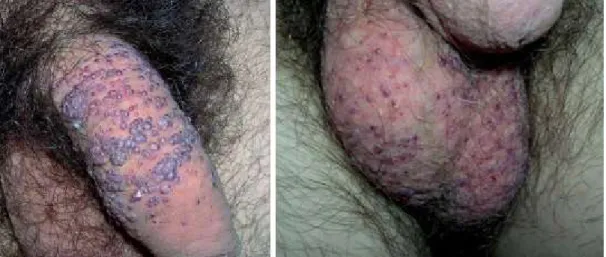
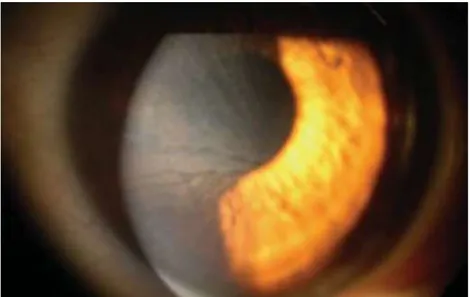
DOENÇA DE FABRY: A PROPÓSITO DE UM CASO
9
0
0
Texto
Imagem
Documentos relacionados