UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA VEGETAL
GENE EXPRESSION DURING PLASMODIUM
TRANSMISSION
Dissertação
Neuza Arrimar Duarte
Mestrado em Biologia Molecular e Genética
2013
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA VEGETAL
GENE EXPRESSION DURING PLASMODIUM
TRANSMISSION
Dissertação orientada por:
Doutor Jorge Miguel L. Marques da Silva, Faculdade de Ciências, Universidade de Lisboa Doutora Patrícia Agostinho Gonçalves Costa da Silva, Instituto Medicina Molecular
Mestrado em Biologia Molecular e Genética
Acknowledgments
To all the UPAMOL team, a sincere thank you for having me during this last year.
To Patrícia who kindly accepted me as her student and diligently helped me in everything I needed. To Jorge, whom I’m already picturing as a P.I….for giving me the opportunity of working with him these last months. Two both for all the time you took teaching me everything I needed… and for this long correction marathon.
To Ana, the “always on top of things” lab manager, who always has a solution for everything, and who would always narrate to you the online news the funniest way.
To Gunnar, who gave me the opportunity of working in his group, whose work is an inspiration for everyone in this field. Sorry for any inconvenience I may have caused you. Most importantly, thank you UPAMOL for showing me how a group is supposed to be like. UPAMOL fighting!
To Prof. Jorge Silva, my internal supervisor, for helping me with all the thesis issues. To everyone in UPAR, our neighbours, for always being hyper, enthusiastic and a mood raiser. In particular to Leonor, former UPAMOL member, and current UPAR lab manager, for being a one-of-kind person and for the promptly assistance you would give me, and everyone else, when needed.
To everyone in UMA, who were always nice and helpful to me. To Vanessa, for the liver samples. To Ana Parreira just because you’re the coolest insectary technician ever! To Joana Dias, wish you great luck in your future!
To our friendly invaders, Miguel Prudêncio’s lab, even though our cohabitation was short, you were always nice to me. Inês, your geekiness and personality always makes you a nice presence to have around.
To Patrícia Meireles, my “sister”, flat mate, and former companion in the trenches of the Biochemistry battlefield in Porto… well, there’s no need for words, you rock! さいこう! Also, to my other “sister” Sarinha, for all these years of friendship. Good luck in your PhD! Most importantly, to my family, mom, dad, sista, grandma and grandpa… for whom I always did my best. Even though things haven’t always turned out the way we wanted…we are gonna make it! Mamy, you are going to be a published writer, for sure!
Abbreviations used 3’UTR: 3’ untranslated region 5’UTR: 5’ untranslated region cDNA: complementary DNA
CITH: CAR-I and fly Trailer Hitch Homolog
DHHC-CRD: Asp-His-His-Cys Cystein rich domain DNA: Deoxyribonucleic acid
DOZI: Development of Zygote Inhibited DTT: Dithiothreitol
EEF: Exo-erythtrocytic forms
EPSF: Essential Protein for Sporozoite Formation Fig.: Figure
GFP: Green fluorescent protein
HIV/AIDS: Human immunodeficiency virus infection / acquired immunodeficiency syndrome
HRP: Horseradish peroxidase Hsp70: 70 kDa heat shock protein IFA: Immunofluorescence assay IP: Immunoprecipitation
IPET: Invasion Protein Essential for Transmission iRBC: infected RBC
kDa: kilodalton
LB medium: Lysogeny Broth medium MG Spz: Midgut sporozoites
mRNA: messenger RNA
mRNP: messenger ribonucleoprotein ook: ookinete
PAT: Protein S-acyl transferase
PbSR: Plasmodium berghei scavenger receptor-like protein PBS: Phosphate buffered saline
p.i.: post-infection
PCR: Polymerase chain reaction PTM: Post-translational modification PUF: Pumilio protein
RNA: Ribonucleic acid RBC: Red blood cell RT: Room temperature SDS: Sodium dodecyl sulfate
SDS-PAGE: Sodium dodecyl sulfate Polyacrylamide gel electrophoresis
SG Spz: Salivary glands sporozoites SOC: Super Optimal Broth
TAE: Tris-acetate-EDTA
TAP: Transcription associated protein TBV: Transmission blocking vaccine TE: Tris-EDTA
TF: Transcription factor
tgdhfr/ts: Toxoplasma gondii dihydrofolate reductase/ thymidylate
synthase
TMD: Transmembrane domain
uis4: upregulated in infectious sporozoites gene 4
Abstract
Malaria is an infectious disease caused by the Plasmodium parasite that is transmitted by the bite of a female Anopheles mosquito to its host. Nowadays, malaria still constitutes to be a major burden despite the enormous progression made in different strategies of prevention and treatment. Plasmodium exhibits a complex life cycle whose mechanisms of regulation are still a matter of intense research and debate. Translational repression is a post-transcriptional mechanism that allows regulation of transcripts translation both spatially and temporally. In the P. berghei female gametocytes, DOZI- (Development of Zygote Inhibited) and CITH- (CAR-I and fly TrailerHitch Homology) defined translational repressor complexes have a vital role in maintaining in a quiescent state certain messenger RNAs (mRNAs) that will be translated later on, during post-fertilization development. On the other hand, PUF2 (Pumilio 2) protein was found to have a similar vital role but at the sporozoite stage in maintaining their latency by translationally repressing certain transcripts. In addition,
P. falciparum PUF2 protein was shown to regulate sexual development and differentiation,
although in P. berghei it has not been possible to confirm such role for PUF2 protein. Our findings suggest that dhhc10 (Asp-His-His-Cys cysteine-rich domain – DHHC-CRD family member), ipet (Invasion Protein Essential for Transmission) and epsf (Essential Protein for Sporozoite Formation) transcripts are translationally repressed by the DOZI- and CITH-defined repressor complexes. In the ookinetes, they are translated and apparently stored in crystalloid bodies. Protein palmitoylation mediated by DHHC10 will have then a vital role in sporozoite formation. Lack of EPSF also leads to the absence of oocyst sporulation. Our results further confirm the dynamic and complex nature of post-transcriptional gene regulation in the Plasmodium life cycle, and its importance in the parasite transmission.
Keywords: CITH, development, DOZI, gametocytes, mRNPs, ookinete, oocyst,
Resumo
A malária é uma doença infecciosa causada por parasitas do género Plasmodium pertencente ao filo Apicomplexa, cujo modo de transmissão requer um vector, um mosquito fêmea do género Anopheles. A espécie responsável pela forma mais mortífera de malária denomina-se Plasmodium falciparum, que possui uma incidência mais prevalente nas regiões de África. De acordo com o World Malaria Report (WMR) de 2012, as populações com um maior risco de contrair malária estão localizadas nas regiões a sul do Sahara. As estimativas indicam que o número de pessoas em risco de contrair malária em 2011 seria de 3,3 biliões de pessoas no mundo inteiro. Nos últimos anos diferentes estratégias foram desenvolvidas para combater esta doença, ainda considerada como uma das três grandes doenças infecciosas, a par com HIV/AIDS (Human Immunodeficiency Virus/ Acquired Immunodeficiency Syndrome) e tuberculose. Exemplos dessas estratégias incluem métodos de prevenção, como é o caso das redes impregnadas com insecticidas de longa duração, e também as várias formas de tratamento com diferentes combinações de fármacos. Actualmente, o problema que se considera difícil de contornar na luta contra a malária é a capacidade que o parasita tem de adquirir resistência aos fármacos antimaláricos, como é o caso da artemisinina; mas para além disso, a aquisição de resistência estende-se também aos insecticidas.
O parasita da malária possui um ciclo de vida complexo que é caracterizado pela presença de um hospedeiro humano, ou no caso do modelo usado no nosso estudo, um morganho, e de um vector. A reprodução sexual ocorre na fase do ciclo de vida em que o parasita se encontra no interior do mosquito e é um evento que ocorre apenas uma vez por ciclo. Os mecanismos de regulação responsáveis pelo desenvolvimento e diferenciação sexual do parasita permanecem por deslindar.
Devido ao facto dos hospedeiros e do vector possuírem ambientes fisiológicos muito discrepantes, é imprescindível que haja uma rápida adaptação a nível celular e molecular por parte do parasita e um controlo exacto da expressão génica ao longo do seu ciclo de vida. No entanto, a forma como é regulada a expressão génica no parasita ainda tem de ser elucidada. Com a sequenciação do genoma de Plasmodium verificou-se que este possui uma menor quantidade de proteínas associadas à transcrição (TAPs) do que seria de se esperar para um genoma do seu tamanho. Este facto levou a que se colocasse a hipótese de que a regulação transcricional em Plasmodium difere da de outros sistemas eucarióticos, como por exemplo, a levedura.
