Coats plus is a highly pleiotropic disorder particularly affecting the eye, brain, bone and gastrointestinal tract. Here, we show that Coats plus results from mutations in CTC1, encoding conserved telomere maintenance component 1, a member of the mammalian homolog of the yeast heterotrimeric CST telomeric capping complex. Consistent with the observation of shortened telomeres in an Arabidopsis CTC1 mutant and the phenotypic overlap of Coats plus with the telomeric maintenance disorders comprising dyskeratosis congenita, we observed shortened telomeres in three individuals with Coats plus and an increase in spontaneous gH2AX-positive cells in cell lines derived from two affected individuals. CTC1 is also a subunit of the a-accessory factor (AAF) complex, stimulating the activity of DNA polymerase-a primase, the only enzyme known to initiate DNA replication in eukaryotic cells. Thus, CTC1 may have a function in DNA metabolism that is necessary for but not specific to telomeric integrity.
Telomeres comprise long TTAGGG nucleotide repeats and associated proteins located at the ends of chromosomes. Telomeres both protect the chromosome terminus from unwanted nuclease and DNA repair activities and provide a mechanism to compensate for the inability of DNA polymerase to replicate the 5′ end of a linear chromosome. Telomeric DNA is packaged by a core group of proteins that bind the DNA duplex and the 3′ G overhang on the chromosome terminus. The associated telomere proteins form a protective cap that pre vents the chromosome end from eliciting a DNA damage response.
Although it is essential to sequester the DNA end from nuclease and DNA repair activities for much of the cell cycle, during S phase the telomere must be made accessible to telomerase and the DNA replication machinery1.
The bone marrow failure syndrome dyskeratosis congenita repre sents a clinically and genetically heterogeneous collection of pheno types arising from telomeric protein dysfunction. Dyskeratosis congenita–associated mutations have been identified in the genes encoding DKC1, TERC, TERT, NOP10, NHP2 and TCAB1, which belong to the telomerase holoenzyme responsible for maintaining telomere length, and a member of the shelterin protein complex TINF2, which is responsible for maintaining the structural integrity of the telomere. Thus, dyskeratosis congenita is a disorder of telomere maintenance and is associated with shortened telomeres2,3.
Coats plus is a rare disorder of which the most characteristic features are retinal telangiectasia and exudates (Coats disease), a distinctive pattern of intracranial calcification with an associated leukodystrophy and brain cysts, osteopenia with a tendency to frac tures and poor bone healing, and a high risk of lifelimiting gastro intestinal bleeding and portal hypertension caused by the development of vasculature ectasias in the stomach, small intestine and liver4–9
(Fig. 1). These extraneurological problems differentiate Coats plus from Labrune syndrome (leukoencephalopathy with calcifications and cysts), in which affected individuals otherwise show an identical neuroradiological appearance10. Of note, some individuals with Coats
plus develop sparse, graying hair, dystrophic nails and a normocytic anemia that might reflect a degree of bone marrow failure. These latter
Mutations in CTC1, encoding conserved telomere
maintenance component 1, cause Coats plus
Beverley H Anderson
1,53, Paul R Kasher
1,53, Josephine Mayer
1,53, Marcin Szynkiewicz
1, Emma M Jenkinson
1,
Sanjeev S Bhaskar
1, Jill E Urquhart
1, Sarah B Daly
1, Jonathan E Dickerson
1, James O’Sullivan
1,
Elisabeth Oppliger Leibundgut
2, Joanne Muter
3, Ghada M H Abdel-Salem
4, Riyana Babul-Hirji
5, Peter Baxter
6,
Andrea Berger
7,8, Luisa Bonafé
9, Janice E Brunstom-Hernandez
10,11, Johannes A Buckard
12, David Chitayat
5,13,
Wui K Chong
14, Duccio M Cordelli
15, Patrick Ferreira
16, Joel Fluss
17, Ewan H Forrest
18, Emilio Franzoni
15,
Caterina Garone
15,19, Simon R Hammans
20, Gunnar Houge
21, Imelda Hughes
22, Sebastien Jacquemont
23,
Pierre-Yves Jeannet
24, Rosalind J Jefferson
25, Ram Kumar
26, Georg Kutschke
27, Staffan Lundberg
28,
Charles M Lourenço
29, Ramesh Mehta
30, Sakkubai Naidu
31, Ken K Nischal
32, Luís Nunes
33, Katrin Õunap
34,
Michel Philippart
35, Prab Prabhakar
36, Sarah R Risen
37, Raphael Schiffmann
38, Calvin Soh
39,
John B P Stephenson
40, Helen Stewart
41, Jon Stone
42, John L Tolmie
43, Marjo S van der Knaap
44, Jose P Vieira
45,
Catheline N Vilain
46, Emma L Wakeling
47, Vanessa Wermenbol
48, Andrea Whitney
49, Simon C Lovell
50,
Stefan Meyer
3,51, John H Livingston
52, Gabriela M Baerlocher
2, Graeme C M Black
1, Gillian I Rice
1&
Yanick J Crow
1A full list of author affiliations appears at the end of the paper.
