34. N. petragnani e J.V. Comasseto, "Synthetic App1ications of Te11urium Reagents". A ser publicado na Revista Synthesis.
35. K.J. Irgo1ic, P.J. Busse e R.A. Grigsby, J. Organometa1. Chem., 88, 175 (1975).
36. J. Liesk, P. Schu1tz e G. K1ar, Z.Anorg.Allg.Chem., 435, 98 (1977).
37. Trabalho em desenvolvimento em colaboração com Prof. J.T.
B . F e r re i r a (O. Q . - U FS C a r) .
38. L. Tschugaeff e W. Chlopin, Chem.Ber., lZ., 1269 (1914). 39. G. Vicentini, Chem.Ber., 2.l, 801 (1959).
40. S. Uemura, 5.1. Fukuzawa e A. Toshimitsu, J.Organometal. Chem., 250, 203 (1983).
41. S. Uemura ,e S. r. Fukuzawa, J.Am.Chem.Soc., 105, 2748 (1983).
42. S.V. Ley, C.A. Meerholz e O.H. Barton, Tetrahedron, lZ,
213 (1981).
43. L. Engman e M.P. Cava, Chem.Commun., 164 (1982). 44. H.J. Reich e S. Wo11owitz, J.Am.Chem.Soc., 104,7051
(1982).
45. N. petragnani e G. Vicentini, Univ. são Paulo, Faculdade
de Filosofia Ciências e Letras, Boletim n9 249 - QUlmi-ca n9 5, 75 (1959).
-46. P. Travornyutikarn e W.R. McWhinnie, J.Organometal.Chem.,
.§.Q, 135 (1973).
47. D. Bendi x, Tese de Doutoramento, Technischen Hochschule Aachen, Alemanha Ocidental
-
1981.48. K.J. Irgolic e R.A. Zingaro, Organometallic Reaction, Volo 2 John Wiley e Sons, Inc., 1971.
49. J.V. Comasseto, J.T.B. Ferreira e J.A. Fontanillas, J. Organometal.Chem., (no prelo).
50. W. Farrar, Research., 4, 177 (1951).
51. C.A. Brandt, J. V. Comasseto, W. Nakamura e N. Petragnani,
J.Chem.Research(S), 156 (1983).
52. J.V. Comasseto, J. Organometa1.Chem., 253, 131 (1983).
53. E.V. Dehmlow e S.B. Naranjo, J.Chem.Research(S)., 142
(1981).
54. a)M. Delmar, Y. Le Bigot e A. Gaset, Tetrahedron Lett.,
4831 (1980);
b)J. Villieras, M. Rambaud-e B. Kirschleger, Phosphorus
and Sulfur., li, 385 (1983).
55. LV. Dehmlow e S.B. Naranjo, J.Chem.Research(S)., 143
(1981 ).
56. W. Tagaki, I. Inone, Y. Yano e T. Okonogi, Tetrahedron
57. J.1.G. Cadogan (Editor) 1I0rganophosphorus Reagents in
Organic Synthesisll, Academic Press, New York, 1979.
58. J.V. Comasseto e C.A. Brandt, J.Chem.Research(S).,
~
(1982).59. J. V. Comasseto e N. Petragnani, J.Organometa1.Chem., 152,
295 (1978).
60. a) C.A. Brandt e J. V. Comasseto, Comunicação ã IIFifth
Internationa1 Conference on Organic Synthesisll
-Freiburg
-
BRD- 1984.
b)C.A. Brandt, Dissertação de Mestrado, prevista para
1985. Orientador : J. V. Comasseto.
61. S.R. Buzi10va, L.I. Vereshchagin, 1.D. Sedekov e V.I.
Minkin, ZH.Obshch.Khim., 46, 932 (1976).
62. S.R. Buzi10va, 1.D. Sedekov, LV. Lipovich, LM.
Fi 1i ppova e L. I. Vere s h c ha 9 i n, ZH. Obs c he i Khi m., 47,
1999 (1977).
63. A. Toshimi tsu, S. Uemura e M. Okano, Chem. Commun., 965
(1982).
CAPITULO11
I - INTRODUÇAO
Reação entre uma ilida de fósforo (I) e um compos-to carbon;lico (11) constitui um dos métodos mais utilizados para a formação de duplas ligações carbono-carbono com aumen-to de cadeia 111. Supõe-se que a reação se processa através do mecanismo mostrado no esquema 1, envolvendo uma beta;na
(111) e uma oxifosfetana (IV). A força propulsora da re a çao
é a formação de ligação fósforo-oxigênio (150 kcaljmol).
Esquema 1
R3P-CRlR2 I~I O~CR3R4
~ R1R2C=CR3R4 + R P=O 3
( I V)
Quando Rl ou R2 é um grupo capaz de estabilizar ca.!:
gas negativas, a ilida de fósforo (ou fosforana) é chamada de fosforana estabilizada e reage com alde;dos fornecendo
olefi-nas de configuraçãopreferencialE. No entanto, quando Rl e
+
+
R3P-CR1R2 + R3 R4 CO
R3P-CRlR2
>
Õ-R3R4
( I ) ( II)
>
R2 são ambos grupos atraentes
de
elétrons, a reação de Wittig normalmente não se processa, pois o carbânion (V) não é sufi-cientemente nuc1eõfi10 para atacar o carbono carboní1ico.+ /X R
P-C-3
"V
( V)
X,V = -C02R, -COR, -CN, etc.
Essas fosforanas, altamente estabilizadas, sao, e~ tretanto, de grande uti 1idade em sintese orgânica, conforme PE. demos observar no esquema 2, onde são mostradas as aplicações sintéticas de fosforanas estabilizadas, tando das que re agem como das que não reagem via reação de Wittig, devido a e1eva-da estabilização. As aplicações sintéticas dessas espécies
foram recentemente revisadas 121.
As fosforanas estabilizadas são, via de regra, pr~ paradas através de uma reação de transi1idação descoberta por Bestman em 1960 13,41, a qual passamos a comentar.
11
Sai s de fosfôni o se comportam como âci dos de Brons-red, sendo as fosforanas suas bases conjugadas (Equação 1).
( 1 ) -HX
+ 1 ...
R3P=CHR1
R3PCH2R
I X-
...-<l>l=CRl 0=t-R2
t:.
1 - 2
R -c =C-R 18,10-161
R1CH2COR2 15,17,71 R1CH2COR2 1181 R1CH2COR2 1"191 C1 I R1_CHCOR2 1201 R3 CH=CHR1 I 0=C-R215,171 R3CH=C-X I 0=C-R2 120,241 R3CH2COCOR2 1251 (Rl = OCH3) R2COCOR1 1261
R2COCOOCH3 127,281' ( R1 = OCH3 ) RCOCOS4>1261 (R 1 = S4»
R1CH2C02H 130,311 R2 = OR
R3CH=CR1(C02H) /31,331 R
+ 1 ~3P-C-R 1I
X-0=C-R2 121
,
H20
Zn
1) HX ,
2) eletrõlise
1) 4>1C12 2) Na2CO/H20
R3CHO ,
1) 4>1C12 .... 2) R3CHO R3CHO , 101 , 101 ... 101 , 1) H20/0H-2) H+
1) R3CHO
-2)A força ácido-base dos sais de fosfônio e das
fos-foranas correspondentes depende principalmente do
substituin-te Rl. Grupos atraentes de elétrons aumentam a acidez do
sal de fosfônio e diminuem a basicidade da fosforana.
Em vista dessa relação ácido-base entre sal de fo~
fônio e fosforana, podemos escrever o seguinte equi l1brio (Equ~
çao 2).
R3P=CHRl +
+ +
I
2
'
---:::"
11-R3P-CH2R
X ~ R3PCH2R Ix + R P=CHR23 ( 2 )A posição de equilibrio serã determinada pela nat~
reza dos grupos Rl e R2. Se a diferença de acidez entre o
primeiro e o segundo sal de fosfônio, ou a diferença de
basi-cidade entre
a
primeira e a segunda fosforana for muitopro-nunciada, o equilibrio sempre favorecerá a formação da fosfo-rana menos básica e do sal de fosfônio menos ácido (Equação 3).
<P3P=CH2 +
+
+-1<P3PCH2CO<PIBr-
~
1<P3PCH3IBr + <P3P=CHCO<p ( 3 )Esse processo é denominado reação de transilidaçã~
A
nucleofilicidade das fosforanas revela-se naoapenas nas reações com compostos carbonilicos, como também com
outros reagentes eletrofilicos, formando s a i s de fosfônio.
Quando o sal de fosfôni o apresentar grupos atraentes de
de ocorrer, originando-se uma fosforana substituida (Equação 4~
O
2
~
CH-NHR - C"'" N/ I
"'-..CH=CH
16, 7I
;O R1
- C~
"-1 O
R -C/
~
O18I
;ha letos organometãl i cos ou não metãli cos 191
Uma das propriedades das fosforanas estabilizadas
e sua decomposição para fornecer acetilenos e trifenilfosfinõ
xi do (Esquema 3) 18,10-161. Supõe-se que a decomposição das fosforanas se processe através de uma reação de Wittig
intra-molecular, conforme mostrado no esquema 3.