A repressão da tradução é um mecanismo no qual um conjunto de mRNAs é movido selectivamente para complexos ribonucleoproteicos (mRNPs) onde são armazenados, sendo apenas traduzidos mais tarde no ciclo de vida. Este processo permite tanto uma
regulação temporal como uma regulação espacial da expressão das proteínas. Estudos demonstraram que uma região rica em uridinas (designada U-rich region) encontrada nas regiões 3’ ou 5’ não traduzidas (UTRs) dos mRNAs reprimidos, é um factor-chave neste mecanismo de regulação. Ao longo dos últimos anos, a repressão da tradução tem vindo a estabelecer-se como um mecanismo essencial no desenvolvimento sexual em Plasmodium. Em gametócitos (as células percursoras sexuais) de P. berghei, demonstrou-se que os mRNAs que dão origem às proteínas de superfície de oocinetos (o estadio seguinte após a fertilização) P25 e P28 colocalizam com a DEAD-box RNA helicase DOZI (Development of Zygote Inhibited) e com a Sm-like factor CITH (CAR-I/Trailer Hitch Homolog). DOZI e CITH definem um complexo ribonucleoproteico composto por 16 factores principais, que incluem eIF4E, um factor de iniciação da tradução e uma proteína de ligação à cauda poli-(A) (PABP). Os complexos repressores definidos por DOZI e CITH reprimem a tradução de certos mRNAs em gametócitos, que só irão ser utilizados mais tarde, após a fertilização.
A família PUF consiste em proteínas de ligação a RNA cujo papel principal é a regulação pós-transcricional dos RNAs aos quais se ligam. A investigação incidente nesta família de proteínas teve uma importância fulcral no desenvolvimento do conhecimento sobre os mecanismos pós-transcrição. Em P. falciparum, PfPUF2 desempenha um papel importante na regulação do desenvolvimento e diferenciação sexual. Em gametócitos, a proteína PUF2 reprime a tradução de determinados transcritos, reprimindo assim a diferenciação sexual. Por outro lado, em P. berghei, esporozoítos puf2-KO desenvolvem-se precocemente dentro das glândulas salivares do mosquito, exibindo uma morfologia característica de estadios posteriores, nomeadamente de estadios do fígado. PUF2 parece ser então de extrema importância para a latência dos esporozoítos de P. berghei. Para além disso, estes resultados demonstram que a repressão da tradução é um mecanismo essencial para o controlo do desenvolvimento dos esporozoítos e da sua transmissão ao hospedeiro.
O principal objectivo deste trabalho é obter uma compreensão mais aprofundada sobre o papel dos complexos de repressão da tradução em gametócitos. Em gametócitos pbdozi-KO, um número elevado de mRNAs estão sob e sobre-expressos em relação ao wild type (WT). Usando uma análise por RIP-Chip foi possível confirmar que muitos desses transcritos estão fisicamente ligados aos complexos repressores definidos por DOZI e CITH, sugerindo que a sua tradução está a ser reprimida. Por outro lado, em P. berghei, a expressão de PUF2 em gametócitos ainda não foi demonstrada. O estudo de mRNAs potencialmente reprimidos pelos complexos definidos por DOZI e CITH, e também o estudo da proteína PUF2, permitir-nos-á compreender mais aprofundadamente qual a importância destes complexos repressores na expressão génica em gametócitos.
A immunoprecipitação (IP) de proteínas é uma técnica que permite o isolamento de uma proteína em particular, através de anticorpos específicos, e de qualquer parceiro (proteína ou ácido nucleico) que esteja fisicamente ligado à proteína alvo. Utilizou-se esta técnica para confirmar a expressão de PUF2 em gametócitos de P. berghei e isolar potenciais parceiros; infelizmente, não fomos bem-sucedidos nesta tarefa.
Foram também alvo de estudo neste projecto um conjunto de proteínas, denominadas DHHC2 e DHHC10, enzimas da família das S-aciltransferase de Proteínas (PATs), IPET (Invasion Protein Essential for Transmission) e EPSF (Essential Protein for Sporozoite Formation). Foi proposto que a tradução destas proteínas seria reprimida pelos complexos repressores definidos por DOZI e CITH, em gametócitos, tal como acontece com P25 e P28. Para além disso, o facto de todas elas possuírem péptidos sinal e domínios transmembranares, é uma forte indicação de que possam ser proteínas de superfície. Esta possibilidade gera interesse adicional neste conjunto de proteínas já que, ao longo dos anos foram descobertas várias proteínas de superfície com função essencial para a transmissão do parasita, tendo muitas delas, tornado-se candidatos promissores a vacinas de bloqueio de transmissão (TBVs). Estas vacinas têm como objectivo principal a diminuição do número de mosquitos infectados, cessando o desenvolvimento do parasita no mosquito.
Os nossos resultados sugerem que os transcritos de dhhc10, ipet e epsf estão num estado quiescente nos gametócitos, sendo a sua tradução reprimida nesta fase. Ao contrário de dhhc2, cuja expressão proteica já existe nos gametócitod, não sendo por isso, a sua tradução reprimida, como inicialmente previsto, pelos complexos repressores definidos por DOZI e CITH. Mais tarde, já no interior do mosquito, a produção das proteínas DHHC10, IPET e EPSF começa nos oocinetos, onde são armazenadas nos corpos cristalóides, e usadas posteriormente, durante o desenvolvimento no mosquito. Os corpos cristalóides são um organelo tipicamente encontrado em oocinetos e oocistos maduros, que se supõe constituir um reservatório de proteínas que serão usadas pelo parasita durante o desenvolvimento de oocistos. Para além disso, sabe-se que DHHC10 tem como função a palmitoilação de proteínas – uma modificação pós-traducional de extrema importância no controlo da actividade, localização e transporte de proteínas. Os parasitas KO desta proteína não desenvolvem esporozoítos apesar do número de oocistos permanece normal quando comparado com wild type (WT). Estes oocistos ∆dhhc10 apresentam contudo uma morfologia aberrante, com oocistos não esporulados e vacuolados. Estes resultados indicam que a palmitoilação de proteínas mediada por DHHC10 é essencial para o processo de formação de esporozoítos. Paralelamente, os parasitas KO de EPSF também não desenvolvem esporozoítos, indicando que esta proteína será essencial para a sua formação. No geral, os nossos resultados confirmam o papel vital do mecanismo de repressão da tradução no desenvolvimento sexual em Plasmodium. Para além disso, fornecem indicações
adicionais sobre a elevada e complexa estruturação dos mecanismos pós-transcricionais e pós-traducionais que regulam o ciclo de vida deste parasita.
Palavras-chave: CITH, desenvolvimento, DOZI, esporozoíto, gametócitos, mRNPs, oocineto, oocisto, Plasmodium, proteínas PUF, repressão da tradução, transmissão
Table of Contents Acknowledgements………...1 Abbreviations…..………2 Abstract……….………..4 Resumo………...…5 1 – Introduction………..………..11 1.1. Malaria disease………..………..11