Received 4 November 2011; accepted 21 December 2011; published online 22 January 2012; doi:10.1038/ng.1084
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
features are also observed in dyskeratosis congenita. Furthermore, individuals with HoyeraalHreidarsson or Revesz syndrome, both of which are associated with telomeric shortening, can have intracranial calcification and, in the case of Revesz syndrome, an exudative retinopathy2.
In view of reports of both male and female affected siblings, we considered it most probable that Coats plus was an autosomal reces sive trait, although we noted that our collection of ten families fulfill ing strict diagnostic criteria (characteristic intracranial calcification with white matter changes, exudative retinopathy with telangiectasia and fractures with poor bone healing and/or gastrointestinal vas cular ectasia) included only one family in which the parents were known to be related. Because we were unable to define a disease associated locus using SNP arrays (data not shown), we undertook wholeexome sequencing of two siblings (F335_P1 and F335_P2) and two additional unrelated individuals (F332 and F336) showing classical features of Coats plus (Supplementary Table 1). We per formed wholeexome capture followed by massively parallel sequenc ing. Over 4.8 Gb of sequence was generated for each subject, such that >76% of the coding bases of the GENCODEdefined exome were represented by at least ten reads (Supplementary Table 2). We identified singlenucleotide substitutions and small insertion and/or deletion variants using our inhouse variant calling pipe line. We analyzed the exome variant profiles under a model of a rare autosomal recessive disorder. Taking into account difficulties in identifying insertion and deletion variants, we looked for genes harboring at least one previously unobserved nonsynonymous or splicesite substitution or a coding insertion or deletion in the same gene in all four individuals, with the same variant(s) required
in the siblings examined. CTC1 (encoding conserved telomere maintenance component 1) was highlighted as the only candi date gene when using this strategy. Sanger sequencing confirmed the variants in these affected individuals, and all parents tested showed appropriate heterozygosity for a single variant. In light of these data, we proceeded to sequence an additional nine affected individuals from seven families showing typical disease characteristics.
We identified biallelic CTC1 variants that were likely to be path ogenic in the affected proband(s) from nine of the ten families with Coats plus that were sequenced (Table 1). We also identified two CTC1 variants in a Norwegian individual (F319) having exu dative retinopathy with a history of recurrent fractures but in whom cranial imaging had not been undertaken (and who there fore did not fulfill our initial inclusion criteria). In only one family with a prior clinical assignment of Coats plus were we unable to identify CTC1 variants. In the ten families with presumed biallelic mutations, six harbored a nonsense lesion in combination with a missense variant, while the consequences of the splicesite vari ant seen in F273 and the inframe deletions observed in F367 and F382 are currently unknown. The probands from F342 carried two missense variants (c.775G>A and c.2518C>T). Of the 14 distinct table 1 Ancestry, pedigree structure and sequence alterations in individuals with Coats plus
Family Ancestry Tested Nucleotide alterations Exons Amino acid alterations
Parental consanguinity F273 Egyptian 1A, M, F Het. c.2831dupC | Het. c.3011+4A>C 17 | intron 18 p.His945Serfs*56 | splice donorb No
F319 Norwegian 1A Het. c.721C>T | Het. c.2923A>G 5 | 17 p.Gln241* | p.Arg975Gly No
F332a English 1A Het. c.724_727delAAAG | Het. c.2959C>T 5 | 18 p.Lys242Leufs*41 | p.Arg987Trp No
F335_P1aScottish 2A Het. c.724_727delAAAG | Het. c.2959C>T 5 | 18 p.Lys242Leufs*41 | p.Arg987Trp No
F335_P2aScottish 2A Het. c.724_727delAAAG | Het. c.2959C>T 5 | 18 p.Lys242Leufs*41 | p.Arg987Trp No
F336a English 1A Het. c.724_727delAAAG | Het. c.2611G>A 5 | 15 p.Lys242Leufs*41 | p.Val871Met No
F339 Canadian 2A – – – Yes (third cousins)
F340 European-American 1A Het. c.19C>T | Het. c.2959C>T 1| 18 p.Gln7* | p.Arg987Trp No
F342_P1 English and Italian 2A, M, F Het. c.775G>A | Het. c.2518C>T 5 | 15 p.Val259Met | p.Arg840Trp No F342_P2 English and Italian 2A, M, F Het. c.775G>A | Het. c.2518C>T 5 | 15 p.Val259Met | p.Arg840Trp No F345 Swiss and French 1A Het. c.724_727delAAAG | Het. c.1507G>C 5 | 9 p.Lys242Leufs*41 | p.Gly503Arg No F367 African and European 1A, 1U, M Het. c.859C>T | Het. c.2954_2956delGTT 6 | 18 p.Arg287* | p.Cys985del No F382 Portuguese 1A, 1U, M, F Het. c.2954_2956delGTT |
Het. c.3586_3606del
18 | 23 p.Cys985del | p.Leu1196_ Arg1202del
No
A, affected; U, unaffected; M, mother; F, father; Het., heterozygous.