Esquema 3
cjJ3P=C-Rl
1
O = C-R
+
-
1cjJ P-C-R
3 1
O=C-R
+ 1
cjJ
P-C-R
3(- "
0- C-R
~cjJ3P-C-R1
I 11
O-C-R
+--+ ~
~ R -C=C-R1 + cjJ P=O
3
1
R = -C02R, Ar, CN
+ <J>lCHR +
-<J>l=CHR+ EX I<J>lCHREIX-
>
l<J>lCHlIX + cjJ3P=CRE( 4)O O
EX = Rl-c' 12,3,4,51; Rl_C 15I;
Esta reação limita-se ã preparação de acetilenos dissubstituídos, nos quais Rl é um grupo atraente de elétrons. Portanto, acetilenos terminais ou dissubstituídos alquílicos não eram acessíveis até há pouco tempo por esse método. Re-centemente descrevemos uma generalização desta metodologia, a qual permite agora a preparação de qualquer acetileno.
Sabe-se que o selênio é capaz de estabilizar carbâ
ni ons em Ct 134 I
.
Ao mesmo tempo, selenofosforanas (VI) ese-lenofosfonatos (VII) (carbânions estabilizados por fósforo e
por selênio) são objeto de estudo de nosso laboratório 19,35,
361.
./" SeAr
4>3P
=
C"
R
o
/I
(E tO) 2-P CHR( Se 4> )
( VI) (VII)
t
de se supor que, em vista da capacidade doselê-nio em estabilizar carbânions, essas espécies possam s of re r
fragmentação pirolítica, fornecendo seleno acetilenos (VIII),
conforme mostrado no esquema 4.
Esquema 4
4> P=C-SeA
3 I
r
O=C-R ~
4> P-C-SeAr
36~~- R 4 R-C=C-SeAr
Se1enoaceti1enos (VIII) podem reagir com a1qui1-1i tio fornecendo os aceti1etos de 1itio correspondentes, atra-ves de uma reação de transmeta1ação. Os aceti1etos de 1itio assim obtidos poderiam reagir com e1etrõfi10s, fornecendo ac~ tilenos terminais ou a1qui1dissubstituidos (IX) (Esquema 5).
Esquema 5
R-C=C-SeAr
n-BuLi
~ R-C=C-Li + BuSeAr
(VIII)
tE+
R-C=C-E( I X)
Isso tornaria a pirõ1ise de fosforanas um me t o do geral de obtenção de aceti 1enos.
11 - DISCUssAo DOS RESULTADOS
As se1enofosforanas de partida (XII) foram prepar~ das por uma reação de transi1idação entre uma fosforana esta-bilizada (X) e um brometo de arilselenenila apropriado
(Equação 5). Essa reação se processa rapidamente com
(X I) bons
*
Os rendimentos se referem aos produtos recrista1izados ou
des ti 1ados.
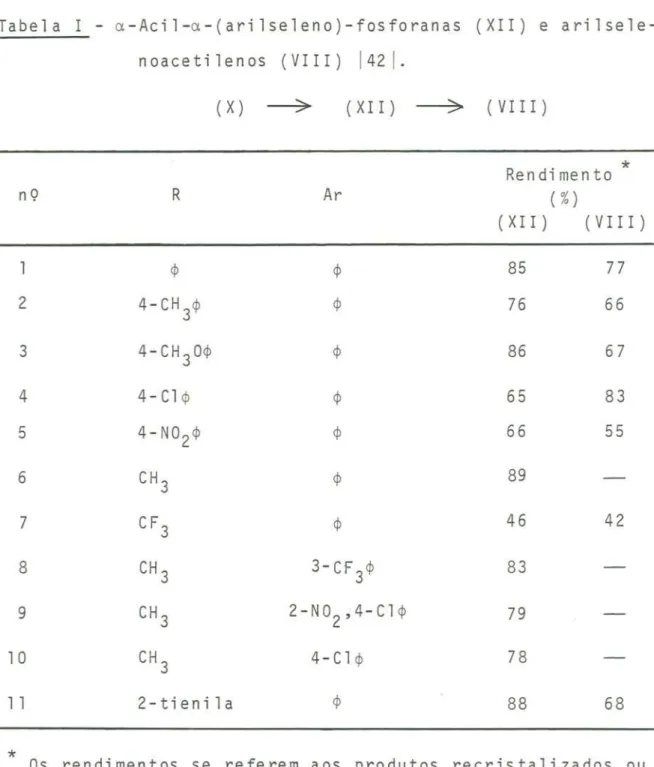
Tabela I - a-Aci1-a-(ari1se1eno)-fosforanas (XII) e ari1se1e-noaceti1enos (VIII) 142 I.
( X) (XII)
>
(VIII)*
Ren di men to
nQ R Ar ( %)
(XII) (VIII)
<p <p 85 77
2
4-CH3<P <P 76 66
3
4-CH 3O<P <P 86 67
4 4-C1<p <P 65 83
5 4-N02<P <P 66 55
6
CH3 <P 89
-7
CF3
<P 46 428 CH3
3-CF3<P 83
9 CH3 2-NO2''4-C1<P 79
10 CH3 4-C1<p 78
2<P3P=CH
I
+ O=C-R( X)
ArSeBr ~ <p3P=C-SeAr
I
+ <P +3
PCH2cORIBr-O=C-R ( 5 )
( XI )
Este metodo não se mostrou adequado para a prepar~
ção da selenofosforana na qual R
e
o grupo trifluorometil (co.!!!posto 7, Tabela I).
Nesse caso, certamente,
o carbânio
não e
suficientemente reativo para atacar o haleto de selenenila. A
fosforana desejada foi obtida reagindo-se a selenofosforana
não estabilizada (XIII) 191 com
(Equação 6) 1421.
o anidrido trifluoroacetico
2<P3P=CHSe<p+ CF3CO)20 ~
<PP=C-Se<p +
-3
I
+ <P PCH2se<PIF3CC02O=C-CF 3
3
Quando submetidas
ã
pirõlise sob vãcuo, àsseleno-fosforanas
estabili
zadas (XII) decompõem-se fornecendo
os aril
( 6 )
seleno acetilenos (VIII) e trifenil fosfinõxido (Equação 7).
Os rendimentos da reação de fragmentação são satisfatõrios (T~
be 1a I).
(XII) 2100C/5 X 10-3 mmHg~
<P P=O
3
(VIII) (7)
Examinando a Tabela I observamos que quando R
e
ogrupo metila, a reação de fragmentação não leva ao acetileno,
be-se que fosforanas estabilizadas (V), onde Y
-COR(R=alquila) e X um substituinte fortemente
e um grupo
atraente de
eletrons, fornecem acetilenos quando submetidas
-
a pirolise.Isso nos levou a crer que a substituição do grupo fenila lig~
do ao sel~nio por grupos arila contendo substituintes
atraen-tes de eletrons poderia promover a fragmentação das
arilsele-nofosforanas (XI I), mesmo quando R fosse um grupo alquila (Equ~
ção 8).
( 8 )
Mesmo nesse caso nao obtivemos os selenetos
aceti-l~nicos alqUllicos.
Conforme já notamos (Esquema 4), os selenetos
ace-ti l~nicos podem sofrer uma reação de. transmeti lação quando re~
gem com butil litio, fornecendo os acetiletos de lltio corres
pondentes, que ao reagirem com eletrofilos apropriados, forn~
cem os acetilenos terminais ou dissubstituldos em bons
rendi-mentos, como mostrado na Tabela 11.
Alem do uso como equivalentes sinteticos de
aceti-letos, os selenoacetilenos podem ser transformados facilmente
em selenetos vinllicos de configuração
Z
(XIV) 137/ ou E (XIV)cp P=C-SeAr
*
3 I
CH3-C:::C-SeAr O=C-CH
3
Tabela II - Acetilenos (IX) 142).
* Rendimento
n9 R Eletrôfilo ( %)
A B
1 cp H20 72 81
2 cp CH3I 68 82
3 cp
CH3COCH3 69 79
4 cp
CH3COCl 51 77
5 cp
C02 68 87
6 4-CH3CP C02 78 85
7
4-CH30CP C02 76 85
8 4- C1cP C02 73 82
9 4-N02CP C02 - 56
10 CF3 C02 - 52
11 2-tienila C02 77 81
a) A = rendimento de acetileno.
B
= rendimento de seleneto de fenil butila.1381 conforme mostrado no Esquema 6. Selenetos vinilicos sao intermediãrios sintéticos muito versãteis
1391.