1.2. Plasmodium life cycle……….12
1.3. Plasmodium regulation of gene expression………13
1.3.1. Translational repression in Plasmodium………..15
1.3.2. Translational repression and Plasmodium PUF2 protein………..16
1.4. Aims of this work………..17
2 – Materials and Methods……….18
2.1. Experimental animals………..18
2.2. Reference P. berghei ANKA lines used………...18
2.3. Immunoprecipitation of PUF2 protein in P. berghei gametocytes………...19
2.3.1. puf2::gfp line……….……… 19
2.3.2. puf2::gfp sequencing………19
2.3.3. IP of PUF2::GFP and DOZI::GFP in gametocytes and mixed blood stages…………..20
2.3.3.1. Nycodenz method……….20
2.3.3.2. Nycodenz method with enhanced gametocytaemia………20
2.3.3.3. PUF2::GFP and DOZI::GFP mixed blood stages IP using GFP-Trap® Kit………….20
2.3.4. Detection of PUF2::GFP and DOZI::GFP proteins in gametocytes and mixed blood stages by Western blot analysis………21
2.4. DHHC2, DHHC10, IPET and EPSF mutant lines characterization, Western blot analysis and expression profiles………..21
2.4.1. Mosquito infections and bite-backs………...21
2.4.2. Genotyping and RT-PCRs of ∆dhhc10, ∆epsf, dhhc2::gfp, dhhc10::gfp, ipet::gfp and epsf::gfp parasite lines………22
2.4.3. Sequencing of ipet-main version and ipet-splice variant cDNA………22
2.4.4. Life cycle RT-PCRs………..23
2.4.5. Western blot analysis of CSP expression in oocysts from dhhc10-KO and epsf-KO parasite lines………23
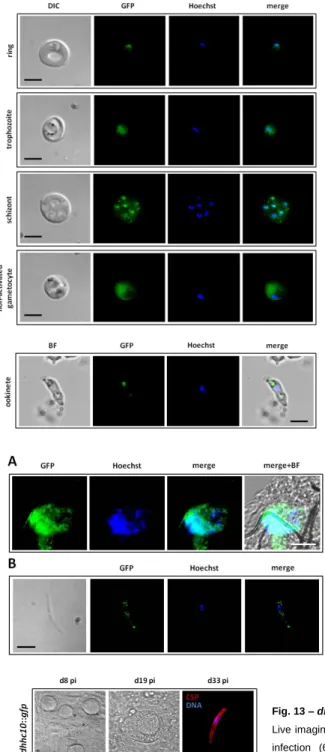
2.4.6. Live imaging and immunofluorescence assays (IFAs) of blood stages, ookinetes, oocysts and sporozoites………24
3 – Results and Discussion………24
3.1. Immunoprecipitation of PUF2 protein in P. berghei gametocytes………24
3.1.1. puf2::gfp sequencing………27
3.1.2. Discussion……….27
3.2. Characterization of a set of proteins translationally repressed by DOZI and CITH defined mRNPs………..29
3.2.1. Confirmation of the targeted disruption of dhhc10 and epsf……….29
3.2.2. Molecular analysis of GFP-tagged parasite lines………29
3.2.3. Alternative splicing in ipet………29
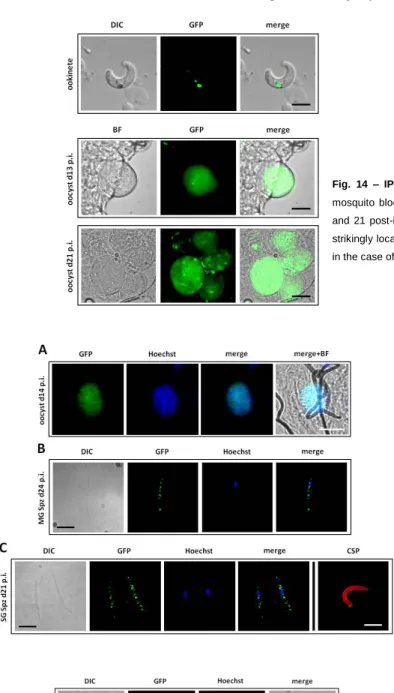
3.2.4. Life cycle mRNA and protein expression profile and protein localization………30.
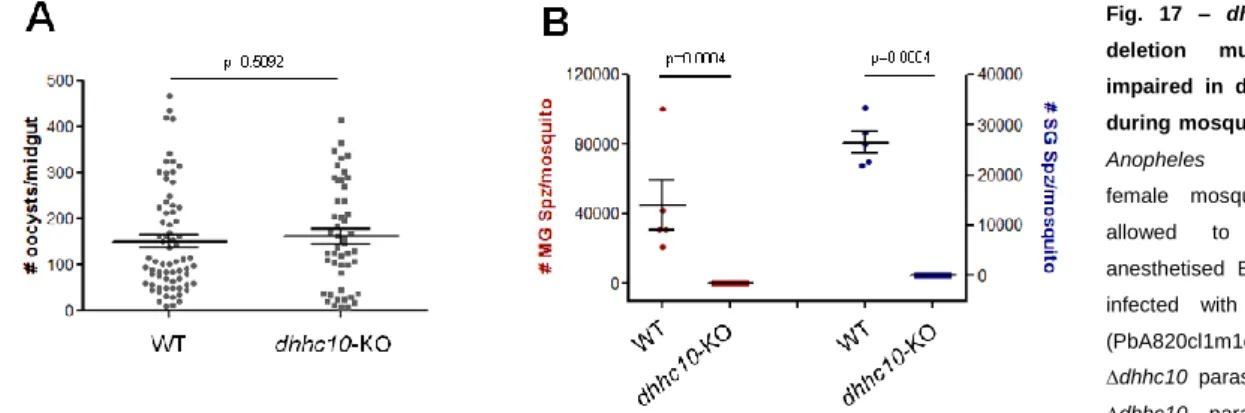
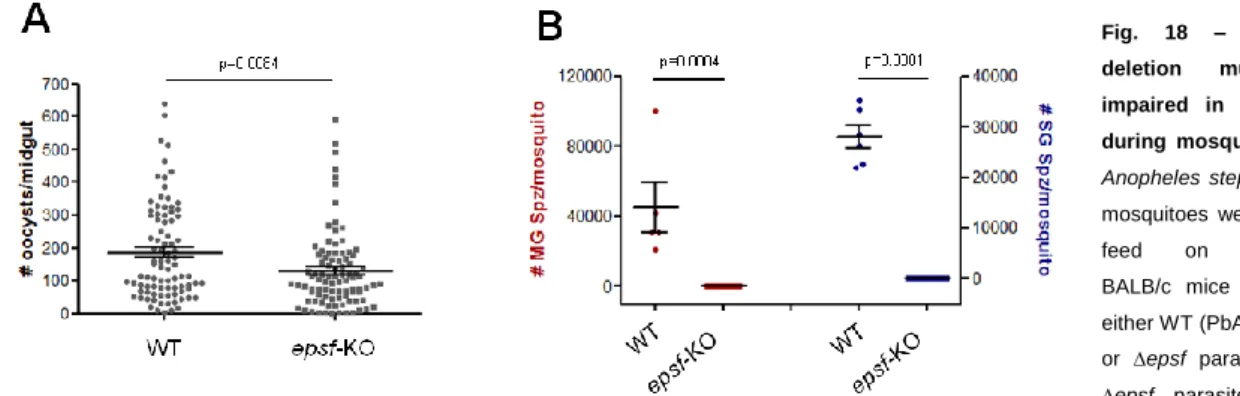
3.2.5. dhhc10-KO and epsf-KO mutant phenotypes………..34
3.2.6. Discussion……….37
3.2.6.1. DHHC10, IPET and EPSF role in Plasmodium life cycle………...37
3.2.6.2. DHHC family in Plasmodium berghei: DHHC2 and DHHC10………38
4 – Conclusions………...39 5 – References……….40 6 – Appendix I………..44 7 – Appendix II……….44 8 – Appendix III………...……… 45 9 – Appendix IV………47
1 – Introduction
1.1. Malaria disease
Malaria is an infectious disease caused by parasites of the genus Plasmodium [1], belonging to the Phylum Apicomplexa [2], that are transmitted by a vector, a female mosquito of the genus Anopheles, to its host [1]. Five Plasmodium species can cause human malaria:
P. falciparum, P. vivax, P. ovale, P. malariae and P. knowlesi [1]. The responsible for the
deadliest form of malaria is P. falciparum, which has a prevailing incidence in the region of Africa [3]. A remarkable feature of the Plasmodium parasite is the restrictedness of the hosts they choose, only closely related vertebrates are infected by the around 5000 Plasmodium species described [4]. Plasmodium host exclusiveness has been supported by molecular phylogenetic studies, which have demonstrated the evolutionary relatedness between the different species that infect mammals [2]. In malaria research, there are several Plasmodium species that, over the years, became essential to its development; among these we have the primate malaria model Plasmodium knowlesi, which is a macaque monkey’s parasite in the wild, and three rodent malaria parasites, Plasmodium berghei, Plasmodium yoelli and
Plasmodium chabaudi, which infect thicket rats in central Africa [5]. The rodent malaria
parasite P. berghei was used as a malaria research model in the present study.
According to the World Malaria Report of 2012, the estimate number of people at risk of acquiring malaria in 2011 was 3,3 billion people worldwide; more particularly, the populations at a higher risk are located in the regions of sub-Saharan Africa [3]. In the last years, many different strategies have been developed to fight the malaria burden; relevant examples include nets treated with long-term insecticides and also combination of drug therapies. These strategies proved their success as a strong reduction of malaria cases was achieved among several regions, while in others complete eradication was successfully accomplished [6]. Nowadays, a central problem in the fight against malaria is the ability of the Plasmodium parasite to acquire resistance to antimalarial drugs, like artemisinin, and also insecticides [7]. In addition, as in any other disease control program, the success also depends largely on a continuous funding [7]. Presently, there are 104 countries marked as endemic areas, 99 of these have ongoing malaria transmission, and the remaining ones are in the prevention or reintroduction phases [3].
Uncomplicated malaria is characterized by sporadic febrile episodes which the host’s immune system is capable of controlling, and eventually, eliminate [8]. In contrast, severe malaria may cause death and is characterized by one or a combination of the following syndromes: cerebral malaria, metabolic acidosis and severe anemia [8]. Cerebral malaria and metabolic acidosis have a mortality rate of 15-20%, even with artemisinin derivatives treatment. Moreover, neurological impairment is a common feature among the survivors [8].
The ones most affected by severe malaria are children under five years and pregnant women [3]. The main aim of today’s antimalarial drugs is to prevent progression to severe disease by targeting the asexual blood stages responsible for the symptoms of the disease [8].
