FACULDADE DE FARMÁCIA
Role of microRNA in microglial phenotype
during the progression of Alzheimer’s disease
Mafalda Aurélio Monteiro
Dissertação
MESTRADO EM CIÊNCIAS BIOFARMACÊUTICAS
2016
UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIA
Role of microRNA in microglial phenotype during
the progression of Alzheimer’s disease
Mafalda Aurélio Monteiro
Dissertação orientada por:
Professora Doutora Adelaide Fernandes
Professora Doutora Dora Brites
MESTRADO EM CIÊNCIAS BIOFARMACÊUTICAS
2016
The studies presented in this thesis were performed at the Neuron-Glia Biology in Health and Disease research group, at the Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, under the supervision of Adelaide Fernandes, Ph.D and Dora Brites, Ph.D
This work was funded by EXPL/NEU-NMC/1003/2013 grant from FCT to Adelaide Fernandes and FCT grant UID/DTP/04138/2013 to iMed.ULisboa.
Publications
The studies included in this thesis were presented in the following publications:
Oral presentations
Monteiro M, Brites D, Fernandes A. Role of microRNA in microglia phenotype during the
progression of Alzheimer’s disease. VIII Cycle of Scienceshops – Alzheimer’s disease. 2015. Lisbon, Portugal
Poster communications
Monteiro M, Caldeira C, Brites D, Fernandes A. Human microglia phenotype and microRNA
profile change over time under Amyloid-beta-enriched conditions. 7th Postgraduate iMed.ULisboa Students Meeting. 2015, Lisbon, Portugal
Monteiro M, Caldeira C, Brites D, Fernandes A. Human microglia phenotype changes in the
presence of Amyloid-beta expressing neuroblastoma cells. XIV Meeting of the Portuguese
Agradecimentos
Gostaria de começar por agradecer à Prof.ª Dr.ª Dora Brites por me ter dado a oportunidade de me juntar ao grupo Neuron-Glia Biology in Health and Disease para realizar esta tese, mesmo com a condicionante de ser trabalhadora-estudante e, por isso, não poder participar em muitas das atividades do grupo. Agradeço também pelo apoio que me deu aquando da candidatura a bolsas, não só pelas cartas de recomendação mas também pelas dicas que, estou certa, me serão sempre úteis a nível profissional.
Tenho também muito a agradecer à Adelaide, pois sem o seu apoio esta tese nunca teria sido feita. Um muito obrigada por me ter dado a oportunidade, desde os projetos do 1º ano do CBF, de trabalhar e aprender consigo. Obrigada por toda a dedicação e paciência, mesmo quando as coisas não correram tão bem ou tive dificuldade em cumprir prazos...É, sem dúvida, um exemplo no mundo da ciência não só por todos os conhecimentos que tem mas também por saber transmiti-los.
Agradeço a todos os que fazem parte do grupo, sem exceção, por me terem acolhido e ajudado nas muitas vezes em que precisei. Obrigada ao Pedro Dionísio por toda a ajuda que me deu durante o CBF, mas também pela companhia no CPM quando as horas já iam avançadas.
Não posso deixar de agradecer o companheirismo da equipa da farmácia de Tercena, e em particular à Dr.ª Martine por ter permitido que eu conciliasse o meu trabalho com a realização do CBF.
Ao meu pai agradeço todo o apoio que me deu ao longo do tempo do mestrado, principalmente quando tive de fazer um esforço maior para cumprir deadlines. Á minha irmã Catarina, obrigada por teres participado ativamente nesta tese ao ajudares-me com as imagens. À Patrícia, desculpa tantas vezes ter-te usado como “caixote do lixo emocional” mas, acredita, foste uma grande ajuda para mim ao longo deste percurso, obrigada! Fica aqui uma última palavra à minha mãe que, mesmo estando tão longe, tenho a certeza que esteve sempre a olhar por mim...
A toda a minha família, obrigada por tudo! E a ti, avó Especiosa, uma palavra especial por todo o carinho que sempre me deste. Espero que esta tese sirva para que outras pessoas cheguem à tua idade, 103 aninhos, com a mesma qualidade de vida.
Não podia deixar de agradecer às minhas amigas “galinhas” por me terem acompanhado ao longo desta jornada.