aUsed in the primary exome sequencing screen. bPredicted by SplicePort to significantly reduce the strength of the canonical donor site.
a
b
d
e
c
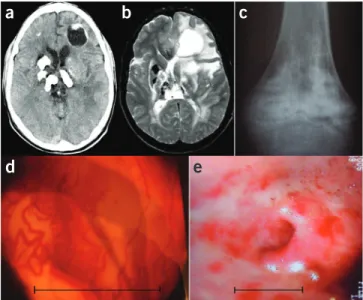
Figure 1 Features of Coats plus. (a) Cranial axial computed tomography
(CT) scan showing characteristic coarse and asymmetrically distributed calcification. Note the cyst in the left frontal cortex. (b) Cranial axial
T2 magnetic resonance imaging (MRI) scan taken at similar level to a,
highlighting the asymmetric, high signal of the deep and subcortical white matter. (c) X ray of knee illustrating mixed lytic and sclerotic lesions
that are mainly metaphyseal. (d) Retinal photograph showing retinal
microangiopathy. Scale bar, ~2.5 mm. (e) Endoscopy image showing
areas of erythema, which represent vascular ectasia involving the gastric antrum. Scale bar, ~2.5 cm. The neuroradiological features of Labrune syndrome (leukoencephalopathy with calcifications and cysts) are identical to those in a and b.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
mutations observed, three were seen in more than one family (c.724_727del, four families; c.2954_2956del, two families; c.2959C>T, three families) (Table 1).
Reflecting apparently rapid evolutionary divergence, some telomeric proteins show poor interspecies conservation11. The human
CTC1 protein sequence shares only 69% iden tity with mouse, 30% with zebrafish and 14% with Arabidopsis. Not unexpectedly, although the residues affected by nonsynonymous missense changes are well conserved across mammals, ClustalW alignment showed a relatively low level of residue conservation in other species (Supplementary Fig. 1). Related to this, 15 of the 20 CTC1 mutations we observed occurred in one of four exons
(Fig. 2). However, although we suspect that this clustering is of func tional significance, it is difficult to model the domain structure of CTC1 because of the high degree of sequence divergence from other proteins12. Of note, none of the 14 distinct, putatively pathogenic
CTC1 variants have been annotated as polymorphisms in dbSNP. Moreover, 13 of these variants were not seen in 1,730 European American and AfricanAmerican subjects collated in the Exome Variant Server database, and the c.19C>T transition seen in F340 was recorded in only 1 of 1,497 EuropeanAmericans (the same ancestral background as the affected proband).
Cerebroretinal microangiopathy with calcifications and cysts (CRMCC) is a term coined to encompass the Coats plus and Labrune syndrome phenotypes, on the grounds that the neuro radiological characteristics of these two disorders are essentially identical7,8. Thus, we sequenced CTC1 in a collection of probands
from 21 families showing stereotypical intracranial calcification and white matter changes in the absence of extraneurological fea tures, but we were unable to identify any likely pathogenic variants (Supplementary Table 3).
Recently, it has been shown that CTC1 interacts with STN1 and TEN1 to form the mammalian homolog of the yeast hetero trimeric Cdc13Stn1Ten1 (CST) telomeric capping complex13,14.
Consequently, we also undertook Sanger sequencing of OBFC1 (STN1) and TEN1 in our collection of individuals with Labrune syndrome and in the proband with Coats plus in whom we identified no CTC1 mutations. Again, sequencing identified no variants of likely pathogenic significance (Supplementary Tables 3 and 4).