1.2. Plasmodium life cycle
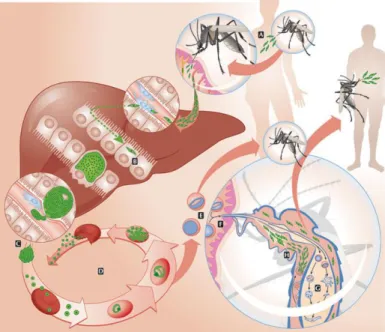
The malaria parasite exhibits a complex life cycle that involves a vertebrate, human host (or a mouse in the rodent model) and a mosquito vector, as depicted in Fig. 1. Sexual reproduction occurs in the mosquito; its regulation and the formation of sexual precursors in infected red blood cells (iRBC) is still unclear, but it was shown for P. falciparum, that the commitment to sexual differentiation occurs before schizont maturation [9]. During a blood meal, the mosquito ingests gametocytes and, in the mosquito midgut lumen, the formation of gametes (gametogenesis) is initiated. Male gametogenesis, termed exflagellation, is triggered by a set of environmental cues: xanthurenic-acid (a molecule present in the mosquito) and temperature and pH changes [10]. When fertilization occurs, a diploid zygote is formed, followed by a tetraploid motile ookinete. The ookinete route includes traversing of the midgut epithelial cells and subsequent exit through the basal lamina of the epithelium. This process activates the ookinete to switch from a cell traversal mode to a sessile mode called oocyst [10]. Oocysts go through a process of plasma membrane invagination, forming sporoblasts, through which sporozoites will bud-off [10]. The process of sporozoite formation takes place during a time period of 2 weeks, and after mature oocysts rupture, sporozoites are released into the haemolymph. From this point on, sporozoites travel to the salivary glands of the mosquito, invade them, and reach the salivary duct, their journey’s destination, where they can be injected into a new host during a subsequent blood feeding by the mosquito [1].
Plasmodium sporozoites are transmitted to a mammalian host during the bite of an
infected mosquito and are deposited in the dermis of the host. However, as it was shown by Amino et al. (2006), only a proportion of the parasites have the capability of entering the blood capillaries, the remaining is drained by the lymphatics [11]. Once inside the circulatory system, sporozoites rapidly reach the liver [10, 12]. Sporozoites traverse a number of hepatocytes before reaching and invading a final hepatocyte, using a membrane-associated actin-myosin motor [10]. This traversal process was shown by Mota et al. (2001) to be essential for the completion of the life cycle, hypothesizing that sporozoites might need to traverse a number of cells, so that signaling pathways essential for entry and development inside the hepatocyte can be activated [13]. Inside the final hepatocyte, sporozoites will develop and multiply, giving rise to thousands of merozoites [12]. The release of Plasmodium merozoites from hepatocytes is a vital step in the life cycle of this intracellular pathogen. The common place idea was that the release of merozoites would occur after hepatocyte rupture.
However, Sturm et al. (2006) showed that parasites induce hepatocyte death, and subsequently bud-off inside vesicles (merosomes) into the sinusoid lumen [14]. After being released from the merosomes, the merozoites invade the host’s red blood cells and the blood stage of infection begins. The intraerythrocytic parasite development includes the ring, trophozoite, and schizont stages, and ultimately the formation of new merozoites. To successfully enter a new red blood cell (RBC), the merozoite must be very quick in selecting a RBC, adhere to it, enter and enclose itself inside it [15, 16, 17]. Some merozoites develop into gametocytes, which when taken up by a mosquito during a blood meal allow for a productive infection of the parasite’s vector [15].
Fig. 1 – Plasmodium life cycle. (A) Sporozoites transmission to a human host during a blood meal of a infected mosquito; (B) Traversal of several hepatocytes before invading a final one; (C) Release of merozoites into the bloodstream; (D) Merozoite infection of red blood cells; (E) Formation of male and female gametocytes; (F) Ingestion of gametocytes by the mosquito during a blood meal; (G) Gametes fertilization in the mosquito midgut and further development into ookinetes, and later on, to oocysts; (H) Migration of sporozoites to the salivary glands of the mosquito and transmission to another human host during the next blood meal (Adapted from [18]).
1.3. Plasmodium regulation of gene expression
How the Plasmodium parasite regulates and manages gene expression has been a matter of intense research and debate [19, 20]. Our malaria study model P. berghei has an estimated genome size of approximately 18Mbp[21], very similar to P. falciparum (22.8 Mbp) [22], and also like P. falciparum, an exceptionally high A+T composition [22, 23]. Both P.
berghei and P. falciparum have their genome split in 14 chromossomes [22, 24]. P. falciparum genome sequencing made available an enormously amount of information about
its composition and characteristics. Particularly, an interesting feature was revealed about this parasite, it possesses a small amount of transcription associated proteins (TAPs
)
[22]. This observation was the starting point for the hypothesis that transcriptional regulation inPlasmodium is most probably unlike that of others well studied eukaryotic systems, like yeast
or mammalian cells [19, 25]. Van Noort et al. (2006) proposed that the few regulatory proteins encountered in Plasmodium regulate gene expression in a combinatorial manner,
relying on a higher number of regulatory DNA sequences per gene than those observed in other eukaryotes [26].
Until 2005, no specific transcription factors in Plasmodium had been reported. Balaji et
al. (2005) were the first to describe a group of conserved proteins containing putative AP2
DNA-binding domains, referred nowadays as the Apicomplexan AP2 (ApiAP2) protein family. The P. falciparum ApiAP2 gene family was reported to have 27 members, with a high degree of conservation between Plasmodium species and with DNA binding domains closely related to the ones found in transcription factors of other eukaryotes [27]. Even though it was initially thought that the Plasmodium ApiAP2 would have a prominent role in asexual blood stages, transcriptional and proteomic studies indicated that possibly they are required throughout the life cycle [28]. Furthermore, it was shown that the Plasmodium transcription factor (TF) AP2-O activates gene expression in ookinetes [29], providing additional evidence for a significant role of this TF family in gene regulation.
The combination of high-throughput techniques and bioinformatics in malaria research were decisive tools to uncover the prominent role of post-transcriptional regulation in the
Pasmodium parasite [20]. A global analysis of transcript and proteins levels in P. falciparum
is an example of such studies, where it was possible to identify the delay between mRNA and protein accumulation as the most common expression pattern. In the last years, it became clear that stage-specific gene regulation is essential for the parasite, which is illustrated by reports in transcriptional regulation during asexual and sexual development. Bozdech et al. (2003) analyzed the transcriptome of P. falciparum complete asexual intraerythrocytic developmental cycle transcriptome and proposed a model for the parasite transcriptional regulation. The authors compare the transcriptome at this stage of development to a “just-in-time manufacturing process”, where a gene induction is a onetime event per cycle and only at the required moment [30]. The existence of sex-specific genes in
Plasmodium was established by Khan et al. (2005), leading to an enormous progress in
uncovering the mechanisms of sexual development regulation. By analyzing the proteome of separate male and female gametocytes, the authors showed that the male proteome contained 36% of male-specific proteins and the female proteome 19% of female-specific proteins [31]. Uncovering the role played by post-transcriptional mechanisms role in
Plasmodium sexual differentiation began when it was shown that p25 and p28 (genes
encoding ookinete surface proteins) mRNAs were kept in a quiescent state in female gametocytes. These stored transcripts were only, later on, translated in gametes and zygotes [32]. This process was termed translational repression, and it will be discussed with further detail in 1.3.1. section. Further studies demonstrated, both in human and rodent malaria parasites, that an additional substantial amount of mRNAs were also stored in gametocytes and only translated in a later stage, namely in gametes [21, 33]. The studies mentioned
above emphasize that precise regulation of transcription is essential for the parasite, since dynamic expression of transcripts seems like a critical mechanism at several points during the intraerythrocytic life cycle.
1.3.1. Translational repression in Plasmodium
Translational repression is a mechanism in which certain mRNAs are selectively moved into cytoplasmic messenger ribonucleoprotein (mRNPs) complexes and are maintained in a quiescent state for translation at a later time. This process allows the control of both temporal and spatial protein expression [20, 34]. The mRNAs that are translationally repressed in this manner usually possess a U-rich RNA region that can be found in the 3’ or 5’-untranslated regions (UTRs), which is a key factor for their regulation [35].
As mentioned above, both p25 and p28 mRNAs are kept untranslated in female gametocytes ribonucleoparticles. It was found that these unstranslated p25 and p28 mRNAs colocalize with the DEAD-box RNA helicase DOZI (Development of Zygote Inhibited) and Sm-like factor CITH (CAR-I/Trailer Hitch Homolog) proteins in P. berghei [34, 36]. DOZI and CITH disruption leads to a drastic reduction of abundance of both p25 and p28 mRNAs, as well as of an additional 370 mRNAs [34, 36]. pbdozi and pbcith-KO female (but not male) gametocytes do not form ookinetes after fertilization, which indicates that most likely a fraction of these translationally repressed mRNAs are essential for this particular step of development [34, 36]. P-granules have an important role in metazoan sexual development, since they are the storage particles where the translationally silent transcripts are kept and stabilized [36]. The idea of P-granules as stable storage places is strengthened by the absence of RNA degradation factors in these granules [36]. A translational repression model in gametocytes was established in which an mRNP complex is composed of CITH, DOZI and 16 major factors, including eIF4E, a translation initiation factor, and poly-(A)-binding protein, as depicted in Fig. 2. This complex translationally represses mRNAs that will have a critical role in the initial stages of the mosquito infection [36]. More recently, a biochemical characterization of the DOZI homologue commonly known as DDX6 from Plasmodium
falciparum was presented, being the first report that shows the direct interaction between
DDX6 and PfeIF4E [37].