Esquema 6
2) CH3C02H
R,
C=C
/Secp
H/
'H
1)
(c-C6Hll )2BH(XIV)
R-C=C-Secp
LiA1H4/THF refl uxo
R,
C=C/H
H/ 'Secp
(XV)
Te n d o e m v i s ta que s e 1 e n o f os f o r a nas (X I I) nas q ua i s R é um grupo alquila não fornecem acetilenos quando submeti das ã pirõlise e que o enxofre possui uma maior capacidade de estabilizar carbãnions relativamente ao selinio 1341,
decidi-mos preparar tiofosforanas estabilizadas (XVII) e submetê-las
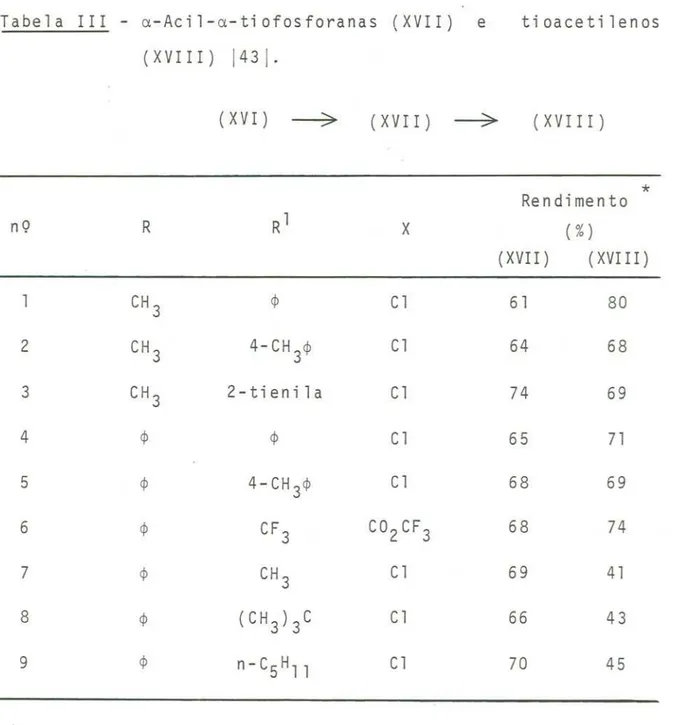
As tiofosforanas estabilizadas (XVII) foram
prepa-radas por uma reação de transilidação entre a tiofosforana
(XVI) e os cloretos ou anidridos de ãcido apropriados (Equa-ção 9). Os rendimentos se encontram na Tabela III.
2~3P=CRSR + R1COX (XVI)
benzeno
~
P=C-SR 3 I 1O=C- R +
~3PCH2-SRIX-(9)
~
temperatura ambiente(XVII)
Quando aquecidas a -2300C sob pressão de 5 X 10-3 mmHg as tiofosforanas (XVII) fornecem tiocetilenos (XVIII) com rendimentos satisfatõrios, mesmo quando Rl e um grupo alquila (Equação 10, Tabela III).
(XVII)
-2300C/5 X 10-3 mmHg ~
-~3P=0
R l_C=C-SR (XVIII)
( 10 )
Tioacetilenos sofrem reação de transmetalação com n-butil-1Ttio, fornecendo acetiletos de lTtio, os quais podem ser capturados com eletrõfilos, fornecendo acetilenos te rm i
-nais ou alquil dissubstituTdos. Por outro lado, sulfonas ace tilênicas, facilmente obtidas por oxidação dos sul fetos
respondentes 1401, reagem com alqui 1 lTtio ou reagentes
cor-de
Grignard, fornecendo diretamente o acetileno dissubstituTdo
Tabela III
- a-Aci1-a-tiofosforanas
(XVII)
e tioaceti1enos(XVIII) 1431.
(XVI)
~
(XVII) ~ (XVIII)*
Os rendimentos se referem aos produtos recrista1izados ou
destilados.
* Ren di men to
nQ R R1 X ( %)
(XVII) ( XVIII )
1 CH3 <P C1 61 80
2
CH3 4-CH3<P C1 64 68
3 CH3 2-tieni1a C1 74 69
4 <P <P C1 65 71
5 <P 4-CH3<P C1 68 69
6 <P
CF3 C02 CF3 68 74
7 <P CH3 C1 69 41
8 <P
(CH3)3C C1 66 43
9 <P
Esquema 7
El
)
~ 1R-C=C-S-R
Ir
O
2
R MgX ou '> R2Li
O
- 2 1II
R-C=C-R + R
S-II
O
101
R-C=C-S-Rl
j
Nu
n-BuLi
:::.. R- C=C- Li + BuS R1
tE+
R-C=C-E
Pelo esquema 7 pddemos notar que o carbono acetil~
nico ligado ao enxofre pode agir formalmente como centro
nu-cleofilico ou eletrof;lico. Esse fato torna os sul fetos ace-tilênicos intermediârios de grande versatilidade para a forma ção de ligações carbono-carbono.
Atualmente uma extensão dos trabalhos descritosaci
ma se encontra em desenvolvimento em nosso laboratório.
Rea-ção de transilidaRea-ção entre cloretos de diâcidos carbox;licos
e seleno- ou tiofosforanas leva is bis-fosforanas (XIX) (Equ~
çã o 11). Pirõlise das mesmas levaria aos bis-chalcogeno - di~
o
,
O
)
-C~
+ <P3P=CHXR/C-(CH2 n
""-Cl Cl
~
o
O
11 . 11
<PP=C-C-(CH ) -C-C=P<P
3 I
2 n
I 3XR XR
(XIX)
-
---->
RX-C=C-(CH ) -C=C-XR2 n(XX)
( 11 )
X = S, Se
Os chalcogenoacetilenos (XX), por tratamento com n-butil-litio gerariam os acetiletos de li ti o correspondentes
~
ri-..
~
(Equação 12). Esses acetiletos poderiam ser utilizados na
sintese de produtos naturais poliacetilênicos 1441.
(XX)
n-BuLi ~
Li-C=C-(CH2)n-C=C-Li + BuXR (1 ê )
X = S, Se
Outra possibilidade seria efetuar a reação de tran
silidação com anidridos de ãcidos dicarboxilicos, capturando
a fosforana
-
carboxilato com sulfato de di metil a (Esquema 8).A fosforana
resultante
(XXII) ao ser submetida
ã
pirõlise fo~ neceria o acetileno (XXIII), o qual, por tratamento com um al quil-litio, seguido de protonação forneceria acetona acetilênica (XXIV). Hidrõlise de (XXIV) seguida por reação com hidrõ
BIBLIOTECA
Instituto de Química
xi do de sõdio levaria ao sistema pentagonal (XXV),
de jasmonõides e prostaglandinas.
Esquema 8
2<P3P=CHXR
/).
~
-<P3P=O
hidrõlis~
precursor
o
+ ~ <P3P=C-XR
I
O=C
~
OMeO
Me2S04
'o
(XXII)
O O
.
~OCH
l)~Li
~l
R
~
32)HO>~
R
'x/
. 2(XXIII) (XXIV)
O
~Rl
O
NaOH ~
R2
(XXV)
Uma discussão mais detalhada das reações que acab~
mos de apresentar neste capitulo foi
1451.
recentemente publicada
-Experimental : a-Acil-a-(arilseleno)-fosforanas (XII) - Pro
ce dimento Ge ra l lflJ.
.
A uma solução do brometo de aril selenenila (XI)
(530 X 10-3 mol) em benzeno (10ml) foi adicionada uma solução
da a-acil fosforana apropriada (X) (10 X 10-3 mol) em benzeno
(90ml) sob atmosfera de nitrogênio e agitação magnética. A
suspensão formada foi filtrada e o sol vente do filtrado evap~
rado.
o
res{duo foi recristalizado de acetato de etila3for-necendQ as a-acil3 a-(arilseleno)-fosforanas (XII) com os ren
dime~tos indicados na Tabela I.
a-Acil-a-(organotio)fosforanas (XVII) - Procedimento j43345L.
A uma suspensão do cloreto de (metiltio) ou (fenil
tio)meti ltrifeni lfosfônio (30
-3
X 10 mol) em benzeno (250ml)
sob atmósfera de nitrogênio3 adicionou-se n-butil-l{tio (30
X 10-3 mol). Em seguida adicionou-se o cloreto ou anidrido
de ácido apropriado (15 X 1O-3 mol). Após duas horas de agi
tação a temperatura ambiente a mistura reacional foi filtrada.
Evaporou-se o solvente e recristalizou-se o res{duo de
aceta-to de etila/éter de petróleo. Os rendimentos dos produtos re
Penilseleno acetilenos (VIII) - Procedimento Geral l.i,tl.
