vetor w. O