i
Table of Contents
Abbreviations ... vii Abstract ... ix Resumo ... xi I. INTRODUCTION ... 1 1. Alzheimer’s disease... 11.1. Diagnostic and treatment ... 1
1.2. Pathogenesis ... 2
1.2.1. Amyloid β-peptide and neurofibrillary tangles ... 2
1.2.2. Neuroinflammation ... 5
2. Microglia: the key players in neuroinflammation ... 6
2.1. Microglial regulation and functions ... 6
2.2. Microglial phenotypic diversity ... 7
2.3. Microglia in the aged brain... 9
2.4. Microglial deregulation in Alzheimer’s disease... 11
3. MicroRNAs: biogenesis and functions ... 13
3.1. Inflammation-related microRNAs in microglia ... 14
3.1.1. MiR-124 ... 14
3.1.2. MiR-155 ... 15
3.1.3. MiR-146a ... 16
3.2. MicroRNA profile in Alzheimer’s disease ... 18
3.3. Deregulation of microglial microRNAs in Alzheimer’s disease ... 20
4. Human versus rodent microglia ... 21
4.1. The human CHME3 microglial cell line ... 22
5. Aims ... 22
II. MATERIALS AND METHODS ... 23
1. Cell culture and treatment... 23
2. Protein extraction and western blot analysis ... 24
3. Enzyme-Linked Immunosorbent Assay (ELISA) ... 25
4. Total RNA extraction, reverse transcription and semi-quantitative RealTime Polymerase Chain Reaction (qRT-PCR) ... 26
5. Evaluation of microglial phagocytic ability ... 27
6. Senescence-associated β-galactosidase assay ... 28
7. Statistical analysis ... 28
ii
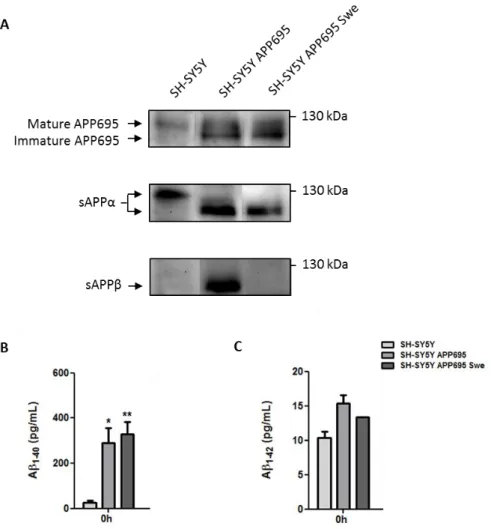
1. APP expression and Aβ secretion in neuroblastoma cells ... 29 2. The presence of CHME3 microglia does not alter APP expression in neuroblastoma cells but reduces sAPPα, sAPPβ and Aβ1-40 levels in co-culture media ... 31
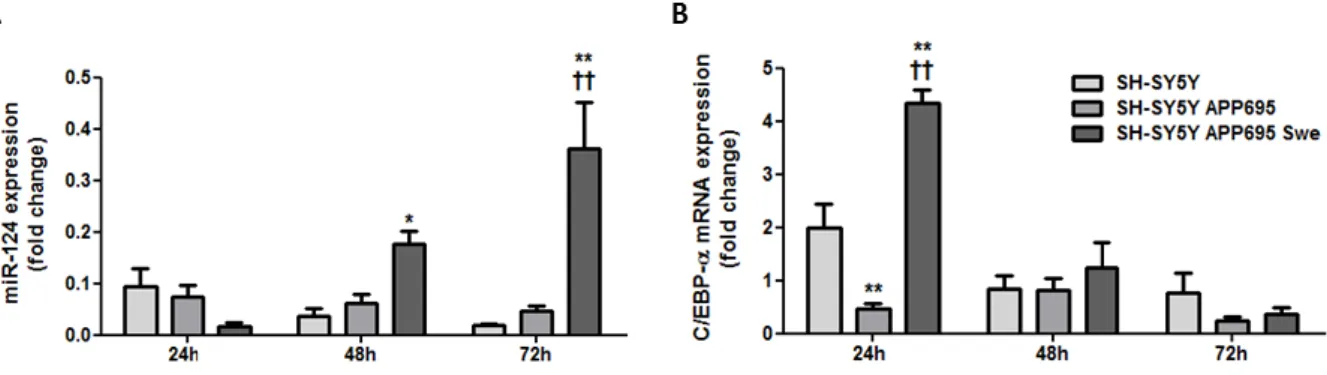
3. Human CHME3 microglial expression of inflammation-related miRNAs and their targets is mainly altered in the presence of SH-SY5Y APP695 Swe cells ... 33
4. The expression of pro-inflammatory cytokines in CHME3 microglia is markedly induced when co-cultured with SH-SY5Y APP695 Swe cells ... 36
5. The expression of CHME3 microglial immune markers is reversed when co-cultured with SH-SY5Y APP695 or SH-SY5Y APP695 Swe cells ... 38
6. The expression of anti-inflammatory markers in CHME3 microglia is markedly induced when co-cultured with SH-SY5Y APP695 Swe cells, with TGF-β exception ... 39
7. CHME3 microglia co-cultured with SH-SY5Y APP695 Swe cells preserve their phagocytic capacity for longer periods ... 40
8. CHME3 microglia show increased SA-β-gal activity when co-cultured with SH-SY5Y APP695 Swe cells ... 42
IV. DISCUSSION ... 45 V. REFERENCES ... 55
iii
Figure Index
I. INTRODUCTION
Figure I.1 | Metabolism of the amyloid precursor protein (APP) and representation of the Swedish mutation.. ... 4
Figure I.2 | Microglial phenotypes in the healthy (M0, M1, M2a, M2b and M2c) and aged (dystrophic and primed) brain.. ...11
Figure I.3 | Regulation of inflammation and immunity by miR-124, miR-155 and miR-146a.. 18
II. MATERIALS AND METHODS
Figure II.1 | Schematic representation of the experimental design ... 24
III. RESULTS
Figure III.1 | APP expression, and sAPPα, sAPPβ and Aβ secretion by neuroblastoma cells.. ...30
Figure III.2 | Presence of CHME3 microglia does not alter APP expression in neuroblastoma cells but reduces sAPPα, sAPPβ and Aβ1-40 in co-culture media.. ...32
Figure III.3 | Expression of miR-124 gradually increases in CHME3 microglia when co-cultured with SH-SY5Y APP695 Swe cells, whereas the mRNA expression of C/EBP-α decays over time.. ...33
Figure III.4 | Expression of miR-155 peaks in CHME3 microglia when co-cultured with SH-SY5Y APP695 Swe cells with a subsequent reduction, whereas the mRNA expression of SOCS1 increases in a time-dependent manner. No evident changes are observed for C/EBP-β mRNA expression.. ...35