As a-acil-a-(arilseleno)fosforanas (XII) (2:J°X 10-3
mol) foram aquecidas num KUBelrohrofen sob vácuo (-5:J0 X 10-3
mmHg) a 2300C por 1 hora. Os fenilseleno acetilenos (VIII) e
trifenilfosfinóxido destilaram:J sendo coletados em um balão
resfriado em banho de gelo seco e acetona. A mistura foi fil
trada numa coluna curta de gel de s{lica usando éter de petr~
leo como eluente (compostos n9 1-4 e 11:J Tabela I) ou éter de
petróleojéter et{lico (8:1) (compostos n9 5 e 7:JTabela I). O
elu{do foi secado com MgS04 e o solvente evaporado:J
fornecen-do os fenilseleno acetilenos (VIII) com os rendimentos indica
dos na Tabe la I.
Feniltio- e metiltio acetilenos (XVIII) - Procedimento Geral
J43:J45L.
Seguiu-se o mesmo procedimento descrito para a
ob-tenção dos fenilseleno acetilenos. A purificação foi
efetua-da por redestilação da mistura de tioacetilenos e trifenilfo~
finóxido a uma temperatura inferior à temperatura da pirólise.
Os rendimentos dos tioacetilenos destilados se encontram na
CZivagem dos seZeno- e tioacetiZenos - Preparação de
acetiZe-tos de Z{tio - Procedimento GeraZ 142,.43,.45 L.
n-ButiZ-Z{tio U",05 X 10-3 moZ) foi adicionado a
uma soZução do feni Zse Zeno- (XIII) ou feni Ztio- acetiZeno(XVIII) (1",0 X 10-3 moZ) em tetra-hidrofurano (5mZ) sob nitrogênio a
-? aO c. Após a adição deixou-se a mistura atingir a temperat~
ra ambiente. A seguir a soZução do acetiZeto de Z{tio assim
preparada foi tratada com eZetrófiZos", fornecendo os
REFERtNCIAS
01. J.I.G. Cadogan (Editor) "Organophosphorus.Reagents in
Organic Synthesis", Academic Press, New York, 1979.
02. H.J. Bestmann e R. Zimmermann, in "Carbon-Carbon Bond Forma t i on ", Vo1. 1. Edited by R.L. Augustine, Marce1 Dekke r, I nc. New York, 1979, p. 353.
03. H.J. Bestmann, Chem.Ber.,~, 58 (1962).
04. H.J. Bestmann, Tetrahedron Lett., 7 (1960).
05. H.J. Bestmann e B. Arnason, Chem.Ber., 95, 1513 (1962). 06. H.J. Bestmann, N. Sommer e H.A. Staab, Angew.Chem.lnt.Ed.,
1, 270 (1962).
07. H.A. Staabe N. Sommer, Angew.Chem. Int.Ed., 1, 270 (1962). 08. P. A. Chopa r d , R. J . G. Se a r 1e e F. H. Devi t t, J. Or 9 . Chem. ,
lQ, 1015 (1965).
09. N. Petragnani, R. ROdrigues e J. V. Comasseto, J.Organometa1. Chem., 114, 281 (1976) e referências citadas.
10. S.T.D. Gough e S. Trippett, J.Chem.Soc., 2333 (1962).
11
11. G. Mark1, Chem.Ber., 2.i, 3005 (1961).
14. H.J. Bestmann, C. Geismann, Justus Liebigs Ann.Chem., 282 (1977).
15. Y. Kobayashi, T. Yamash ita, K. Takahashi, H. Kuroda e I. Kumasaki, Tetrahedron. Lett., ~, 343 (1982).
1 6. Y. Z. .H u a n g, Y. S h e n, W. O i n 9 e J. Z h e n g, Te t r a h e d r o n Le t t.,
22,5883 (1981).
17. F. Ramirez e S. Oershowitz, J.Org.Chem.,~, 41 (1957). 18. S. Trippett e O.M. Wa1ker, J.Chem.Soc., 1266 (1961).
19. L. Horner e A. Mentrup, Liebigs Ann.Chem., 646, 65 (1961).
20. E. Zbira1 e M. Rasberger, Tetrahedron., ~, 1871 (1969).
11
21. G. Mark1, Chem.Ber., 94, 2996 (1961).
n
22. G. Mark1, Chem.Ber., 95, 3003 (1962).
23. O.B. Oenneye St. 1. Ross, J.Org.Chem., ~, 998 (1962). 24. H.J. Bestmann.e R. Armsen, Synthesis., .§.2. (1970).
25. E. Zbira1, Tetrahedron Lett., 1483 (1965).
26. E. Zbira1 e E. Werner, Monatsh.Chem., 97, 1797 (1966).
27. E. Zbira1 e Rasberger, Tetrahedron., 3.i, 2419 (1968). 28. H.J. Bestmann, R. Armsen e H. Wagner, Chem.Ber., 102,
2259 (1969).
11
30. H.J. Bestmann e H. Schu1tz, Liebig.Am.Chem., 674, 11 (1964).
31. H.J. Bestmann e H. Schu1tz, Chem.Ber., 22, 2921 (1962). 32. O. Is1er, H. Gutmann, M. Montavon, R. Ruegg, G. Ryser e
P. Zeller, He1v.Chim.Acta., 40, 1242 (1957).
33. H.O. House e G. Rasmusson, J.Org.Chem., ~, 4278 (1961). 34. H.J. Reich, F. Chow e S.K. Shah, J.Am.Chem.Soc., 101,
6638 (1979).
35. N. Petragnani, J.V. Comasseto, R. Rodrigues e LJ. Brock-som, J.Organometal.Chem., 124, 1 (1977).
36. J. V. Comasseto e N. Petragnani, J. Organometal. Chem., 152,
295 (1978).
37. S. Raucher, M.R. Hansen e M.A. Co1ter, J.Org.Chem., ~, 4885 (1978).
38. J.V. Comasseto, J.LB. Ferreira e N. Petragnani,
J.Orga-nometa1.Chem., 216, 287 (1981).
39. J.V. Comasseto, J.Organometal.Chem., 253, 131 (1983).
40. LG. Back, S. Collins e R.G. Kerr, J.Org.Chem., 48,
3077 (1983).
41. R. L. Smora da e W.E. Tr uce, J. Or 9. Chem., .ii, 3445 (1 979 ) .
42. A.L. Braga, J.V. Comasseto e N. Petragnani, Synthesis.,
43. A.L. Braga, J.V. Comasseto,e N. Petragnani, Tetrahedron.
Lett., 25, 1111 (1984).
44. F. Bohlmann, 1. Burkhardt e C. Zdero, IINaturally Occur-ring Acetylenesll, Academic Press, London, 1973.
45. A.L. Braga, "Slntese de Acetilenosll, Dissertação de
Mes-trado, apresentada ao Instituto de QUlmica da Universi
CAPÍTULOIII
TRICLORETOS
DE ARIL TELORIOCOMOAGENTESDE CICLIZACÃO DE
I - INTRODUçAO
Reações envolvendo um reagente eletrofllico e um
substrato insaturado que possui um nucleõfilo interno, sao c~ nhecidas hã muito tempo, encontrando sempre grande interesse, pois levam a sistemas tais como êsteres, ê te re s , sul fetos,
aminas e amidas clclicas .(Esquema 1). Essas reações são atual mente conhecidas como reações de ciclofuncionalização.
Esquema 1
Rl
RlrNUX
R2~ R3
+ Nu 1 ~
R3
NuX = C02H, OH, NH2' SH, SAc, NHC02R, CH2snMe3
+1 +
Nu = H 11 I; H g X2 12 I; C 12' Br 2' I2 13 I; RS H 141 ;
RSeXI5,6,7,81; RTeXnI5,91.
A rigor não podemos considerar as ciclizações pro-movidas por ãcidos como sendo uma ciclofuncionalização, pois esse termo define reações "que envolvem fechamento intramole-cular de anel, em que uma das extremidades da ligação dupla
envolvida no fechamento do anel liga-se a um grupo que
111
.
Como podemos observar no esquema 1, um grande num~
ro de reagentes e1etrofi1icos pode ser usado para essa final i
dade. o mecanismo dessas reações tem sido muito discutido
16,3,10,111. Por esse motivo, e por não ser o estudo do meca nismo das reações de cic1ofunciona1ização o objetivo de s te trabalho, não faremos maiores considerações a esse respei to. o mecanismo mais aceito para explicar essas reações e mostra-do no esquema 2.
Esquema 2
",
",
.
"
""~NUX
N~l
~
Nu1 "- ,', ti.
Tbux
Nu1
~
,>" ", "
..---Nu
+
O agente nucleofi1ico Nu1 forma um complexo w
a ligação dupla, dando origem a um intermediário cic1ico
com
de
3 memb r os. Ataque do nuc1eõfilo interno NuX a esse intermediã
trans" 1121. Por esse motivo, sempre
que se
formam an~isfun-di dos atrav~s de reações desse tipo, a fusão dos aneis
senta a configuração eis (Esquema 3).
apre-Esquema 3
H
6=:'!J1
XNu
+1
Nu ~
N 1 H
~'l~
L-~~
XNu~)
nn
H
:::.