Fig.2 – DOZI and CITH defined mRNP structure in female gametocytes.
1.3.2. Translational repression and Plasmodium PUF2 protein
The PUF family consists of RNA-binding proteins whose major role is post-transcriptional regulation. The first two members of the PUF family to be analyzed in detail were Drosophila melanogaster Pumilio and Caenorhabditis elegans FBF, so consequently, the group was termed PUF or PUM-HD proteins [38, 39] PUF proteins regulate mRNAs by binding to specific regions in the 3’-UTRs [40, 41, 42]. All members of the PUF family contain a PUM-HD (Pumilio homology domain)-type RNA binding domain [39], that can physically interact with both RNAs and proteins [40, 41]. PUM-HD-type RNA binding domain usually has 8 consecutive Puf repeats, that normally consist of around 40 aminoacids with two short flanking regions [38, 39]. Puf repeats are located in the C-terminus region of the protein and are characterized by having a “core consensus” containing aromatic and basic residues and [40]. Examples of this family functions are: development, differentiation, germline function, neuronal function, memory, and mitochondrial and cell cycle biogenesis [40, 41, 42, 43]. The preservation of stem cells mitotic potential has been proposed as an ancestral role for the PUF family [40].
Studies focused on the PUF family had a major role in uncovering post-transcriptional mechanisms [41]. The mechanism of mRNA repression was first described by Goldstrohm et
al. (2006), where the authors suggest that yeast PUF protein recruits factors that assist on its
role in mRNA repression and/or decay, namely proteins of the deanylase complex [45]. Moreover, it was also found that in many cases PUF proteins regulate specific sets of transcripts whose encoded proteins have related functionalities [45]. For example, mRNAs involved in organellar biosynthesis were found to be regulated spatially and temporally by PUF proteins [45]. Since PUF proteins dynamically coordinate many transcripts, its own regulation has to be extremely tight. Both their activity and expression are regulated at all levels of gene expression, from transcription to post-translation [42].
In Plasmodium falciparum, PfPUF2 plays an important role in arresting sexual development in gametocytes [46]. PfPUF2 was found to be expressed in both male and female gametocytes, and deletion of this gene resulted in increased gametocyte formation and a considerable higher male/female sex ratio [46]. In contrast, such role for PUF2 in parasite sexual development and sex differentiation was not observed with the rodent P.
berghei. Interestingly though, P. berghei PUF2 was found to play a critical role in regulating
parasite development during transmission from the mosquito to the mammal host [47]. P.
berghei puf2-KO sporozoites exhibit a precocious development into exo-erythrocytic liver
stage forms (EEFs) inside mosquito salivary glands, showing that translational repression by PUF2 is an essential control mechanism in sporozoite development [47], as depicted in Fig. 3. Other recent findings also support the idea of PUF2 as a key player in parasite
transmission between the mosquito and the mammalian host [48]. Their results suggest that
P. berghei PUF2 regulates IK2 (eIF2α kinase) inhibiting translation of certain transcripts in
salivary gland sporozoites [48]. The rodent P. yoelii PUF2 protein was also analyzed and, despite some differences when compared with P. berghei, found to play a similar crucial role in maintaining the homeostasis of specific transcripts and therefore ensuring a successful parasite transmission from the mosquito to the mammalian host [49].
Fig.3 – Model for the regulation of development during transmission between mosquito and mammalian host having PUF2 protein as a key player (Adapted from [47]).
1.4. Aims of this work
Translational repression is an essential mechanism in sexual differentiation and gametogenesis. DOZI and CITH roles at this stage have been well established, and evidence is growing for a PUF2 role in the repression of transcripts in gametocytes. In pbdozi-KO gametocytes, a great number of transcripts are up and downregulated [37], and RIP-Chip analysis has been done to confirm that those transcripts are indeed being physically bound to DOZI- and CITH-defined mRNPs (data not published), thus suggesting that they are translationally repressed. In P. berghei, PUF2 expression in gametocytes has not yet been demonstrated.
The main goal of this work is to further dissect the role of these translational repression complexes in gametocytes. To do so, two different strategies were applied:
a) Perform immmunoprecipitation of PUF2::GFP in P. berghei gametocytes, to confirme its expression at this stage, and subsequently use the IP-eluates to analyze the candidate transcripts to be translationally repressed. In addition, the puf2::gfp parasite line can be used to assess PUF2 localization using microscopy tools.
b) A set of transcripts predicted to be translationally repressed by the DOZI- and CITH- defined complexes in P. berghei gametocytes (based on the previous data obtained from the pbdozi-KO and pbcith-KO mutant parasite lines) were chosen using defined criteria
(please read below), and GFP-tagged and KO mutant parasite lines were constructed for each selected gene. Using molecular biology tools, microscopy and Western blot analysis we were able to characterize them in terms of protein expression, localization, transcription profile throughout the life cycle and function. The selected genes, with their predicted features, designations attributed and respective mutant parasite lines constructed in the lab, are summarized in Appendix IV Table S1.
In the beginning of the selection process, a bigger set of genes were chosen that had the common characteristic of possessing either a signal peptide, transmembrane domains (TMDs) or both, indicating that they could potentially be surface proteins, and like many others discovered in the last years, be of relevance for Plasmodium development in the mosquito. Preliminary data of the KO-parasite lines for dhhc10, ipet (Invasion Protein Essential for Transmission) and epsf (Essential Protein for Sporozoite Formation), rendered them as promising for further studies. DHHC10, as well as DHHC2, another protein studied in this work, belong to a family of proteins called Asp-His-His-Cys cysteine-rich domain (DHHC-CRD) S-acyltransferase (PAT) family. This group of enzymes catalyzes the transfer of a palmitate from palmitoyl-CoA to a protein, a post-translational modification termed protein palmitoylation. A recent report states that some apicomplexan-specific DHHCs are essential for parasite growth [50], leading us to an additional interest in this family of proteins, aiming at a better understanding of the role of these enzymes in P. berghei life cycle, specially within its mosquito vector.
2 – Material and Methods
2.1. Experimental animals. Female Balb/c ByJ mice (6–8 weeks bred at Charles River, France) were used. All animal experimentation protocols were approved by the IMM Animal Care Committee and performed according to EU regulations.
2.2. Reference P.berghei ANKA lines used. Four reference P. berghei ANKA parasite lines were used in the present work: line HPEcy1m50cl1, a wild-type non-gametocyte producer clone (Janse et al., 1989); line 820cl1m1cl1 (WT Fluo-frmg; RMgm-164) expressing RFP under the control of the female gametocyte specific promoter of lap4(ccp2) gene (PBANKA_131950) and GFP under the control of the male gametocyte specific promoter of dynein heavy chain gene (PBANKA_041610); line 259cl1 (WT PbGFPcon; RMgm-5) expressing GFP under the control of the constitutive eef1a promoter; and line cl15cy1 (WT). Lines 820cl1m1cl1 and contain the transgene integrated into the silent 230p gene locus (PBANKA_030600) and do not contain a drug-selectable marker. Line 259cl1 contains the transgene integrated into the small subunit ribosomal rna gene (c-type unit) and does contain the tgdhfr/ts drug selectable marker.
2.3. Immunoprecipitation of PUF2 protein in P. berghei gametocytes
2.3.1. puf2::gfp line. The mutant parasite line that expresses a C-terminally GFP-tagged version of PUF2 (PBANKA_071920) was previously constructed in the lab (parasite line 1750cl4; data not published).