In view of the finding of shortened telomeres in an Arabidopsis CTC1 mutant13 and the phenotypic overlap between Coats plus and
the telomeric maintenance disorders comprising dyskeratosis con genita, we measured telomere lengths15 in F332, F367 and F382 at the
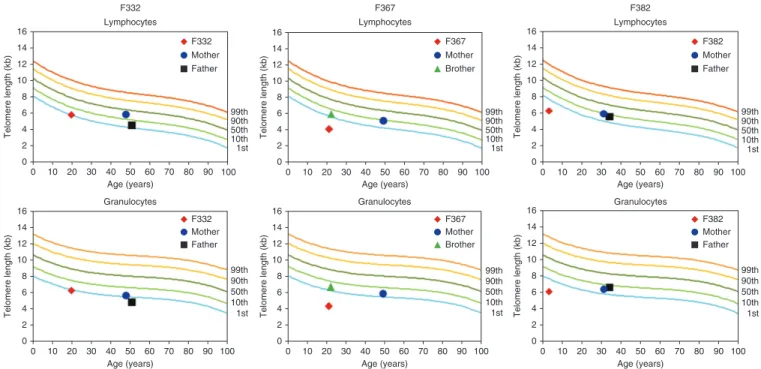
ages of 20, 20 and 3 years, respectively. Samples were also available from the mother and heterozygous brother of F367 and from both parents of F332 and F382. Testing showed markedly shortened telo meres in lymphocytes and granulocytes from the peripheral blood
of F367 and F382 and telomeric lengths at the lower range of normal in the heterozygous relatives of these individuals (Fig. 3). In F332, telomere length was recorded on the first percentile for age in both lymphocytes and granulocytes, with parental telomere lengths also at the lower range of normal.
Considering the enrichment for CTC1 variants in individuals with a predefined clinical and radiological diagnosis, we are confi dent that mutations in CTC1 represent the major cause of Coats plus. The observation of a single family, F339, in whom we were unable to identify mutations suggests possible genetic heterogeneity or a pheno copy. Of note, the parents of this child are related as third cousins, but there was no evidence of homozygosity around any of the three CST protein–encoding genes (data not shown).
Although Coats plus is rare and inherited as an autosomal recessive trait, none of the ten mutationpositive families we identified are con sanguineous. In keeping with this, all of the affected individuals in these families are compound heterozygotes for two different CTC1 variants (with six of ten families harboring a nonsense and missense mutation in combination). This observation helps to explain our inability to define a disease locus using autozygosity mapping and leads us to speculate that biallelic null mutations might be incompatible with development, whereas homozygosity for (most) missense variants may be associated with a normal phenotype or a different pathogenic one.
We and others have noted the highly stereotyped neuroradiological features common to Coats plus and Labrune syndrome, leading to the introduction of the umbrella term CRMCC. However, a recent analysis of our cohort (data not shown) suggested that the two groups might be dis tinguished according to the presence or absence of extraneurological fea tures, as, in our experience, individuals without retinal abnormalities have never shown any skeletal or gastrointestinal manifestations over extended time periods. The genetic data presented here support this conclusion, indicating that Coats plus and Labrune syndrome are not allelic.
CTC1 comprises 23 exons and encodes a 134.5kDa protein. Because of rapid evolutionary divergence, the human CST complex was only defined recently13,14. This led to the recognition that mammalian
Chromosome 17 p13.3 Tel.
a
b
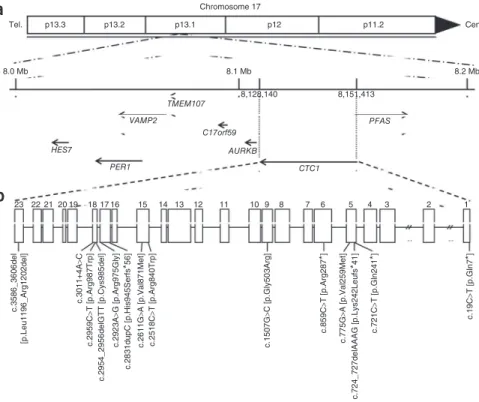
8.0 Mb 8.1 Mb TMEM107 C17orf59 AURKB PFAS CTC1 8,128,140 8,151,413 8.2 Mb p13.2 p13.1 p12 p11.2 Cen. VAMP2 PER1 23 22 c.3586_3606del [p.Leu1196_Arg1202del] c.3011+4A >C c.2959C >T [p.Arg987Trp] c.2954_2956delGTT [p.Cys985del] c.2923A >G [p.Arg975Gly] c.2831dupC [p.His945Serf s * 56 ] c.2611G >A [p.Val871Met ] c.2518C >T [p.Arg840Trp] c.1507G >C [p.Gly503Arg ] c.859 C >T [p.Arg28 7 * ] c.19C >T [p.Gln7 * ] c.775 G >A [p.Val259Met] c.721 C >T [p.Gln24 1 * ] c.724_727delAAAG [p.Lys242Leufs * 41] 21 20 19 18 17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 HES7Figure 2 Schematic representation of the
human CTC1 gene. (a) CTC1 spans 23,273 bp of genomic sequence on chromosome 17p13.1 (8,128,140–8,151,413). Neighboring genes are also shown. Tel., telomere; cen., centromere. (b) Position of identified mutations within
the CTC1 gene. Protein alterations are given in brackets.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
CTC1 is identical to one subunit of αaccessory factor (AAF132), whereas a second subunit of AAF (AAF44, also known as OBFC1) corresponds to mammalian STN1 (ref. 16).