H "l
~
Nu
H NuC
n
""
Nu
Nosso laboratõrio
há
muito tempo se dedica ao estudo de reações de ctclofuncionalização, especialmente reaçoes
de lactonização 15,131.
Em
1960 foi descoberto nestelabora-tõrio
que
brometos de aril selenenila, bem como haletos de telu rio reagem com acido difenil alil acetico (I), fornecendo
as seleno- ou telurolactonas correspondentes (II) (Equação 1)
<P <P
~C02H ( I )
:::::..
o
O
X~~
( 1 )
x-v
x = ArSe
v = Br
ArTeC12
Te C13 NaftilTe
Cl
Cl
I
Com a descoberta de métodos oxidativos 1141 ou
re-dutivos 141 de remoção do selênio de moléculas orgânicas, a
selenocicl ofunci onal i zação, passou a constituir uma transfor
mação de grande importância em slntese orgânica, tendo sido
aplicada na slntese de vãrios produtos naturais e não naturais 18,11,14/. Em vista da facilidade com que a
selenociclofun-cionalização
é
efetuada, bem como com a facilidade com que oselênio é removido do resto orgânico da molécula, essa reaçao constitui hoje o método por excelência para efetuar
ciclofun-c i o na 1 i za ç õe s
.
vã ri os reagentes de selênio tem sidoQuadro 1
Reagentes utilizados em selenociclofuncionalizações
Embora descobertas na mesma época que a selenocicl~ funcionalização, reações anãlogas envolvendo reagentes de
te-lurio 151 não receberam a mesma atenção.
Recentemente reagentes de te lu rio passaram a se r
intensamente explorados pelos químicos orgânicos sintéticos
117,181. Entre as reações estudadas se encontra a teluroci-clofuncionalização de alcoois insaturados 1191. Essa reaçao
f o i e f e tua da r e a gi n do
-
s e o ã 1coo1 i ns at ur ado (I I I) com umami ~tura de diõxido de telurio e cloreto de litio em ãcido
aceti-co. Os éteres correspondentes (IV) foram obtidos com bons re.!!.
dimentos, sendo a ligação telurio- carbono reduzida com hidre
to de trifenil estanho, o que levou aos éteres cíclicos 1i
-vres de telurio (V) (Equação 2).
o
O
<l>SeCl
O(N-se$
C>-se$
<l>SeBr
O O<l>SeOH +
-
+2
~OH
(lI!)
Te02/Li Cl
AêOH ~
o
~
o
Te~
')
C1 Cl
'0r--1
n
n
(2 )
<P3SnH ~ 2
à
( V)
11
-
TRICLORETOS DE ARIL TELDRIO COMO AGENTES DECICLOFUNCIO-NALIZAÇAO
Em vista do que expusemos no inicio deste trabalho
decidimos retomar as investigações acerca da utilização de
reagentes orgânicos de telurio como agentes de
ciclofunciona-1i za çã o
.
A primeira reação a ser investigada foi a telurolactonização de ácidos carboxilicos insaturados. Para tal utili
zamos o tricloreto de p-metoxifeniltelurio. A escolha desse
reagente se deu em vista de sua estabilidade, fácil
aquecer-mos os reagentes em clorofórmio ã temperatura de refluxo (Equa
ção 3).
R
2 CH O~TeCl /CHC13 O~Te
R1 R 3 3 :> CH3 /
\
.
R
~
n.' C02H -HCl Cl Cl ("n Rl(3)
( VI )
tativas.
Os rendimentos brutos da transformação são
quanti-No entanto perde-se material durante a recristaliza
çao. As diclorotelurolactonas (VI) são sólidos cristalinos incolores, estãveis, não higroscõpicos, podendo ser facilmente manuseados ao ar. Como podemos observar na Tabela I, a pre-sença de substituintes na posição a
ã
carbonila do ãcidocar-boxilico insaturado leva a um aumento na velocidade da rea-çao. Isso pode ser explicado como sendo devido
ã
aproximaçãodo grupo carboxila ao sitio catiõnico, causada pelas re pu1
-sões provoca das pelos grupos volumosos na posição a j31. As estruturas propostas (Tabela I) foram confirmadas pela anãli-se dos espectros de absorção no infravermelho
magnética protõnica.
e ressonância
A
segúir comentaremos os dados espectrais mais6
10Hz ArTe
/\
C1 C1
(VI.6) (ver espectro na pãgina 145)
o espectro de absorção no infraverme1ho de (VI.6)
apresenta uma banda intensa em 1780 cm-1, característica do estiramento do grupo carbonila em lactonas de 5 membros. O e~
pectro de absorção no infravermelho de todos os demais compo~
tos relacionados na Tabela I apresentam essa absorção caracte rística. Da mesma forma, os espectros de absorção no
infra-vermelho dos produtos de redução (teluretos), apresentados na
estira-mento da carbonila em anéis de 5 membros.
o espectro de ressonância magnética protônica do
composto (VI.6) apresenta dois dubletos, em 7,048 e 8,128 res pectivamente, com uma constante de acoplamento de 8Hz. Esses sinais foram atribuídos ao grupamento aromático ligado ao te lurio e são comuns a todos os compostos preparados. Em 5,288 aparece um duplo dubleto atribuído ao prõton ligado ao
carbo-*
no 8 . Esse prõton estaria acoplando com os prõtons 1 i gados aos carbonos 3 e 7. Irradiação em 3,008 (centro da banda atri buida ao prõton ligado ao carbono 3) transformou o duplo
du-bleto a 5,28 em um dubleto com constante de acoplamento de
10 Hz. Essa seria a constante de acoplamento entre os
pro-tons ligados aos carbonos 8 e 7. De posse desse dado deduzi-mos, por análise do espectro, ~ue a constante de a c o p 1 a me n to
entre os prôtons ligados aos carbonos 8 e 3 é de 8 Hz.
lores das constantes de acoplamento variam, dependendo
Os va
dos substituintes no carbono 2, bem como do estado de oxidação do
te 1 u ri o. Quando passamos de Te (IV) (composto VI.6,Tabela I)
para Te (11) (composto X.6, Tabela 11) as constantes de
aco-plamento entre os prôtons 8 e 3 e entre os prôtons 8 e 7
pas-sam a ser praticamente iguais (-5 Hz), o que está bastante
*
próximo dos valores da literatura para a lactona corresponde~
te (sem substituinte no carbono 7) com fusão de anel eis \41.
Essa mudança nas constantes de acoplamento, certam~nte
relacionada com o volume do grupo que contem telurio, o
es tã que se reflete .na conformação dos aneis, variando os ~ngulos die-dros entre os prótons e, consequentemente, afetando as cons-tantes de acoplamento. Vemos, portanto, que uma conclusão de finitiva com respeito
a
estereoquímica da fusão dos aneis soserã possível por transformação da lactona (VI.6) em composto conhecido, ou por determinação de sua estrutura por di fração
de ra i os X 119 \
.
No entanto, conforme discutimos na parte introdutõ ria, as reações de ciclofuncionalização devem ocorrer via ata que do nucleõfilo interno ao intermediário carregado positiv~ mente (VII), formado pelo ataque do agente de
ciclofunciona-lização eletrofilico (neste caso tricloreto de
p-metoxifenil-te 1Ur i o ) ~ ligação dupla carbono-carbono do substrato (Esqu~
Esquema 4
H
H
~
~~H
(VIII)
ArTeC13
Cl-I I
,
ArTeC12
~~
-oA
OH
(VII)
~
OH
-
HCl H~
HArTe ..
c{'b
H O "<::O====-"""
( I X)
Esse ataque leva a uma "abertura trans diaxialll do intermediâ
rio (VII), levando ã lactona (VIII), com fusão eis do
a qual deve adotar a conformação mais estâvel (IX).
Tabela I - Te1urocic1ofunciona1ização de ãcidos carbox;licos
insaturados 191.
a
Composto (VI) Ren di men tob( %) p.f
°c
tempo de reaçao( h )8. 84 152-154 2 , 5
a) Y
=
p-CH30~TeC12; b) Rendimento do produtorecrista-1izado (c1orofõrmio + eter de petrõ1eo 30-60).
1.
YO
87
116-120 1, O2.
y"O:o
80 120-124 1, O
3. yO 80 134-136 1, O
4. 85 176-178 1 , O
5.
10
80 124-126 4,5
Y 6.
"(-{
85 154-155 7,0Y
III - TRANSFORMAÇAODAS DICLOROTELUROLACTONAS(VI) CIES LIVRES DE TELURIO
EM
ESPt-A remoção
de
telurio de moleculas orgânicas tem si do pouco estudada. Esse aspecto'e, no entanto, fundamental quando temos em mente a utilização de um elemento nãometãli-co em síntese orgânica. A remoção poderia ocorrer por meto-dos oxidativos ou redutivos.
o
primeiro metodo envolveria a formação de um telurõxido, o qual sofreria eliminação IIS yn 11,levando a uma olefina (Equação 4).