2.3.2. puf2::gfp sequencing. PUF2::GFP genomic DNA and gametocytes cDNA of were used. Primers used for both gDNA and cDNA were: g0477 X g0459c; TRAP gDNA was used as a positive control with the primers g0432 X g0433c. PUF2::GFP PCR products were obtained using a 50µL PCR mix reaction containing: 10X Taq Buffer with 500mM KCl (Thermo Scientific), 2mM MgCl2 (Thermo Sientific), 15mM of each primer, 10mM dNTPs, 1µl 5U Taq DNA Polymerase (Thermo Scientific) and 2µL of DNA sample. Cycling conditions used were: initial denaturation at 94°C for 3 minutes, followed by 45 cycles of denaturation at 94°C for 10 seconds, annealing at 55°C for 30 seconds and extension at 62°C for 30 seconds. A final round at 62°C for 10 minutes allowed complete elongation of the PCR product. 5µL of a 1:50 dilution of the PCR products were loaded in a 1% agarose gel to confirm amplification. The PCR products obtained were cloned using the kit mentioned in Appendix I Table S2. and transformation was performed using 50µL of DH5α competent cells and 1µL of plasmid. Samples were incubated on ice for 20 minutes, followed by a thermal shock of 45 seconds at 42°C and by 2 minutes on ice. 1mL LB medium was used to incubate the bacteria for 1 hour at 37°C. Afterwards, 100µL were plated directly in LB agar plates supplemented with ampicillin (100µL/mL) and left incubating overnight at 37°C. Colonies were picked from the plates and mini-prep cultures were grown in 2mL LB medium supplemented with ampicillin overnight at 37°C. Plasmid DNA extraction was performed used the kit mentioned in Appendix I Table S2. Correct ligation of the insert into the plasmid was confirmed with a restriction reaction using the enzymes XhoI (3U; from Promega, Madison, WI U.S.A.) and XbaI (30U from Jena Bioscience), 1X NEBuffer 4 (BioLabs, Inc. New England), 100 X Purified BSA (BioLabs, Inc. New England), mili-Q water (Millipore®) and 15µlL of the DNA previously extracted (in a total volume reaction of 30µl) and incubated for 1 hour and 30 minutes at 37°C. The restriction product was loaded in a 1% agarose gel, and the PCR products with the expected insert size were chosen. pJET plasmid containing the correct inserts were used to prepare cultures for midi-preps in LB medium supplemented with ampicillin and grown overnight at 37°C. DNA concentrations obtained were determined with a Nanodrop-1000® Spectrophotometer. DNA samples were sequenced by Stabvida using pJET forward and reverse primers. The sequences obtained were afterwards analyzed using Finch TV Version 1.4.0 © and Clone Manager © Software.
2.3.3. Immunoprecipitation of PUF2::GFP and DOZI::GFP in gametocytes and mixed blood stages
2.3.3.1.Nycodenz method. After sacrificing one mouse per each parasite line, heparinized syringes were used to subtract the blood from each mouse by cardiac puncture. PBS was added up to a volume of 2mL. Purified gametocytes were obtained by loading the blood on a 5mL of 49% Nycodenz solution in PBS, followed by a centrifugation of 20 minutes at 450 rcf room temperature (RT) with acceleration 3 but no brake. The brownish layer was collected from the gradient and centrifuged for 5 minutes at 450 rcf at RT. The supernatant was discarded. The purified gametocytes obtained were washed two times in ice-cold “enriched” PBS with spins of 5 minutes each at 450rct at 4ºC. Lysis of the parasites was done with 210 μl of NET-2++ buffer for 30 minutes at 4°C using a shaker. After lysis the extract was centrifuged for 10 minutes at 14000 rpm and the supernatant collected. 50 μl extract were used per IP with mouse anti-GFP-antibody (1µg Roche Diagnostics, Inc.), anti-c-myc-antibody (1 μg, Sigma-Aldrich, St. Louis Missouri, USA) or beads-only and incubated for 1 hour at 4°C using a shaker. Another 50µL were saved for the control input sample. Protein G Sepharose™ 4 Fast Flow beads (GE Healthcare), 20μL packed bead volume per IP, previously washed 5x with NET-2 buffer and 2x with NET-2++ buffer were then added to each sample and incubated for 1 hour and 30 minute at 4°C in a shaker. IP samples were afterwards washed three times with 200μL NET-2 buffer with an additional final wash with 300µL NET-2 buffer. One third of the final wash was resuspended in 500µL TRIzol® (Reagent for RNA from Ambion®), while the other two thirds were centrifuged and the beads resuspended in 50µL 2X SDS-PAGE loading buffer and used for western blot analysis.
2.3.3.2. Nycodenz method with enhanced gametocytaemia. Enhanced gametocytemia was obtained as described in [52]. Nycodenz purification and Immunoprecipitation were performed as described in the previous section.
2.3.3.3. PUF2::GFP and DOZI::GFP mixed blood stages IP using GFP-Trap® Kit. After sacrificing one mouse per each parasite line, heparinized syringes were used to subtract the blood from each mouse by cardiac puncture. 1X RBC lysis buffer was added to the blood the lysis step performed on ice until the solution became translucent. After a centrifugation of 8 minutes at 2000 rpm, an extra lysis step was performed using 0,1% saponin in PBS supplement with protease inhibitors (1 tablet of Complete Mini, Roche) 30 minutes on ice. After another 8 min 2000 rpm spin, the pellet was washed three times with enriched PBS. Parasite lysis step was performed with 210 µL NET-2++ buffer during 30 minutes at 4°C using a shaker and a 10 minute spin at maximum speed. 50µL of the lysate were saved as
the input fraction to which 50µL 2X SDS-PAGE loading buffer were added. From this point on, GFP-Trap® _A Kit for Immunoprecipitation of GFP Fusion Proteins (Chromotek) Protocol (2012-12-07 version) was used starting from step 4.
2.3.4. Detection of PUF2::GFP and DOZI::GFP protein in gametocytes and mixed blood stages by Western Blot analysis. For Western Blot analysis 15 to 25 µL of IP samples from purified PUF2::GFP and DOZI::GFP gametocytes as well as mixed blood stages were used. DTT (200mM final concentration) was added to the samples before the denaturing step at 95°C for 10 minutes. Samples were loaded in a precast gel (BioRad, Any-kD™) and ran the required time to obtain a desirable band separation at the molecular weight of interest (PUF2::GFP and DOZI::GFP expected molecular weight were ~83kDa and ~76kDa, respectively). Using a wet transfer system, the proteins were transferred to a nitrocellulose membrane for 1 hour and 45 minutes at 200mA. The nitrocellulose membrane was then blocked for 1 hour at RT with 5% skim milk in 0,05% PBS/Tween 20 and afterwards probed with anti-GFP antibody in blocking solution overnight at 4ºC. Rabbit anti-GFP (Invitrogen) and rabbit anti-GFP (Sigma-Aldrich, St. Louis Missouri, USA) were used in a dilution of 1:500 and anti-GFP Roche was used in a dilution of 1:5000. After washing the membrane two times, 10 minutes each with 0,05% PBS/Tween 20, it was probed with a secondary antibody for 3 hours at 4°C. HRP-conjugated donkey anti-rabbit (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was used in a 1:500 dilution and HRP-conjugated goat anti-mouse (Santa Cruz Biotechnology Inc.) was used in a 1:5000 dilution. The membrane was then washed with 0,05% PBS/Tween 20 developed using Immobilon® Western Chemiluminescent HRP Substrate (Millipore®).
2.4. DHHC2, DHHC10, IPET and EPSF mutant lines characterization, Western blot analysis and expression profiles
2.4.1. Mosquito infections and bite-backs. For mosquito passage of the different parasite lines used in this study, female Balb/c ByJ mice (6–8 weeks years old) were infected intraperitoneally (IP) with 106 iRBCs of each line. On days 4-5 post-infection (p.i.), these mice were anesthetised and Anopheles stephensi female mosquitoes allowed to feed for 30 min. Twenty-four hours after feeding, mosquitoes were anesthetized by cold shock and unfed mosquitoes were removed. To determine oocyst burdens, mosquito midguts were dissected at days 12 or 13 p.i., stained with mercurochrome and imaged using a Leica DMR microscope. To determine oocyst diameters over time mosquito midguts were dissected at days 12, 14, 16, 19 and 21/22 p.i. and imaged using a Leica DMR microscope. Oocysts were counted and measured from these images using ImageJ 1.47n software (imagej.nih.gov/ij). For confirmation of normal development of GFP-tagged parasite lines in mosquitoes, midguts
were dissected at different time points and directly imaged in a Leica DM5000B microscope. For sporozoite (Spz) counting, midguts and salivary glands were dissected at days 20-22 p.i., mashed and Spz counted in a Neubauer chamber. For bite-back experiments, 10 starved female infected mosquitoes were allowed to feed for 30 min on anesthetised naïve female Balb/c ByJ mice (6–8 weeks years old) on days 20-21 p.i. (10 mosquitoes per mouse). Successful feeding was confirmed by the presence of blood in the abdomen of mosquitoes. Parasitemias in these mice were followed up to 32 days post-bite.
2.4.2. Genotyping and RT-PCRs of ∆dhhc10 and ∆epsf and dhhc2::gfp, dhhc10::gfp; ipet::gfp and epsf::gfp lines Parasite genomic DNA extraction was done using the kit mentioned in Appendix I Table S2. RNA extraction and cDNA synthesis was performed as stated in the Appendix II (7.1 and 7.2). Genomic DNA samples were used in a 10 ng/µL concentration. For cDNA samples, several dilutions were tested, and the most satisfactory was chosen. PCR mix was done as described in the Appendix II (7.3) and PCR cycling conditions were the following: initial step of 3 minutes at 95°C, followed by denaturation step of 10 seconds 95°C, annealing step during 30 seconds using an appropriate annealing temperature, and an extension step at 68°C for the required amount of time depending on the size of the fragments amplified. Finally, an extension step of 10 minutes at 68°C was done. The variable PCR conditions used for each PCR are summarized in the Appendix III Table S3. RNA polymerase II was used as control genes; PbAcl15cy1 and PbA820cl1m1cl1 were used as WT control lines for GFP-tagged lines and KO lines, respectively.