The AAF complex stimulates the activity of DNA polymeraseα primase, the only enzyme known to initiate DNA replication in eukaryotic cells17. AAF functions by binding singlestranded DNA
(ssDNA) and enhancing DNA polymeraseα primase association with a DNA template. The finding that human CTC1STN1 modulates DNA polymeraseα primase activity indicates that, as for budding yeast CST, mammalian CTC1STN1 provides a link to the lagging strand replication machinery. Thus, it can be proposed that a con served function of CST is to promote efficient priming of telomeric Cstrand synthesis. In addition, given the role of Cdc13 in budding yeast, another conserved function of the CST complex could be to regulate telomerase. All of these processes may contribute to main taining telomeric DNA integrity and could couple the conventional replication process to telomerespecific priming and telomerase dependent elongation steps18 (see Supplementary Fig. 2).
Experiments in budding yeast and Arabidopsis have identified a functional role for the CST complex in maintaining telomeric structural integrity, and our finding of shortened telomeres in individuals with CTC1 mutations confirms an important role for the CST complex in mammalian telomere metabolism. However, the CST complex only partially localizes to telomeres and binds to ssDNA in a sequenceindependent manner (through the oligonucleotide/oligosaccharidebinding (OB) fold domains predicted to be present in all three subunits)13,14. Of note, knockdown
of CTC1 in human cells resulted in an increase in the number of γH2AX foci not confined to telomeres13. Although we did not study chromo
somal localization, we observed a significant increase in spontaneous γH2AXpositive cells in cell lines from the two affected individuals (F332 and F382) that we were able to examine (P = 0.0001 and 0.0007, respec
tively), indicative of an ongoing DNA damage response (Supplementary
Fig. 3). Consequently, it is possible that the CST complex may have a more
general role in DNA replication and repair that is frequently required by but not specific to telomeres.
Our interest in Coats plus and Labrune syndrome derives from ongoing research into disorders associated with the presence of intra cranial calcification. As such, our cohort is biased toward individuals with obvious neurological involvement. It is possible then that the phenotypic spectrum associated with CTC1 mutations may be broader than that presented here. Of note, phenotypic overlap with dyskeratosis congenita was appreciated at the time of the first clinical description of Coats plus, leading us to speculate that cohorts of indi viduals with a diagnosis of dyskeratosis congenita may be enriched for pathogenic variants in CTC1, OBFC1 (STN1) and/or TEN1.
URLs. National Heart, Lung, and Blood Institute (NHLBI) Exome
Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington. edu/EVS/; SplicePort, http://spliceport.cs.umd.edu/; GENCODE, http://www.gencodegenes.org/; ClustalW, http://www.ebi.ac.uk/tools/ msa/clustalw2/; Primer3Plus, http://www.primer3plus.com/.
MeTHoDS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Accession codes. Nucleotide sequences are available from Entrez
PubMed for human CTC1 (NM_025099.5), human OBFC1 (STN1) (NM_024928.4) and human TEN1 (NM_001113324.2), as is the human CTC1 protein sequence (NP_079375.3).
Note: Supplementary information is available on the Nature Genetics website. F332 Lymphocytes Age (years) Telomere length (kb) 16 F332 Mother Father 99th 90th 50th 10th 1st 0 10 20 30 40 50 60 70 80 90 100 14 12 10 8 6 4 2 0 Granulocytes Age (years) Telomere length (kb) 16 F332 Mother Father 99th 90th 50th 10th 1st 0 10 20 30 40 50 60 70 80 90 100 14 12 10 8 6 4 2 0 F367 Lymphocytes Age (years) Telomere length (kb) 16 F367 Mother Brother 99th 90th 50th 10th 1st 0 10 20 30 40 50 60 70 80 90 100 14 12 10 8 6 4 2 0 Granulocytes 99th 90th 50th 10th 1st Age (years) Telomere length (kb) 16 F367 Mother Brother 0 10 20 30 40 50 60 70 80 90 100 14 12 10 8 6 4 2 0 Granulocytes 99th 90th 50th 10th 1st Age (years) Telomere length (kb) 16 F382 Mother Father 0 10 20 30 40 50 60 70 80 90 100 14 12 10 8 6 4 2 0 F382 Lymphocytes Age (years) Telomere length (kb) 16 F382 Mother Father 99th 90th 50th 10th 1st 0 10 20 30 40 50 60 70 80 90 100 14 12 10 8 6 4 2 0
Figure 3 Telomere length analysis in subsets of leukocytes as measured by automated multicolor flow-FISH. Telomere lengths were assessed in
lymphocytes and granulocytes from F332, F367 and F382 at the ages of 20, 20 and 3 years, respectively, and from unaffected family members harboring a single CTC1 mutation. Telomere lengths were markedly shortened in two affected individuals (F367 and F382) and were at the lower range of normal in their heterozygous relatives. In F332, telomere length was recorded on the first percentile for age in both lymphocytes and granulocytes, with parental telomere lengths at the lower range of normal (being less than the first percentile in the father in granulocytes). The reference range for telomere length over age in percentiles was derived from telomere length analyses in lymphocytes and granulocytes from 400 healthy individuals.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
ACKNOWLEDGMENTS
We are very grateful to the affected families for their involvement in our research. Y.J.C. acknowledges the Manchester National Institute for Health Research (NIHR) Biomedical Research Centre. This work has received support from the Great Ormond Street Hospital Children’s Charity and from the Newlife Foundation. We thank the NHLBI Grand Opportunity (GO) Exome Sequencing Project and acknowledge its ongoing studies that produced and provided exome variant calls for comparison, including the Lung GO Sequencing Project (HL102923), the Women’s Health Initiative (WHI) Sequencing Project (HL102924), the Broad GO Sequencing Project (HL102925), the Seattle GO Sequencing Project (HL102926) and the Heart GO Sequencing Project (HL103010). We also thank G. Forte for organizing courier delivery of patient materials.