-I <PTeOHI~
( 4 )
Essa reação, no entanto, não fornece bons resulta-dos com toresulta-dos os telurôxiresulta-dos. Telurõxidos não funcionaliza-dos fornecem mistura complexa de produtos
1201.
Recentemente foi observado que a pirõlise de telurõxidos contendo um grupo alcõxido na posição a, leva aos eteres alílicos corresponde~te s, ge r a1men te com bons r endi mentos (E sque ma 5) I 21 I .
R R
Y
NaOH>
y
H20<pTeX2 <pTe=0.H20
X=Cl,Br Rl
F
+Esquema 5
Rl R3 Rl
R R5
(<pTe)21Br2
>
CH30H
NaOH/H20 R5
Rl R3
R2,
OCH3
!::,
-
I<pTeOHI
~
R2 R5R5
OCH3
~
çao com hidretos de estanho 122,231.
A remoção por processos redu ti vos consiste na rea-No entanto-essas
ções foram todas efetua das em sistemas bastante simples, não contém grupos funcionais sensíveis.
re du
-Um estudo dessa natu que
reza seria de grande utilidade.
Com vistas a obtenção de lactonas livres de rio investigamos a reação das diclorotelurolactonas (VI) tetrahidrofurano com hidreto de boro e sadio aquoso.
te
1Li-em
de 1 eq. de (VI) com 4 eq. de hidreto de boro e sôdio leva ao
te lu reto correspondente (X) (Equação 5, Tabela II). Esses co.!!!
postos se mostraram pouco estáveis, regenerando o ácido carbo
xílico insaturado de partida e ditelureto de diarila.
NaBH4/H20 ~
I
T;HF, t. a., 10'
Ar
=
p-:-CH Ocp3
O
Rl
( 5 )
(X) R2
Esse resultado pode ser compreendido, ao conside-rarmos que cada ligação Te-Cl reduzida gera uma molecula HC1, o q ua1 de s t r ôi um i on hi dre to.
de
Cisão da ligação telurio-carbono foi possível usa~
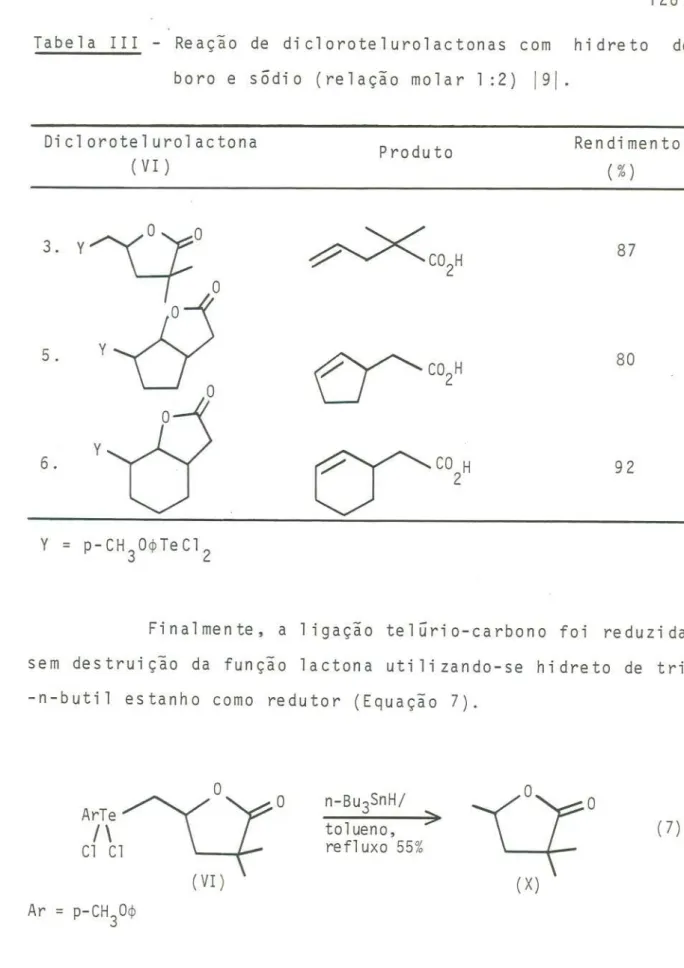
do-se 8 eq. de hidreto de boro e sadio por eq. de diclorotelu
rolactona (VI). No entanto, nesse caso, a função lactona foi
destruída, regenerando o ácido carboxílico insaturado de
par-tida (XI), juntamente com ditelureto de diarila (Equação 6 ,
Tabela III). Portanto, a telurolactonização pode ser conside
rada como um metodo de proteção de ácidos carboxilicos insaturados.
r
oo
.Rl (
R2 (VI)
R
1
2NaBH4/H20
~
THF, t.a., 1O'
R1 R2
~C02H
( XI )
+(ArTe)2 ( 6 )
Ar = p-CH30cjJ
Tabela 11
-
Reação de diclorotelurolactonascom
hidreto de boro e sadio (relação molar 1:1) 191.
Produto de redução (X) Ren di men to (%)
3.
°
y~yo
Lj-,o
91
5.
y 906. y 88
Y
=
p-CH3 OcjJTeR
ArTe
A.
/°'--_0
ClI '(1 \
f-R2
Tabela rrr - Reação de diclorotelurolactonas com h i d re to de
boro e sôdio (relação molar 1 :2)
191-Diclorotelurolactona
( Vr ) Produto
Rendimento
( %)
Finalmente, a ligação telurio-carbono foi reduzida
sem destruição da função lactona utilizando-se hidreto de tri
-n-butil estanho como redutor (Equação 7).
ArTe
:\;{
o
°
1\
Cl Cl
(vr ) Ar
=
p-CH3Ocpn-Bu3SnH/
...
tolueno, refl uxo 55%
40
(X)
(7)
3.
yO
O
C02H
87
y
80
5.
C02H
°
yJj
C02H
92
6.
Tabela IV - Dados espectrais das diclorotelurolactonas (VI).
1 1790
lHRMP
CDC13' ô(ppm), J (Hz), TMS ref.interna
1,7-3,0 (m,4H), 3,7-4,0 (m,5H)a, 5,1-5~5(m,lH),
7,03 (d,J=6,2H), 8,07 (d,J=6,2H)b
11,30 (d,J=6), 1,74 (d,J=10), 3HI, 2,1-3,2 (m,
3H), 3,5-4,1 (m,5H)a, 5,0-5,5 (m,lH), 7,02 (d,
J=8,2H), 8,02 (d,J=8,2H) VI i. v. (KBr)
\>C=O(cm-l)
2 1785
3 1790 1,30 (s,3H), 1,32 (s,3H); 1,98 (dd,J=14,9,lH),
2,38 (dd,J=12,6, lH), 3,7-4,0 (m,5H)a, 5,1-5,4
c
(m, lH), 7,02 (d,J=8,2H), 8,04 (d,J=8,2H)
2,86 (dd,J=9,7,lH), 3,36 (dd,J=9,4,lH),3,7-4,0
a .
(m,5H) ,5,0-5,4 (m,lH),7,06 (d,J=6,2H), 7,36,
b
(s,5H), 7,40 (s,5H), 8,10 (d,J=6, 2H)
4 1765
5 1775 1,2-2,0 (m,lH), 2,0-3,1 (m,6H), 3,86 (s, 3H),
4,18 (sistema ABMX,J=8,9,3; lH), 5,32 (dd, J =
7,3, lH), 7,06 (d,J=8,2H), 8,06 (d,J=8, 2H)c
1,2-2,2 (m,6H), 2,4-2,7 (m,2H), 2,7-3,2(m,lH),
3,6-4,0 (m,4H)a, 5,28 (dd,J=10,8,lH), 7,04 (d,
c
J=8, 2H), 8,12 (d,J=8, 2H)
6 1780
7 1780 1,0-1,4 (m,3H), 1,4-2,2 (m,6H), 2,2-3,4(m,2H),
3,6-4,4 (m,4H)a, 4,96 l(dd,J=4,4); 5,26 (dd,
J=10,6) lHI, 7,04 (d,J=8,2H), 8,12(d,J=8,2H)c
1,26 (s,6H), 1,4-1,9 (m,4H), 1,9-2,3 (m,2H),
2,4-2,5 (m,lH), 3,86 (s,3H), 4,14 (q,J=8,lH),
6,22(dd,J=8,8,lH),7,04(d,J=8,2H),8,12(d,J28, 2H)c
8 1775
a) Os multipletos nos espectros dos compostos VI a, b, c, d,
f e g i n c o r po r am um s i ng 1e t o a 3, 83; 3, 84 ; 3, 9O; 3, 88 e
3,86 ô respectivamente;
b) Espectro registrado em espectrômetro Varian
EM
390;pectro registrado em espectrômetro Varian XL 100.