Figure III.5 | Expression of miR-146a increases in CHME3 microglia when co-cultured with SH-SY5Y APP695 Swe cells decreasing over time. Conversely, whereas mRNA expression of IRAK1 progressively increases, the mRNA expression of TRAF6 does not show any significant variation along time.. ...36
iv
Figure III.6 | CHME3 microglial mRNA expression of the pro-inflammatory cytokines TNF-α, IL-6 and IL-1β is enhanced over time when co-cultured with SH-SY5Y APP695 Swe cells.. 37
Figure III.7 | CHME3 microglial mRNA expression of iNOS is rapidly induced when co-cultured with SH-SY5Y APP695 Swe cells whereas MHC class II mRNA expression is slowly enhanced.. ...39
Figure III.8 | mRNA expression of the anti-inflammatory markers Arginase 1 and IL-10 is rapidly induced in CHME3 microglia when co-cultured with SH-SY5Y APP695 Swe cells and progressively enhances, though TGF-β mRNA expression does not show a gradual variation pattern.. ...40
Figure III.9 | Average of phagocytosed beads per CHME3 microglial cell tends to reduce in all co-culture systems, though CHME3 microglia co-cultured with SH-SY5Y APP695 Swe cells retain their capacity to uptake increased number of beads along time in co-culture.. ...41
Figure III.10 | CHME3 microglia co-cultured with SH-SY5Y APP695 Swe cells show increased levels of senescence-associated β-galactosidase (SA-β-gal) activity along time in co-culture.. ...43
IV. DISCUSSION
Figure IV.1 | Human CHME3 microglia shift from a pro-inflammatory to a more anti-inflammatory/regulatory phenotype when co-cultured with SH-SY5Y APP695 Swe cells.. ....54
v
Index of Tables
I. INTRODUCTION
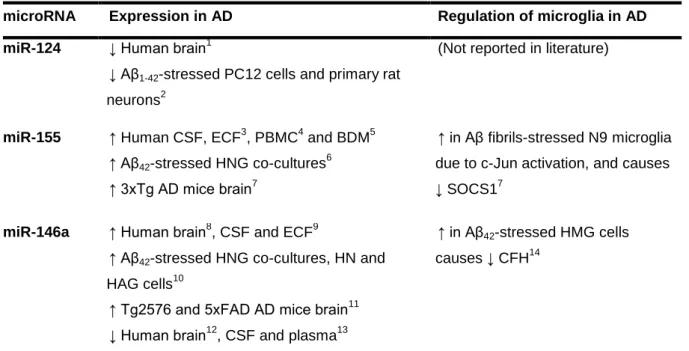
Table I.1 | Expression of miR-124, miR-155 and miR-146a in samples of Alzheimer’s disease (AD) patients, in vivo and in vitro AD models, and evidence of their role in the regulation of microglia in AD.. ...21
II. MATERIALS AND METHODS
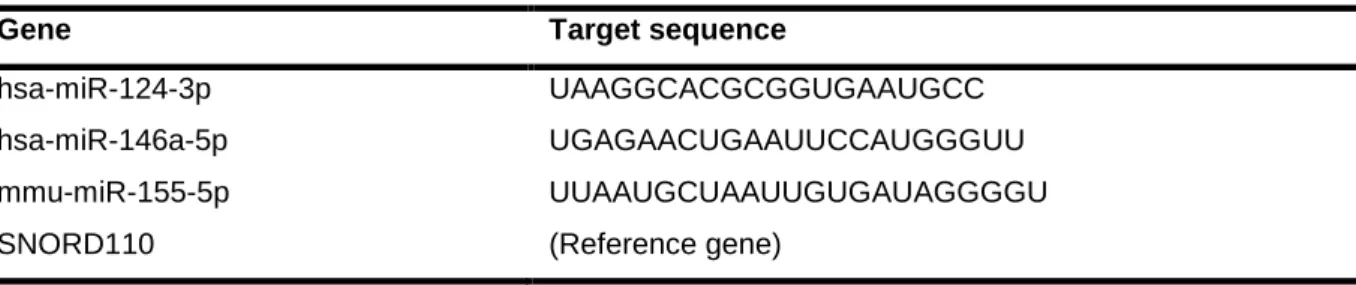
Table II.1 | Sequences used as primers for detection of mRNA expression in CHME3 microglia ...26
Table II.2 | Target sequences of predesigned primers used for detection of miRNAs expression in CHME3 microglia ...27
vii
Abbreviations
AB/AM Antibiotic antimycotic
AD Alzheimer’s disease
AICD Amyloid precursor protein intracellular domain
APP Amyloid precursor protein
Arg1 Arginase 1
Aβ Amyloid β-peptide
Aβ1-40/1-42 40/42 residues length Amyloid β-peptide
BACE1 β-site amyloid precursor protein cleaving enzyme 1
BDNF Brain-derived neurotrophic factor
C/EBP-α CCAAT/enhancer-binding protein α
CD206 Mannose receptor
CFH Complement factor H
CNS Central nervous system
CREB Cyclic AMP response element-binding
CSF Cerebrospinal fluid
CSF-1 Colony stimulating factor 1
CTF C-terminal fragment
DMEM Dulbecco’s Modified Eagle’s Medium
ECF Extracellular fluid
ECM Extracellular matrix
ELISA Enzyme-Linked Immunosorbent Assay
EOFAD Early onset familial Alzheimer’s disease
FBS Fetal bovine serum
FIZZ1 Resistin-like α
GM-CSF Granulocyte macrophage colony stimulating factor
HAG Human astroglial
HLA-DR Human leucocyte antigen
HMG Human microglial
HNG Human neuron-glial
h-tau Hyperphosphorylated tau
IFN-γ Interferon γ
IL Interleukin
iNOS Inducible nitric oxide synthase
viii
IRF Interferon regulatory factor
JAK Janus kinase
L-glu L-glutamine
LPS Lipopolysaccharide
MAPK Mitogen-activated protein kinase
M-CSF Macrophage colony stimulating factor
MHC class II Major histocompatibility complex class II
miRNA/miR MicroRNA
NFTs Neurofibrillary tangles
NF-κB Nuclear factor κB
NO Nitric oxide
qRT-PCR Semi-quantitative RealTime Polymerase Chain Reaction
RA Retinoic acid
ROS Reactive oxygen species
RPMI Roswell Park Memorial Institute
sAPP Soluble amyloid precursor protein
SA-β-gal Senescence-associated β-galactosidase
SOCS1 Suppressor of cytokine signaling 1