AUTHOR CONTRIBUTIONS
B.H.A. and J.O. performed exome sequencing. B.H.A., M.S., G.I.R. and E.M.J. were responsible for Sanger sequencing. J. Mayer, S.S.B. and J.E.D. undertook analysis of the exome sequence data. J.E.U. and S.B.D. performed SNP analyses. J.H.L. was responsible for neuroradiological phenotyping. G.M.B. and E.O.L. performed the telomeric length analysis. S.M. and J. Muter assessed γH2AX positivity. S.C.L. analyzed CTC1 structural domains. G.I.R. and G.C.M.B. provided critical input into project development and manuscript preparation. Y.J.C. designed and supervised the project and wrote the manuscript with the support of P.R.K. All other authors identified subjects with Coats plus and/or Labrune syndrome and performed related clinical and laboratory studies. Phenotypic overlap with dyskeratosis congenita was originally appreciated by J.L.T. and J.B.P.S. at the time of the first clinical description of Coats plus.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests. Published online at http://www.nature.com/naturegenetics/.
Reprints and permissions information is available online at http://www.nature.com/ reprints/index.html.
1. Jain, D. & Cooper, J.P. Telomeric strategies: means to an end. Annu. Rev. Genet.
44, 243–269 (2010).
2. Savage, S.A. & Bertuch, A.A. The genetics and clinical manifestations of telomere biology disorders. Genet. Med. 12, 753–764 (2010).
3. Nelson, N.D. & Bertuch, A.A. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat. Res. published online, doi:10.1016/j.mrfmmm.2011.06.008 (2 July 2011).
4. Tolmie, J.L., Browne, B.H., McGettrick, P.M. & Stephenson, J.B. A familial syndrome with coats’ reaction retinal angiomas, hair and nail defects and intracranial calcification. Eye (Lond.) 2, 297–303 (1988).
5. Crow, Y.J. et al. Coats’ plus: a progressive familial syndrome of bilateral Coats’ disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument. Neuropediatrics
35, 10–19 (2004).
6. Nagae-Poetscher, L.M. et al. Leukoencephalopathy, cerebral calcifications, and cysts: new observations. Neurology 62, 1206–1209 (2004).
7. Linnankivi, T. et al. Cerebroretinal microangiopathy with calcifications and cysts.
Neurology 67, 1437–1443 (2006).
8. Briggs, T.A. et al. Cerebroretinal microangiopathy with calcifications and cysts (CRMCC). Am. J. Med. Genet. A. 146A, 182–190 (2008).
9. Briggs, T.A. et al. Treatment of gastrointestinal bleeding in a probable case of cerebroretinal microangiopathy with calcifications and cysts. Mol. Syndromol. 1, 159–162 (2011).
10. Labrune, P. et al. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: a new progressive disorder due to diffuse cerebral microangiopathy.
Neurology 46, 1297–1301 (1996).
11. Linger, B.R. & Price, C.M. Conservation of telomere protein complexes: shuffling through evolution. Crit. Rev. Biochem. Mol. Biol. 44, 434–446 (2009).
12. Price, C.M. et al. Evolution of CST function in telomere maintenance. Cell Cycle
9, 3157–3165 (2010).
13. Surovtseva, Y.V. et al. Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol. Cell 36, 207–218 (2009).
14. Miyake, Y. et al. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol. Cell
36, 193–206 (2009).