Es-Tabela v Da dos espectrais das telurolactonas ( X) .
Composto
( X)
i. v. (fi 1
me
) vc=o (cm-l)1HRMP, CC14'
o
(ppm),J(Hz), TMSre fe rê n c i a i n te rn a
3 1770 1,16 (s,3H), 1,20 (s,3H), 1 ,63
(dd, J=10,6, lH), 2,26 (dd, J = 9,4, lH), 2,83 (dd, J=8,6, lH); 3,16 (dd, J=8,4, lH), 3,76 (s, 3H), 4,3-4,8 (m, 1H), 6,73 (d, J=6, 2H), 7,70 (d, J=6, 2H)c
5 1780 1,2-3,2 (m, 7H), 3,6-4,0(m,4H) b,
4,86 (d (largo) J-6), 6,63 (d, J=9, 2H), 7,60 (d, J=9, 2H)e
6 1770 1 ,0-3 ,9 (m, 9 H), 3,5 - 4, O ( m ,4 H )b ,
4,50 (dd, J=5, -5, 1H), 6,73(d, e J=9, 2H), 7,66 (d, J=9, 2H)
a) Espectro registrado em espectrômetro Varian EM 390; b) O multipleto incorpora um singleto a 3,730;
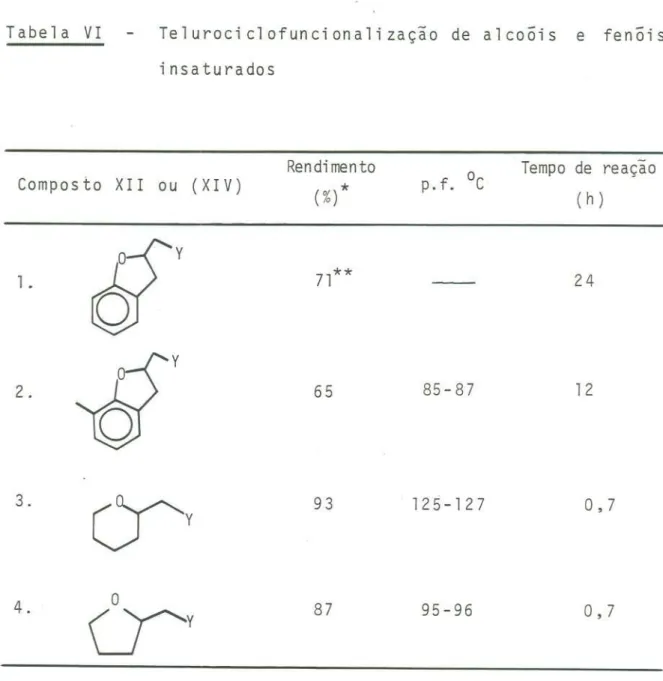
Alcoõis (XI) e fenõis (XIII) insaturados tambem reagem com tricloreto de p-metoxifenil telurio, levando aos dicloroteluro eteres correspondentes (XII) e (XIV) (Equação 8 e 9, Tabela VI).
~OH ArTeCl/CHC13
-HC1, refl uxo
~
° ArTe
~
I \
Cl Cl )n
(XI I)
(8)
( X I )
Ar = P-CH30cp
:;;. R
TeAr
1\
Cl Cl
OH
Y"
R
ArTeCl/CHC13(9)
-HC1, refl uxo
(XII I ) Ar
=
P~CH30cp(XI V)
A reação com alcoõis ocorre bem mais rapi damente do que com ácidos carbox;licos. Isso pode ser entendido fa-cilmente, considerando-se a nucleofelicidade das duas classes de compos tos. Já a reação com fenõis é mais lenta do que a reação com alcoõis, isso devido
ã
menor disponibilidadepar eletrônico do oxigênio fenôlico para atacar o centro
Os dicloroteluro éteres ciclicos são todos sólidos cristalinos incolor, com excessão do composto (XIV.l), que
não cristaliza. Todos os produtos de ciclização podem ser re
duzidos aos teluretos correspondentes (XV) usando-se hidreto de boro e sõdio (relação molar 1:1) (Equação 10, Tabela VII).
O
ArT,"'Y
/
C{ Cl
~
NaBH4/H2
O
:::-TH F , t. a., 10 I
ArT;-Q
( 1 O )
(XII)
.(XIV)
(XV)
Ar = p-CH 3Oij)
Esses compostos se mostram bem mais estáveis doque os análogos lactõnicos, podendo ser destilados e armazenados.
Reação entre os dicloroteluro éteres ciclicos e hi dreto de boro e sódio (relação molar 1 :2) não leva ao álcool
de partida Icompostos (XII.3) e (XII.4), Tabela VII. No caso dos derivados aromáticos, a reação leva ao fenol de partida e ditelureto de diarila. Isso se deve, certamente,
ã
estabili-dade do ion fenolato que se forma intermediariamente (EquaçãoTabela VI Telurociclofuncionalização de alcoõis e fenõi
s
insaturados2.
y
65 85
-
87 12*
Rendimento do produto recristalizado (clorofõrmio +
de petrõleo 30-60).
ete r
**
Óleo ã t.a. Não foi possivel recristalizar.
y = P-CH30<jJTeC12
Rendimento o Tempo de reaçao
Composto XII ou (XIV)
(%)* p. f. C ( h )
Y
1. L / 71**
-
24
3.
()v
93
125-127
0,74. ° 87 95-96 0,7
Tabela VII Reação de teluroéteres com hidreto de boro e
sõdio (relação molar 1 :1)
Produto de redução p.e.
p. f. °c
°C/mmHg* Rendimento (%)y
82-83 63
y
79
()v
150/0,025
83
(y'v
140/0,05
84
Y = p-CH O<PTe3
*
H+
~
(11 )
Ar = p-CH3Orp
No caso de derivados alifáticos, o íon alcoolato,
não sendo estabilizado, não tem tendência em se formar.
Um estudo mais completo sobre o uso de tricloretos
de ariltel~rio como agentes de ciclofuncionalização, bem como
sobre a redução de ligações telurio-carbono se encontra atual mente em desenvolvimento em nosso laboratório.
Por serem as telurolactonas e os teluroeteres
com-postos pouco descritos na literatura, apresentamos no
apêndi-ce seus espectros de ressonância magnetica protõnica e de
ab-sorção no infravermelho. Um estudo sistemático das
caracte-risticas espectrais dos compostos descritos neste capitulo se encontra em desenvolvimento 1191.
ArTeH +
Experimental: Preparação das aril telurolactonas (VI) -
Pro-cedimento geral Ltl.
Uma mistura do ácido carbox{lico y-o insaturado (5..0
X 10-3 mol) e tricloreto de p-metoxifeniltelúrio (2..0g..5..8X
10-3 mol) em clorofórmio (80ml) foi mantida sob refluxo pe lo
tempo indicado na Tabela I. o solvente foi evaporado e o
re-s{duo filtrado em gel de s{lica.. usando-se clorofórmio como
eluente. A solução foi secada com sulfato de magnésio e evapE..
rada. o óleo resultante foi recristalizado de clorofórmiol
éter de petróleo (Tabela I).
Reação entre telurolactonas (VI) e hidreto de boro e sódio
(relação molar 1:1) Ltl.
-
-3
Uma soluçao da telurolactona (1 X 10 mol) em
te-trahidrofurano (10ml) foi tratada gota a gota com uma solução
de hidreto de boro e sódio (0..04g.. 1..0 X 10-3 mol) em
J
agua
(5m l) . Observou-se reação imediata com evolução de gás e ap~
recimento de coloração amarela. Após 10 minutos de agitação
a temperatura ambiente a mistura reacional foi dilu{da com
éter (30ml) e lavada sucessivamente com água.. solução
satura-da de c~oreto de amSnio e de cloreto de sódio. A fase orgânf
ca foi secada com sulfato de magnésio e evaporada fornecendo
(Tabe la II).
Reação entre telurolactonas (VI) e hidreto de boro e sódio (relação molar 1:2)
Ltl.
-3
A telurolactona (1~0 X 10 mol) em tetrahidrofur~
no (10ml) a temperatura ambiente~ foi tratada com hidreto de boro e sódio (031g3 236 X 10-3 mol) em água (Sml). Observou-se reação imediata com desprendimento de gás e aparecimento& coloração vermeZho escuro. Após 10 minutos de agitação a tem peratura ambiente a mistura de reação foi dilu{da com éter
(SOml) e extra{da duas vezes com hidróxido de sódio 2N (Sml).
A fase orgânica foi lavada com água3 secada com sulfato de magnésio e evaporada3 fornecendo ditelureto de
di-p-metoxife-nila com rendimento quantitativo.