STAT Signal transducer and activators of transcription
Swe Swedish
TGF-β Transforming growth factor β
TLR Toll-like receptor
TNF-α Tumor necrosis factor α
TRAF6 Tumor necrosis factor receptor-associated factor 6 TREM2 Triggering receptor expressed on myeloid cells 2
T-TBS Tween 20 (0.1%)-Tris buffered saline
ix
Abstract
Alzheimer’s disease (AD) is the most prevalent form of dementia and its impact in society has been aggravating throughout years. Due to its progressive nature and lack of marked treatment benefits, many efforts have been done to unveil AD pathogenesis seeking for novel therapeutic targets or biomarkers.
Current view on AD pathogenesis attributes significant importance to neuroinflammation, where microglia play a pivotal role. Under normal conditions, microglia exhibit a quiescent/vigilant state and perform the brain surveillance. After an injury, microglia initiate the immune defense of the brain and acquire a pro-inflammatory or anti-inflammatory phenotype depending on stimuli. After the inflammation resolution, the brain homeostasis is restored. Various conditions such as the presence of amyloid β-peptide (Aβ) and aging can deregulate microglial response, though it remains unclear how microglial deregulation affect the course of AD. Furthermore, it was established that some microRNAs (miRNAs or miRs) that are known to promote microglial quiescence (miR-124) or regulate microglial activation states (miR-155 and miR-146a) are deregulated in AD. However, it has not been established whether the deregulation of these miRNAs can influence microglial phenotype and response in AD, particularly concerning human microglia.
With this work, we proposed to analyze the temporal response of human CHME3 microglia when co-cultured with two Aβ-expressing human neuroblastoma cells, SH-SY5Y APP695 or SH-SY5Y APP695 Swe cells. We assessed microglia for miRNAs (124, miR-155 and miR-146a) and their targets, as well as for pro-inflammatory (IL-1β, IL-6 and TNF-α), anti-inflammatory (TGFβ, IL-10 and Arginase 1) and immune (iNOS and MHC class II) markers, and additionally for phagocytic capacity and senescence.
We found that when CHME3 microglia are co-cultured with SH-SY5Y APP695 Swe cells they exhibit a more pronounced response than when co-cultured with other neuroblastoma cells. Indeed, in the presence of SH-SY5Y APP695 Swe cells CHME3 microglia initially exhibit a miR-124low/miR-155high/miR-146ahigh profile like activated cells but gradually switch to a miR-124high/miR-155low/miR-146alow profile typical of a gradual shift towards an alternative activated/deactivated phenotype that ultimately give rise to quiescent cells. The pro-inflammatory markers are robustly expressed in microglia during the whole time, but the expression of the anti-inflammatory markers is gradually enhanced suggesting an immunoregulatory response. With regards to immunity, microglia rapidly express the innate immune marker iNOS followed by a later induction of the adaptive immune marker MHC class II.
x
Altogether, we demonstrated that the CHME3 / SH-SY5Y APP695 Swe co-culture is the most adequate in vitro AD model to study human microglial response and possibly to assay new microglia-targeted therapeutic strategies.
xi
Resumo
A doença de Alzheimer (AD) é a forma de demência com maior prevalência no mundo e o seu impacto na sociedade tem vindo a agravar-se ao longo dos anos. Dada a sua natureza progressiva, mas também devido à falta de eficácia dos medicamentos utilizados atualmente na prática clínica, têm sido feitos vários estudos na tentativa de desvendar os mecanismos patogénicos da AD com o objetivo de descobrir novos biomarcadores ou alvos terapêuticos que permitam melhorar o diagnóstico da doença e atrasar a sua progressão.