15. Baerlocher, G.M., Vulto, I., de Jong, G. & Lansdorp, P.M. Flow cytometry and FISH to measure the average length of telomeres (flow FISH). Nat. Protoc. 1, 2365–2376 (2006).
16. Casteel, D.E. et al. A DNA polymerase-α·primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J. Biol.
Chem. 284, 5807–5818 (2009).
17. Goulian, M. & Heard, C.J. The mechanism of action of an accessory protein for DNA polymerase α/primase. J. Biol. Chem. 265, 13231–13239 (1990).
18. Giraud-Panis, M.J., Teixeira, M.T., Geli, V. & Gilson, E. CST meets shelterin to keep telomeres in check. Mol. Cell 39, 665–676 (2010).
1Manchester Academic Health Science Centre, University of Manchester, Genetic Medicine, Manchester, UK. 2Experimental Hematology, Department of Clinical
Research, University Hospital and University of Bern, Bern, Switzerland. 3Stem Cell and Leukaemia Proteomics Laboratory, University of Manchester, Manchester, UK. 4Clinical Genetics Department, Human Genetics and Genome Research Division, National Research Centre, Cairo, Egypt. 5Division of Clinical and Metabolic
Genetics, The Hospital for Sick Children, University of Toronto, Toronto, Canada. 6Ryegate Centre, Sheffield Children’s National Health Service (NHS) Foundation
Trust, Sheffield, UK. 7Department of Pediatric Neurology, University of Mainz, Mainz, Germany. 8Department of Pediatric Neurology, Children’s Hospital Harlaching,
Munich, Germany. 9Division of Molecular Pediatrics, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland. 10Department of Neurology, Washington
University School of Medicine, St. Louis Children’s Hospital, Saint Louis, Missouri, USA. 11Department of Pediatrics, Washington University School of Medicine,
St. Louis Children’s Hospital, Saint Louis, Missouri, USA. 12Ärztlicher Leiter, Sozialpädiatrisches Zentrum am Evangelisches Krankenhaus (EVK) Düsseldorf,
Düsseldorf, Germany. 13Department of Obstetrics and Gynecology, The Prenatal Diagnosis and Medical Genetics Program, Mount Sinai Hospital, University of
Toronto, Toronto, Canada. 14Department of Radiology, Great Ormond Street Hospital for Children NHS Trust, London, UK. 15Child Neuropsychiatry Unit, University
of Bologna, S. Orsola-Malpighi Hospital, Bologna, Italy. 16Division of Medical Genetics, Alberta Children’s Hospital, Calgary, Canada. 17Pediatric Neurology, Geneva
Children’s Hospital, Geneva, Switzerland. 18Glasgow Royal Infirmary, Glasgow, UK. 19Department of Neurology, Columbia University Medical Center, New York, New
York, USA. 20Wessex Neurological Centre, Southampton General Hospital, Southampton, UK. 21Center for Medical Genetics and Molecular Medicine, Haukeland
University Hospital, Bergen, Norway. 22Royal Manchester Children’s Hospital, Central Manchester University Hospitals NHS Foundation Trust, Manchester,
UK. 23Service de Génétique Médicale, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland. 24Pediatric Neurology Unit, Department of Pediatrics,
Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland. 25Dingley Specialist Children’s Centre, Royal Berkshire Hospital, Reading, UK. 26Department of
Neurology, Alder Hey Children’s NHS Foundation Trust, Liverpool, UK. 27Klinik für Allgemeine Pädiatrie und Neonatologie, Kliniken für Kinder- und Jugendmedizin,
Universitätsklinikum des Saarlandes, Homburg, Germany. 28Department of Women’s and Children’s Health, Uppsala University, Uppsala, Sweden. 29Neurogenetics
Unit, Department of Neuroscience, Faculdade de Medicina de Ribeirão Preto, University of Sao Paulo, Sao Paulo, Brazil. 30Bedford Hospital NHS Trust, Bedford,
UK. 31Neurogenetics Unit, Hugo Moser Research Institute, Kennedy Krieger Institute, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA. 32University of Pittsburgh Medical Center (UPMC) Children’s Hospital of Pittsburgh and Eye Center, Pittsburgh, Pennsylvania, USA. 33Department of Medical
Genetics, Hospital Dona Estefânia Centro Hospitalar de Lisboa Central (CHLC), Faculdade de Ciências Médicas de Lisboa Universidade Nova de Lisboa (UNL), Lisbon, Portugal. 34Department of Genetics, United Laboratories, Tartu University Hospital, Tartu, Estonia. 35Brain Research Institute, David Geffen School of
Medicine, University of California, Los Angeles, Los Angeles, California, USA. 36Department of Neurology, Great Ormond Street Hospital for Children, London,
UK. 37Department of Neurology and Developmental Medicine, Kennedy Krieger Institute, Johns Hopkins University School of Medicine, Baltimore, Maryland,