2.4.3. Sequencing of ipet-main version and ipet-splicing variant cDNA. Gel extraction of ipet cDNA from a pool of several PCR products from RT+ PbAcl15cy1 WT mRNA (g1196 X g1201c), with a final volume of 192µL (160µL of PCR product and 32 µL of 6X loading dye) and RT+ 2180 GFP-tagged (g1196 X g0408c), with a final volume of 36 µL (30µL of PCR product and 6 µL of 6X loading dye) was performed using the kit in Appendix I Table S2. The cDNA extracted was eluted in 30 µL of Elution Buffer and quantified in a Nanodrop-1000® Spectrophotometer, followed by cloning of the PCR products obtained in a commercial vector. In the ligation reaction, 7µL of PCR product were used in a 20µL ligation reaction during 25 minutes. The resulting plasmid was transformed in E. coli DH5α chemically competent cells, using 2 µL of product per 50 µL of cells and incubated on ice for 20 minutes. Then thermal shock for 45 seconds at 42°C was done, followed by 2 minutes on ice. 950µL of SOC medium (per tube) were used to incubate the cells for 1 hour at 37°C. Afterwards, 100µL were plated directly in an agar plate supplemented with ampicillin (100µg/mL), the remaining culture was centrifuged 3 minutes at 5000 rpm, and most of the supernatant was discarded leaving around 200µL that were resuspended and plated. Cells were grown at 37°C overnight. Mini-prep cultures were grown using 3 mL of LB medium supplemented with
ampicillin overnight at 37°C. Plasmid DNA extraction was done using the kit in Appendix I Table S2 and the DNA obtained was diluted in a final volume of 50µL. Correct ligation of the insert into the vector was confirmed with a restriction reaction using 0,5 µL of BgI-II 10 U/µL (Fermentas), 1X Buffer Orange (Fermentas) and 5µL of the previously extracted DNA (in a total volume reaction of 20µL) and incubated for 1 hour at 37°C. Five µL of 6X loading dye was added to the restriction reaction and 10µl were in a 1% agarose gel in TAE Buffer. Correctly cloned plasmids were chosen for sequencing using primers flanking the inserts (provided with the kit) by Stabvida. The sequences obtained were analyzed using BioEdit Sequence Alignment Editor Version 7.1.11© and Clone Manager© Software.
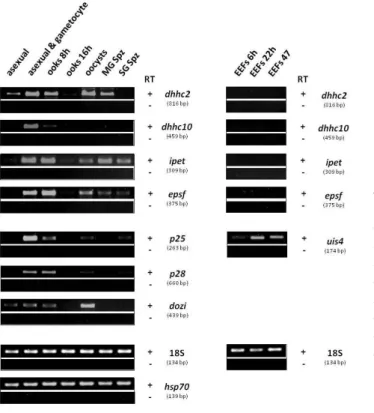
2.4.4. Life Cycle RT-PCRs. Asexual mixed blood stages originated from P. berghei HPE line and asexual and gametocytes stages originated from P. berghei ANKA 820cl1m1cl1 line. In
vitro culture ookinetes 8-hours and 16-hours originated from P. berghei ANKA cl1515cy1 line.
Oocysts sample was obtained from oocysts day 12 p.i. P. berghei ANKA 820cl1m1cl1 line-infected midguts. Midguts and salivary glands sporozoites samples were both obtained from
P. berghei ANKA 259cl1 parasite line at day 21 p.i.. In vivo exoerythrocytic forms 6 hours, 22
hours and 47 hours samples were obtained from P. berghei ANKA 259cl1 parasite line. RNA extraction, cDNA synthesis and PCR mix were done as stated in Appendix II (7.1, 7.2. and 7.3). PCR cycling conditions were the following: initial step of 3 minutes at 95°C, followed by denaturation step of 10 seconds 95°C, annealing step during 30 seconds using an appropriate annealing temperature, and an extension step at 68°C for the required amount of time depending on the size of the fragments amplified. Finally, an extension step of 10 minutes at 68°C was done. The variable PCR conditions used for each PCR are summarized in the Appendix III Table S4. Five µL of each PCR product was loaded in a 3% agarose gel in TAE Buffer. 18S and hsp70 were used as loading controls and p25, p28, dozi and uis4 were used as control genes.
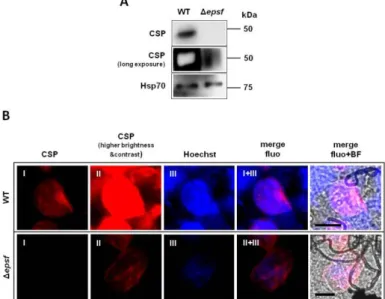
2.4.5. Western Blot analysis of CSP expression in oocysts from dhhc10-KO and epsf-KO parasite lines. To determine circumsporozoite protein (CSP) expression in oocysts from
dhhc10-KO and epsf-KO parasite lines, WT (820cl1m1cl1), dhhc10-KO (PbA2097cl1) and
epsf-KO (PbA2099cl1m7) infected midguts were dissected at day 13 p.i. and resuspended in 1X Laemmli Buffer. DTT was added to a final concentration of 200 mM and samples were boiled for 10 minutes at 95°C. Samples were loaded and ran the necessary time to obtain the desired band separation at the molecular weights of interest, CSP and Hsp70 (loading control) expected molecular weights were about 50kDa and 70kDa, respectively. Afterwards, the proteins were transferred to nitrocellulose membranes for 1 hour and 45 minutes at 200mA. Nitrocellulose membranes were then blocked for 1 hour at RT with 5% skim milk in PBS/Tween 20 0,05% and probed overnight at 4°C with 1:9000 CSP and 1:1000
Anti-Hsp70 in blocking solution in the case of dhhc10-KO line, and in the case of epsf-KO line, probed with 1:18000 Anti-CSP and 1:1000 Anti-Hsp70 in blocking solution. After 6 short washes with 0,05% PBS/Tween 20, membranes were probed with HRP-conjugated anti-mouse antibody for 1 hour at RT, 1:5000 for Hsp70 and 1:10000 for CSP, in the case of
dhhc10-KO line, and in the case of epsf-KO line, 1:5000 for Hsp70 and 1:20000 for CSP.
Finally, membranes were washed with PBS/Tween 20 0,05%, and developed using Immobilon® Western Chemiluminescent HRP Substrate (Millipore®).
2.4.6. Live imaging and immunofluorescence assays (IFAs) of blood stages, ookinetes, oocysts and sporozoites. Live imaging of blood stages and blood meal ookinetes of the different GFP-tagged parasite lines was done by collecting infected red blood cells (iRBCs) from infected mice and by collecting blood meals from infected mosquitoes, incubating with 1 ug/mL of Hoechst-33342/PBS and visualizing under a Leica DM5000B fluorescence microscope. IFAs to detect GFP-tagged protein expression and localization in blood stages, oocysts and sporozoites of the different GFP-tagged parasite lines used in this study were done using rabbit polyclonal anti-GFP 1:100-1:500 as primary antibody and goat anti-rabbit IgG-Alexa Fluor®488 1:400-1:500 as secondary antibody. IFAs to detect CSP expression and localization in WT (820cl1m1cl1) and dhhc10-KO oocysts, as well as in dhhc2 and dhhc10::gfp sporozoites were done using mouse anti-CSP as primary antibody and goat anti-mouse IgG-CyTM3 1:400-1:500 as secondary antibody. In all IFAs, the RBCs were previously washed twice with 1X RPMI (GIBCO) with 8-min spins at 2000 rpm at 37°C, and resuspended in 1X PBS at 37°C. Samples were fixed with 4%PFA/PBS for 10 min at RT, permeabilised with 0.1-0.5% TritonX-100/PBS (for 10 minutes at RT) and blocked for 1h at RT with 1-3% BSA/PBS. All antibody incubations were done in blocking solution for 1h at RT and 1-5 ug/mL of Hoescht-33342/PBS was used to stain nuclei; 3X washes with 1X PBS were done between incubations. Fluoromount-G™ (SouthernBiotech, U.K.) was used to mount the coverslips. Images were taken with a Leica DM5000B or Zeiss Axiovert 200M fluorescence microscope and processed using ImajeJ 1.47n software (imajej.nih.gov/ij). 2.4.7. Statistical methods. Statistical analysis of oocyst numbers per mosquito midgut, sporozoite numbers per mosquito and oocyst diameters was performed using Mann-Whitney test as part of Prism software package 5 (GraphPad Software).