Atualmente sabe-se que a neuroinflamação tem um papel importante na patogénese da AD, pelo que a microglia se destaca dada a sua relevância como reguladora da neuroinflamação. Em condições normais, as células da microglia apresentam um estado quiescente, vigiando a homeostase cerebral. Quando ocorre um dano, a microglia rapidamente inicia a resposta imunitária por forma a neutralizá-lo e proteger o cérebro, adquirindo um fenótipo pro-inflamatório ou anti-inflamatório dependendo do estímulo. Após a resolução da inflamação, as células da microglia voltam ao estado quiescente/vigilante permitindo que a homeostase do cérebro seja reposta. Sabe-se também que em algumas situações tais como na presença de agregados proteicos do péptido β amiloide (Aβ) – que é originado a partir do seu precursor, APP –, mas também durante o próprio envelhecimento, as funções e resposta da microglia estão alteradas. Porém, não foi ainda esclarecido como é que a desregulação das células da microglia devida a esses estímulos pode afetar o curso da AD.
Por outro lado, há certos microRNAs (miRs) reconhecidos pela sua capacidade de modular a expressão de genes que afetam os fenótipos da microglia, nomeadamente o miR-124, o miR-155 e o miR-146a. Enquanto que o miR-124 é expresso maioritariamente nas células da microglia no estado vigilante, sendo responsável pela sua manutenção, o miR-155 e o miR-146a regulam a expressão de genes envolvidos em vias de sinalização que levam à ativação celular. Vários estudos demonstram que a expressão destes microRNAs está desregulada na AD baseando-se não só em amostras de doentes mas também em diferentes modelos animais da AD. Contudo, não foi ainda esclarecido de que forma essa desregulação pode afetar a resposta e o fenótipo da microglia, nomeadamente no que diz respeito às células humanas.
Com este trabalho, pretendemos explorar a resposta da linha de microglia humana CHME3 quando em co-culturas com linhas de neuroblastoma humano que expressam Aβ, sendo estas as células SH-SY5Y APP695 ou SH-SY5Y APP695 Swe. As primeiras são células que expressam a isoforma 695 da APP, e as outras são células que expressam uma forma da APP mutante originada pela mutação Sueca (Swe). A análise da resposta da
xii
microglia foi feita com base na determinação da expressão de microRNAs (124, miR-155 e miR-146a) e respetivos genes alvo, assim como marcadores de fenótipo pro-inflamatório (IL-1β, IL-6 and TNF-α) e anti-pro-inflamatório (TGFβ, IL-10 and Arginase 1) e marcadores de resposta típica imunitária (iNOS and MHC class II). Adicionalmente, avaliámos a capacidade fagocítica da microglia e senescência, que estão associados às funções da microglia na AD.
Os nossos resultados demonstram que a linha de microglia humana CHME3 tem uma resposta particularmente alterada na presença das células SH-SY5Y APP695 Swe face à co-cultura com a outra linha de neuroblastoma, possivelmente pela maior acumulação de Aβ1-40 detetada nessa situação. Nesse modelo, observámos que a microglia inicialmente apresenta níveis elevados do miR-155 e do miR-146a e níveis reduzidos do miR-124, semelhante às células ativadas. Com o decorrer do tempo ocorre um aumento gradual da expressão do miR-124 em detrimento da expressão do miR-155 e do miR-146a, o que sugere que a microglia progressivamente adquire um fenótipo de ativação alternativo, ou desativação, que terminará num retorno ao estado de vigilância. Durante todo o tempo de co-cultura verificámos que a microglia expressa marcadores pro-inflamatórios, em paralelo com um aumento gradual da expressão dos marcadores anti-inflamatórios sugerindo que a microglia progressivamente desenvolve uma resposta imunorreguladora. Por outro lado, observámos que a resposta imunitária inata da microglia foi rapidamente induzida neste modelo, demonstrada pelo pico imediato da expressão da iNOS, enquanto que a resposta imunitária adaptativa foi induzida mais tardiamente, traduzida pelo aumento gradual da expressão do MHC class II.
Deste modo, demonstrámos que as co-culturas compostas pela linha de microglia CHME3 e pelas células SH-SY5Y APP695 Swe são o modelo in vitro da AD mais adequado para estudar a resposta da microglia humana e possivelmente utilizar em ensaios de avaliação de novos agentes terapêuticos que tenham como alvo a microglia.
Palavras chave: Microglia humana, doença de Alzheimer, miR-124, miR-155, miR-146a,
1
I. INTRODUCTION
1.
Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive, non-reversible neurodegenerative disorder that consists in the most common form of dementia by accounting for 50-70% of all diagnosed cases. In 2015, dementia was estimated to affect more than 46 million people worldwide, which is predicted to double every 20 years causing a huge economic impact in health care (Prince et al. 2015).
The global prevalence of AD increases with life expectancy affecting more than one-third elderly over 85 years old. Following advanced age, family history is the second major risk factor for AD whereas the presence of specific genetic mutations correlate with enhanced susceptibility to develop rare early onset familial AD (EOFAD) (45-65 years old) or most commonly late onset AD (> 65 years old) (Reitz and Mayeux 2014, Tanzi 2012). Additional non-genetic factors that can predispose to the development of sporadic AD include some pathological conditions such as the incidence of traumatic brain injury, cerebrovascular disease and diabetes, and lifestyle aspects such as inadequate diet and lack of physical and intellectual activity (Reitz and Mayeux 2014).
1.1.
Diagnostic and treatment
Initial symptoms of AD include episodic loss of memory characterized by difficulty in storing and retrieving new information, termed mild cognitive impairment. During the course of AD, patients progressively evidence memory and cognitive decline, while behavioral impairment can occur in later AD stages (Reitz and Mayeux 2014). Accordingly, cerebral damage in early AD is most prominent in areas responsible for the formation and retrieval of memories such as the entorhinal cortex and hippocampus, though it progressively spreads to the remaining cortical regions. In parallel, the cholinergic neurons in the basal forebrain are also commonly injured in AD (Braak and Braak 1991, 1995).
I. Introduction
2
There are no medicines available to prevent AD onset, and the only pharmacologic options approved are limited to patients ranging from moderate to severe AD stages such as the cholinesterase inhibitors donepezil, galantamine and rivastigmine, and the N-methyl-D-aspartate receptor antagonist memantine. These medicines provide some symptomatic benefits by ameliorating neurologic activity, though they have modest impact on AD progression as they fail to modify the pathologic process (Kumar et al. 2015). On the other hand, the lack of reliable biomarkers remains an obstacle for the determination of AD risk, as well as for establishing AD diagnosis and prognosis. Current approaches for AD diagnosis include a combination of cognitive and memory tests with brain imaging techniques such as Positron Emission Tomography, which allows determining the hippocampus volume or detecting the presence of amyloid plaques when using the Pittsburgh Compound-B. These are accurate methods for AD diagnosis, though they are not routinely used in clinical practice for AD stratification (Reitz and Mayeux 2014).
Therefore, innovative biomarkers are critically required contributing to early AD detection and pharmacologic intervention. Furthermore, unveiling the pathogenesis of AD is essential to identify novel mechanisms and targets with potential to originate alternative therapeutic strategies to delay AD progression.
1.2.
Pathogenesis
The two major hallmarks of the AD brain consist in the presence of extracellular amyloid β-peptide (Aβ)-containing senile plaques and intraneuronal deposition of neurofibrillary tangles (NFTs) composed by hyperphosphorylated tau (h-tau). Since AD is a multifarious disorder, other events cannot be dissociated from the neurodegenerative process in particular neuroinflammation.
1.2.1. Amyloid β-peptide and neurofibrillary tangles
Aβ is a small (~ 4 kDa) peptide which is originated from the cleavage of the amyloid precursor protein (APP), a transmembrane glycoprotein that is ubiquitously expressed in mammalian tissues. Following protein synthesis, APP is modified by glycosylation leading to the formation of immature APP, that is predominantly N-glycosylated, and mature APP, that is N- and O-glycosylated. There are three major isoforms of APP each containing 770, 751 or 695 amino acids, the APP770, APP751 and APP695 respectively, the last being the most predominant in neurons.
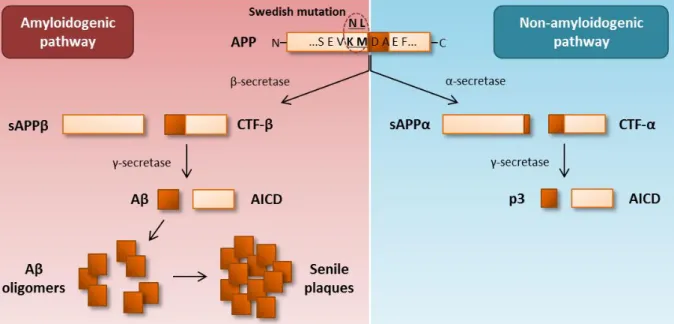
3 APP can be processed through two pathways, the prevalent non-amyloidogenic or the alternative amyloidogenic pathway (Figure I.1). In the non-amyloidogenic pathway, APP is cleaved by α-secretase within the Aβ region generating the C-terminal fragment α (CTF-α) and sAPPα, a large soluble N-terminal ectodomain that is secreted. The CTF-α is further cleaved by γ-secretase, an enzymatic complex composed by presenilin 1 and 2, nicastrin, anterior pharynx defective and presenilin enhancer 2, producing the p3 fragment and an APP intracellular domain (AICD). In the amyloidogenic pathway, APP is primarily cleaved by β-secretase (also denominated β-site APP cleaving enzyme 1, BACE1) resulting in the release of sAPPβ and formation of CTF-β. The CTF-β is further cleaved by γ-secretase generating an AICD and the Aβ monomer. The main form of Aβ produced is 40 residues length (Aβ1-40), although there is a small proportion of Aβ that is 42 residues length (Aβ1-42), the most prone to self-aggregate. Aβ aggregation states range from soluble oligomers to insoluble fibrils, the last being the main component of the senile plaques that typically deposit in the AD brain (O'Brien and Wong 2011).