A fase aquosa foi resfriada em banho de gelo~ ac~-dificada com ácido clor{drico concentrado e extra{da com éter
(3 X 10 ml). o extrato orgânico foi secado com sulfato de
magnésio~ o solvente evaporado e o res{duo destilado em KugeI
rohrofen3 fornecendo os ácidos y-o insaturados com os
Redução de telurolactonas (VI) com hidreto de n-butil
esta-~.
A uma solução da telurolactona (VI.;§) (0~43g~ 1~0
X 10-3 mol) em tolueno (3ml). sob refluxo~ adicionou-se n-Bu3SnH
(0~58g~ 2~0 X 10-3 mol). Observou-se reação imediata. Após
10 minutos sob refluxo todo o material de partida foi
reduzi-do ao telureto correspondente~ conforme indicado por
cromato-grafia em camada de 19ada. Nesse ponto adicionou-se uma nova
- -3
porçao de n-Bu3SnH (0~3g~ 1~0 X 10 mol) e continuou-se o r~
fluxo por mais 30 minutos~ quando uma nova porção de n-Bu3SnH
(O~43g~ 1~0 X 10-3 mol) foi adicionada. Após 30 minutos sob
refluxo o solvente foi evaporado e o res{duo sublimado~ forn~
cendo 70mg (55%) da lactona X.
1
HN.MR (60MHz~ CCl4) 8 1~23 (s~ 6H)~ 1~40 (d~ J
=
7Hz~ 3H);1~66 (dd~ J = 14Hz~ 1H); 2~16 (d~d~ J = 14; 7Hz~ 1H);
(siste--1
ma AM2 X7~ J
=
10; 7~ 7Hz~ 1H). LR. (CCl) 1770 cm .v 4
Preparação dos dicloroteluroéteres
Uma mistura de tricloreto de p-metoxifeniltelúrio
-3 -3
(1~1 X 10 mol) e o ~lcool insaturado (1~0 X 10 mol) em
clorofórmio (16ml) foi mantida svb refluxo pelo tempo
do em gel de s{lica usando clorofórmio como eluente. o
elu{-do foi secado sob sulfato de magnésio~ o solvente foi evapor~
doe o res{duo recristalizado de êlorofórmiojéter de petróleo
(30-60). Os rendimentos estão indicados na Tabela VI.
Reação entre os diclorotelureéteres e hidreto de boro e sódio
(relação molar 1:1).
Seguiu-se o mesmo procedimento empregado na
redu-ção das telurolactonas. Os produtos foram purificados por
destilação a pressão reduzida~ fornecendo os teluroéteres na
forma de sólido incolor ou de óleos amare los com os rendimentos
RE FE Rt Nc I AS
1.
M.F. Ansell e M.H. Pa1mer, Quart.Rev., ~, 211 (1964).2.
R.C. Laroek, Angew.Chem.Int.Ed.Eng1., .1.2, 27 (1978).3.
M.O. Oow1e e 0.1. Oavies, Chem.Soe.Rev., §., 171 (1979).4.
K.C. Nieo1aou, S.P. Seitz,
W.J. Sipio e J.F.
B1ount,
J.
Am.Chem.Soc., 101, 3884 (1979) e referêneiaseitadas.
5..
M. de Moura Campos e N. Petragnani, Chem.Ber., 93, 317(1960).
6. O.L.J. C1ive, C.G. Russe1, G. Chittattu e A. Singh, Tetrahedron, 36, 1399 (1980).
7.
K.C. Nieo1aou, Tetrahedron, 37,4097 (1981).8.
O. Li otta, Aee. Chem. Res., .!.Z., 28 (1984).9 .
J. V. Comasseto e N. Petragnani, Synth.Commun., 13, 889(1983).
10. V.I. Staninets e E.A. Shi1ov, Russian Chem.Rev., 40,272 (1971).
11. S.W. Ro11inson, R.A. Amos e J.A. Katzene11enbogen, J.Am. Chem.Soe., 103,4114 (1981).
12. H.O. House, IIModern Synthetic Reaetionsll, W.A. Benjamin,
13. a)M. de Moura Campos, J.Am.Chem.Soc.,!..i, 4480 (1954);
b)M. de Moura Campos, Chem.Ber., 93, 1075 (1960);
c)N. Petragnani e H.M.C. Ferraz, Synthesis., 476 (1978).
14. H.J. Reich in "Oxidation in Organic Chemistry" Editado
por W~S. Trahanovsky, Academic Press, New York, 1978.
15. a) K.C. Nico1aou, W.E. Barnette e R.L. Mago1da, J.Am.Chem.
Soc., 103,3486 (1981);
b) K.C. Nico1aou, G.P. Casic e W.E. Barnette, Angew.Chem. Int.Ed.Eng1., 12, 293 (1978).
16. W.P. Jackson, S.V. Leye A.J. Whitt1e, Chem.Commun., 1173
(1980).
17. N. Petragnani e J. V. Comasseto in IIproceedings of the
Fourth Internationa1 Conference on the Organic Chemistry
of Se1enium and Te11urium", Eds. F.J. Berry e W.R. McWhinie,The University of Aston in Birmingham, 1983, p.
98-214.
18. N. Petragnani e J.V. Comasseto, "Synthetic App1ications of
Te11urium Reagents", a ser publicado em "Synthesis".
19. Trabalho em desenvolvimento, em colaboração com Prof. R. Zingaro da Universidade do Texas.
20. a) M.W. Sharp1es, K.M. Gordon, R.F. Lauer, D.W. Patrick,
b)H. Lee e M.P. Cava, Chem.Commun., 277 (1981).
21. S. Uemura e S. Fukuzawa, J.Am.Chem.Soc., 105,2748 (1983).
-APENDICE
"ESPECTROSDE RESSONÂNCIAMAGNÉTICA PROTÔNICA E -NO
.+
J ;f
I
I ~t§~-g ~
: ~
~~~lJ.. .. ~ ..
"=---=-'
>
.~
~
",-
'!
<:r
...
~
Jt!
<=>
N
...
3
2
t
..
~g-~-8 ~ O O 0--""; ..!'.. .. ~ .. .. !!. .. ..
o ",-" .. <li
<> ~. '"
o
2 .'t
... :>
o
(Y)
o
(Y)
::t: W
I
I
'1'-'" ..'" '" ).
~
g-~-§~s:-~
! ::" .. "
~ ~OJ ~. ~
-~
I
_.~
.. .
LD
... :>
-} í ;;
~
"
.- .L.8_&_8 g o o o
J:' ~ :: ~ .. ~ '" ..-::.
~
~~~
..
~ :::.... GJT
"
"
..:;
.>t!!
~ ;,-,
>-o
(V) ::r:
-'o " í >!
"\
~'
~"
+~t\~\
~ @ e~
@--~
~
G~
-::y-. -::y-.J-~-g g ~g o
, , ... ~ .. ... ~ "-N'
)
.'1r
... ... :>
o M :J:
U
i
=:;= @ ....
,,
\
---
--.,.-=-
---
. g
lU
.to
! :
'" t
~@
'I I:t§_~_§ g : ~ ~~.! t".. N " ..
l
i *" €)
.~@ .
~~
'"
~. ~ ~~- \
8
~ ., ~'"
~ 0
\
-.~..
"I:!!
-- ...
> o
2-a:
\li
O-\li ::;; O'a: 0-UlU
o.
rtI c: ::;; z
X
::;;
O
'"
O'"
.. :. ...
10ppIII
---'5PPIII
2--- 5 - 6
7 O
CH30-<O)-;Teill~VI
C1rTTT'T,. C1 . 1 )10PPIII
5- fr~t~L
~~->~TI:-~"--r~ - -J
..
.
C1
SR
~(' -Q-OCH3
ppmC" ~,
o O
CH O
-<O~Te::--(
y
~3 C1 C1
~
cp
( VI.4)
,-- .- u ' , ;" '" " , '~ " , ",u...:.'~" " , " " ~" ,
CH30
-fQ;Te~o
20130I lOfO
:t
<éO.p,m '-,-'--30PO-' --rm- -i , ,--2(),OO
1'011 100U
i ' !
- -,750- -,- -, 500
I : ,
500 .i 400
: . I
45
,
0 ! 3YO
, .-- ... _L I
.. . ; I
r-:.!--:-:r---I
I ., ..,. '. ' I
I~--~:!'.-~~r. ~::::c':.~;r': : ... i" -: -, . -. -: . -- -L-; ,---~_.-~..
. -} -:-+.-,.-,: ::.,,::i.
--I .._;. . !'i':. .!
I . "
, I
, , I
,
I ! I ! .,..- :I
I I I : 10':) "V<, ., ?~O-I 2~~ :60 I,. : i
; : i.
-~
-~~._-+--i
I .,,0 . <o -o'.; i'.'- -T ;-. .; ; -! i i +-I i -Io
CH3o-Q-Tau
2.... .-10... ''''''o
CH°-<0)
Je:::Y-')
3 C, '(lU