3 – Results and Discussion
3.1. Immunoprecipitation of PUF2 Protein in P. berghei gametocytes
To study stage-specific expression of P. berghei PUF2 protein in gametocytes, immunoprecipitations of PUF2::GFP fusion protein from Nycodenz-purified gametocytes
were performed. Western blot analysis of the total gametocyte lysate input, the specific fraction (GFP) and control eluates (c-myc and beads fractions), showed that the protein could only be detected in the input (Fig. 4). A band was observed in the input fraction between 72 kDa and 100 kDa that could potentially correspond to PUF2::GFP detection (which is expected to be around 83 kDa). No such putative PUF2::GFP protein could be detected however in the specific immunoprecipitated samples (GFP fraction). A total of 5 independent IP experiments were performed to verify the consistency of the results obtained using the Nycodenz method to purify gametocytes. In two out of five experiments (data not shown), a putative PUF2::GFP protein was obtained in the input fraction, but not on the specific eluate of the GFP fraction, similar to the result on Fig. 4; in the remaining three experiments, PUF::GFP was not detected neither on the input fraction nor on the GFP-fractions. The unsatisfactory results obtained led to the following hypotheses: a) faulty execution of the protocol, or b) the IP method is not suitable for PUF2 protein in gametocytes.
Fig. 4 – Western blot analysis using rabbit anti-GFP (Invitrogen) of one independent PUF2::GFP IP experiment in P. berghei gametocytes purified by the Nycodenz method
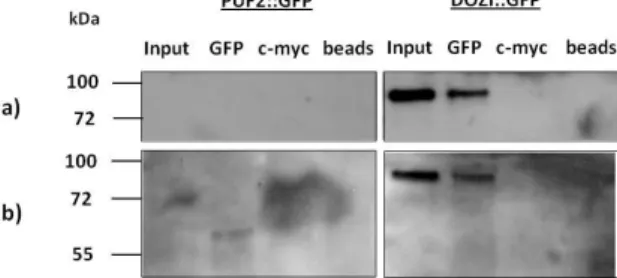
To verify if the protocol was being correctly performed, a control IP was done in parallel with PUF2::GFP, using the Nycodenz method (Fig. 5. a)). DOZI::GFP protein has been demonstrated to be successfully immunoprecipitated in gametocytes [36]. As depicted in Fig. 5. a), no PUF2::GFP protein could be detected in neither the input nor the GFP fractions, while our control line immunoprecipitated successfully, with DOZI::GFP being identified both in the input fraction and in the specific GFP eluate (~80 kDa) (Fig. 5. a)). This led us to hypothesize that maybe the gametocyte purified fraction obtained using the Nycodenz method had an insufficient amount of material for us to be able to consistently detect PUF2::GFP by Western blot analysis. To enhance gametocytemia in the PUF2::GFP infected mouse, and subsequently in the IP starting material, phenylhydrazine was injected 2 days before infecting the mice with puf2::gfp and dozi::gfp (as control) parasite lines, as previously described [51]. Starting from day 3 post-infection sulfadiazine was added to their drinking water [51]. Phenylhydrazine stimulates erythropoiesis and consequently, enhances parasitemia early after infection. Sulfadiazine was used to suppress proliferation of asexual stage parasites. The Western blot results show that again DOZI::GFP protein was identified both in the input fraction and in the specific immunoprecipitate GFP eluate (Fig. 5. b). No PUF2::GFP corresponding band was detected in any of the fractions (Fig. 5. b)). The results suggest that this method, used with the intent of enhancing gametocytemia, did not improve
the amount of IP starting material. This strategy was therefore not successful in increasing the detection of PUF2::GFP expression in purified gametocytes.
Fig. 5 – Western blot analysis of PUF2::GFP IP experiment in parallel with DOZI::GFP IP (control line) in P. berghei gametocytes purified by the Nycodenz method. a) Nycodenz method only; b) Nycodenz method with enhanced gametocytemia.
Another approach used to overcome the unsuccessful immunoprecipitation of PUF2::GFP in gametocytes, was to perform the IP using the commercially available GFP-Trap® Kit, a kit used specifically for immunoprecipitation of GFP-fusion proteins. This kit contains a small GFP-binding protein covalently coupled to the surface of agarose beads. In the previous method, the parasite material was first incubated with the anti-GFP antibody, and afterwards the resulting complex PUF2::GFP/anti-GFP was incubated with beads. In this kit, the GFP-binding protein is already coupled to beads, so the complex GFP-binding protein/beads is directly incubated with the parasite material, saving time, extra handling of the samples and ideally increasing the binding events and their stability. Immunoprecipitations of PUF2::GFP and DOZI::GFP fusion proteins were performed using mixed blood stages lysates (without gametocyte Nycodenz purification). Western blot analysis of total mixed blood stages input, bound fraction (to the beads) and non-bound fraction was performed using a mouse anti-GFP antibody (Roche). Unfortunately this antibody gave a strong unspecific band pattern especially around 55 kDa (Fig. 6), which most likely corresponds to the heavy chain of the antibody. Still, from this western blot, it can be concluded that DOZI::GFP could easily be identified in input and bound fraction, while no specific PUF2::GFP protein could be detected. The strong unspecific band pattern (Fig. 6) masks the signal that could potentially correspond to PUF2::GFP protein in the input sample, as a band between 72-100 kDa (the expected size for this protein is 83 kDa) can be detected; however the fact that this band is not present in the bound fraction, while it is detectable in the non-fractions of both IPs, argues that it is most likely non-specific and unrelated to PUF2::GFP. Having a GFP-binding protein/beads complex would theoretically improve the IP efficiency, as it significantly raises the chances for PUF2::GFP to “find” its binding partner. However, this was not the case as PUF2::GFP was not successfully immunoprecipitated using this strategy.
Fig. 6 – Western blot analysis of PUF2::GFP and DOZI::GFP IPs in mixed blood stages using GFP-Trap® Kit. Non bound fraction abbreviated as Non-B.
3.1.1. puf2::gfp sequencing
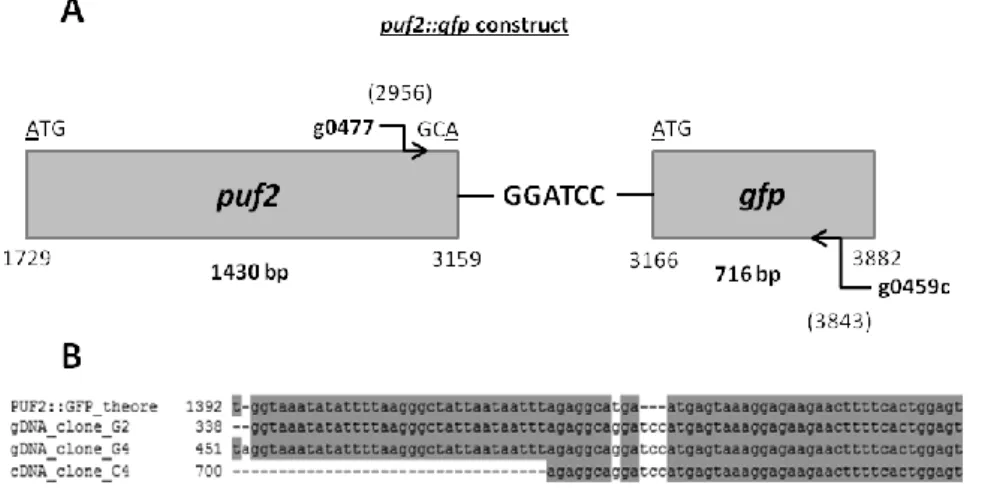
While dealing with the inability to immunoprecipitate PUF2::GFP in gametocytes and mixed blood stages, another hypothesis emerged: what if puf2::gfp parasite line was not expressing a functional PUF2::GFP fusion protein as expected? To verify this hypothesis, two different types of samples were sequenced, puf2::gfp genomic DNA (gDNA) and cDNA prepared from RNA isolated from mouse infected with the PUF2::GFP parasite line. The primers used and their annealing positions are depicted in Fig. 7. A. puf2::gfp construct had been designed changing the TGA linker to GGATCC, creating a BamHI restriction site (Fig. 7). Two single mismatches were found in the gfp gene, one synonymous at position 3529 and one at position 3396 that changes a methionine (ATG) to an isoleucine (ATA). Since the
gfp reading frame is still maintained and the mutations found most likely have a minor, if at all,
effect in the overall folding/functioning of the protein, we concluded that the puf2::gfp parasite line used so far (1750cl4) should express a functional PUF2::GFP fusion protein.
Fig. 7 – puf2::gfp construct sequencing. A. Schematic diagram depicting puf2::gfp construct, showing the sequencing primers (g0477 X g0459c), their annealing position (2956 and 3840, respectively), and the GGATCC linker region between
puf2 and gfp. B. Alignment of
puf2::gfp theoretical construct
(denoted as PUF2::GFP_theore), and the sequenced clones (two gDNA clones (denoted as gDNA_clone_G2 and gDNA_clone_G4) and one cDNA clone (cDNA_clone_C4), focusing only on the linker region.
3.1.2. Discussion
Sexual development is a decisive point in the Plasmodium life cycle. In contrast to the rigid determined progression in all the other stages [52], commitment to gametocytogenesis is a process characterized by plasticity and in addition regulated by several environmental factors [46]. Still, the molecular mechanisms governing sexual development and differentiation remain to be elucidated.