The toxicity of extracellular Aβ oligomers and senile plaques mainly relies on their ability to trigger neuroinflammation (Heppner et al. 2015, Meraz-Rios et al. 2013, Shadfar et al. 2015). On the other hand, intraneuronal accumulation of Aβ oligomers, either produced intracellularly or reuptaken from the extracellular environment, also play a role in AD pathogenesis by facilitating the formation of h-tau, disrupting calcium homeostasis and causing synaptic, proteasome and mitochondrial impairment, thus compromising overall neuronal function leading to cell death (Cavallucci et al. 2012, LaFerla et al. 2007). The imbalance between Aβ production and clearance that result in exacerbated accumulation of Aβ in different assembly states supports the “amyloidogenic cascade hypothesis” of AD pathogenesis (O'Brien and Wong 2011).
Mutations in the APP gene as well as in genes encoding presenilin 1 and presenilin 2, the PSEN1 and PSEN2 genes respectively, are known to modify the APP metabolism towards Aβ generation. These mutations mostly predispose to the incidence of EOFAD, which represents approximately 5% of all diagnosed AD cases. On the other hand, the major genetic risk factor to develop late onset AD is the presence of the ε4 allele of the apolipoprotein E gene, which correlates with deficits in Aβ clearance (Tanzi 2012). Recent genome-wide association study analysis of sporadic AD cases identified variants in genes encoding innate immune molecules including the triggering receptor expressed on myeloid cells 2 (TREM2) (Guerreiro et al. 2013, Jonsson et al. 2013) and the myeloid cell-surface antigen CD33 (Naj et al. 2011), thereby supporting the involvement of phagocytes namely microglia, the brain phagocytic cells, in AD pathogenesis.
The Swedish (Swe) mutation, which was first described in 1992, is a specific modification in the APP gene that correlates with EOFAD (Mullan et al. 1992). This mutation
I. Introduction
4
is characterized by a double amino acid KMNL change in the N-terminal of the APP β-secretase cleavage site (codons 595 and 596 in APP695), making APP a preferable substrate for β-secretase. The consequence of the Swe mutation is the enhancement of the amyloidogenic processing of APP leading to the secretion of exacerbated amounts of Aβ forms and abnormal intracellular Aβ accumulation (Citron et al. 1992, Martin et al. 1995)
(Figure I.1).
Figure I.1 | Metabolism of the amyloid precursor protein (APP) and representation of the Swedish mutation. Most APP is
processed through the non-amyloidogenic pathway, whereas cleavage by α-secretase generates sAPPα, that is secreted, and
C-terminal fragment α (CTF-α), which is secondly cleaved by γ-secretase originating the p3 fragment and an APP intracellular
domain (AICD). In the amyloidogenic pathway, APP is cleaved by β-secretase resulting in the production of sAPPβ, that is secreted, and CTF-β, which is further cleaved by γ-secretase leading to the generation of the amyloid β-peptide (Aβ). Once formed, Aβ aggregates towards higher complex molecules ranging from oligomers to fibrils, the main components of the senile plaques. All these Aβ assembly states are found in the AD brain and contribute to AD pathogenesis. The Swedish mutation consists in a specific variant of the APP gene defined by a double amino acid KMNL change in the β-secretase cleavage site, making APP a preferable substrate for β-secretase. This mutation results in enhanced APP processing through the amyloidogenic pathway and consequent production of increased amounts of Aβ forms.
The other hallmark of AD is the accumulation of NFTs. The microtubule-associated protein tau is responsible for the maintenance of the axonal structure by stabilizing the microtubules, affecting axonal transport of vesicles. There are six tau isoforms ranging from 352 to 441 amino acids in the adult brain deriving from alternative splicing, all containing a high number of phosphorylation sites. The presence of genetic mutations and covalent modifications of tau, as well as external events including Aβ-mediated toxicity, oxidative stress and inflammation, have been postulated to trigger tau hyperphosphorylation and consequent dissociation from the microtubules. Once tau disengaged, microtubules undergo conformational changes that promote h-tau aggregation into NFTs which compromise microtubule polymerization and consequently axonal transport leading to synaptic dysfunction and neurodegeneration (Ballatore et al. 2007).
5
1.2.2. Neuroinflammation
The involvement of inflammation in AD pathogenesis is assumed for more than two decades, primarily supported by studies reporting the presence of pro-inflammatory chemokines and cytokines including interleukin (IL)-1 and IL-6 in the brain, plasma and cerebrospinal fluid (CSF) of AD patients (Bauer et al. 1991, Blum-Degen et al. 1995, Griffin et al. 1989). Previous data also postulated that glial activation was a late event in AD, suggested by the presence of activated microglia and astrocytes in the vicinity of senile plaques (Dickson et al. 1988, Itagaki et al. 1989).