USA. 38Institute of Metabolic Disease, Baylor Research Institute, Dallas, Texas, USA. 39Neuroradiology Department, Salford Royal NHS Foundation Trust, Salford,
UK. 40Fraser of Allander Neurosciences Unit, Royal Hospital for Sick Children, Glasgow, UK. 41Clinical Genetics Department, Oxford Radcliffe Hospitals NHS
Trust, Oxford, UK. 42Department of Clinical Neurosciences, Western General Hospital, Edinburgh, UK. 43Department of Clinical Genetics, Royal Hospital for Sick
Children, Yorkhill, Glasgow, UK. 44Department of Child Neurology, VU University Medical Center, Amsterdam, The Netherlands. 45Department of Neurology, Hospital
Dona Estefânia, Centro Hospitalar de Lisboa Central, Lisbon, Portugal. 46Department of Medical Genetics, Université Libre de Bruxelles (ULB) Center of Human
Genetics, Université Libre de Bruxelles, Brussels, Belgium. 47North West Thames Regional Genetics Service, North West London Hospitals NHS Trust, Harrow,
UK. 48Department of Pediatric Neurology, Université Libre de Bruxelles, Hôpital Erasme, Brussels, Belgium. 49Southampton University Hospital Foundation Trust,
Southampton, UK. 50Faculty of Life Sciences, University of Manchester, Manchester, UK. 51Paediatric Oncology, Central Manchester University Hospitals NHS
Foundation Trust, Manchester, UK. 52Department of Paediatric Neurology, Leeds General Infirmary, Leeds, UK. 53These authors contributed equally to this work.
Correspondence should be addressed to Y.J.C. ([email protected]).
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
leagues in pediatric neurology and clinical genetics. Written informed consent was obtained for all participants. The study had full ethical approval from the Leeds Multicentre Research Ethics Committee (07/Q1206/7).
Exome sequencing. Genomic DNA was extracted from lymphocytes from
affected individuals, parents and siblings by standard techniques. For whole exome analysis, targeted enrichment and sequencing were performed on DNA extracted from peripheral blood from four individuals (F332, F335_P1, F335_P2 and F336). Enrichment was performed using the SureSelect Human All Exon Kit v.1 (Agilent) for the Applied Biosystems SOLiD system. Emulsion PCR (ePCR) was conducted on the resultant sample library, and products were then sequenced on a SOLiD 4 sequencer (Life Technologies). Sequence data were mapped using SOLiD Bioscope software (Life Technologies), with the hg18 human genome as a reference. SNPs were called using the diBayes tool in the BioScope software suite with the medium stringency setting and then filtered to remove those SNPs with less than 5× coverage. A total of 4.8 Gb of sequence mapped uniquely to the hg18 genome reference, with 76.4% of the targeted exome covered at tenfold or higher (Supplementary Table 2).
Sanger sequencing. Mutation analysis was performed by direct sequencing
of purified genomic PCR products using the BigDye Terminator v3.1 cycle sequencer system (Applied Biosystems). Primers were designed for indivi dual exons and intron boundaries of CTC1, OBFC1 (STN1) and TEN1 using
available on request).
Telomere length measurement. Telomere length was measured in white blood
cell subsets by automated multicolor flowFISH analysis15 of granulocytes,
CD45RA+ lymphocytes (naive T cells), CD45RA− lymphocytes (memory
T cells), CD20+ lymphocytes (B cells), CD57+ lymphocytes (NK/NKT cells)
and total leukocytes, and compared with agematched controls. ‘Markedly short’ was defined as less than the first percentile for age.
FACS for measurement of gH2AX positivity. Lymphoblastoid cells from
two affected individuals (F332 and F382) and three control individuals were seeded at a density of 5 × 105 cells per well of a sixwell plate (Sigma) and
maintained at 37 °C overnight. Cells were pelleted and washed in PBS and fixed in 500 µl of 1× Fixation Solution (Millipore γH2AX Phosphorylation Assay Kit) for 20 min at 4 °C. Cells were pelleted and washed in 1 ml of PBS, pelleted again and resuspended in 50 µl of 1× permeabilizing solution (Millipore γH2AX Phosphorylation Assay Kit), and 3.5 µl of appropri ate antibody was added (FITCconjugated antibody to γH2AX or control FITCconjugated rabbit IgG) and incubated at 4 °C for 20 min. We added 100 µl of 1× washing solution to each sample, and cells were pelleted and resuspended in 400 µl of PBS. Samples were analyzed using the BD Biosciences FACSCalibur using CellQuest Pro software, and data were analyzed using FlowJo.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.
All rights reserved.
npg
© 2012 Nature
America, Inc.