However, it is now established that the neuroinflammatory response in AD is not exclusively attributed to the presence of exacerbated amounts of pro-inflammatory and oxidative species, as deregulated anti-inflammatory mediators are also found in the brain and circulation of AD individuals (Colton et al. 2006, Cribbs et al. 2012, Sudduth et al. 2013, Swardfager et al. 2010). Furthermore, current view on AD pathogenesis sustain that glial activation is not exclusively a consequence of plaque deposition but can preclude Aβ accumulation, for instance due to the incidence of genetic mutations or due to local/systemic inflammation. Upon activation, microglia and in a lower fashion astrocytes secrete a wide range of molecules such as glutamate, cytokines, chemokines, reactive oxygen species (ROS), nitric oxide (NO), complement factors and byproducts of cyclooxygenase 2 (e.g. prostaglandins) that can directly promote neuronal apoptosis, or cause marked functional and structural neuronal impairment leading to death. Besides neurons, the oligodendrocytes can also be compromised in AD which in turn supports neurodegeneration. Glial activation can also promote tau phosphorylation and enhance Aβ burden either by supporting APP amyloidogenic processing or due to inefficient Aβ degradation. In turn, the presence of Aβ in different assembly states interacts with glia enhancing the neuroinflammatory response, culminating in a vicious pathological cycle driving AD pathogenesis (Heppner et al. 2015, Meraz-Rios et al. 2013, Shadfar et al. 2015).
Besides glia, other cells such as brain endothelial cells, infiltrating T lymphocytes, macrophages and monocytes can also support neuroinflammation in AD. However, along with astrocytes, these cells might have reduced participation in AD pathogenesis compared with microglia, since microglia are fundamental for the regulation of neuroinflammation and maintenance of the brain homeostasis.
I. Introduction
6
2. Microglia: the key players in neuroinflammation
Microglia are the brain-resident myeloid cells that arise during the first wave of hematopoiesis in yolk sac blood islands (Ginhoux et al. 2010, Mizutani et al. 2012). Microglial differentiation from myeloid progenitors is particularly driven by the granulocyte macrophage colony stimulating factor (GM-CSF) and the macrophage colony stimulating factor (M-CSF), as well as the transcription factors CCAAT/enhancer-binding protein α (C/EBP-α) and PU.1 (Ponomarev et al. 2013).
Microglia are highly dynamic and multipurpose cells, playing fundamental role in the maintenance of the brain homeostasis. One of the most important functions of microglia involves their ability to participate in both innate and adaptive immunity, since microglia produce inflammatory/oxidative agents, have phagocytic capacity and perform antigen presentation. Microglia are also responsible for the clearance of neurotransmitters and debris, as well as extracellular matrix (ECM) remodeling and immunoregulation which are important in the resolution phase of inflammation (Boche et al. 2013).
2.1.
Microglial regulation and functions
Microglial regulation is dependent on their interaction with the whole brain microenvironment where the neuron-microglial crosstalk emerges as the most relevant axis. This communication involves neuronal secretion of molecules, commonly termed signals, that are recognized by specific receptors on microglial surface and regulate their activity and functions.
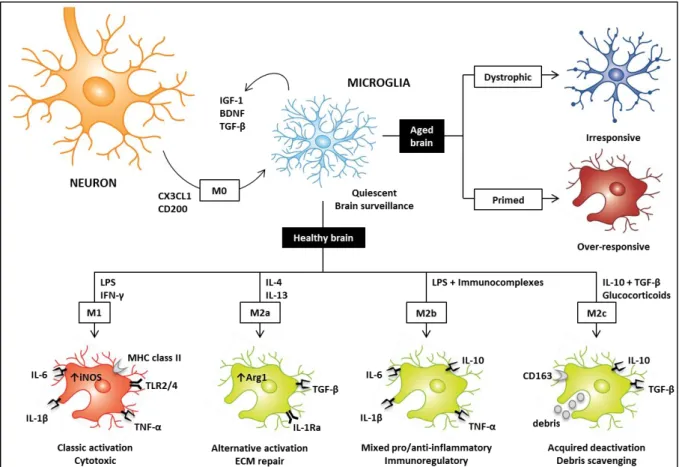
Under normal conditions, neurons secrete colony stimulating factor 1 (CSF-1) and IL-34 that act as “survival” signals upon binding the CSF-1 receptor on microglial surface, supporting microglial development and survival. Furthermore, neurons secrete “resting” signals mainly CD200 and CX3CL1 (also termed fractalkine) that bind the respective targets CD200R and CX3CR1 (Brown and Neher 2014, Kierdorf and Prinz 2013). These signals are particularly important for microglia to remain in their quiescent/vigilant state (M0 phenotype), in which cells are extensively ramified (Figure I.2). Quiescent/vigilant microglia are characterized by low levels of expression of typical markers of activated cells including antigen-presenting proteins (CD45, CD80, CD86 and major histocompatibility complex class II, MHC class II) and integrins (CD11c). Due to this phenotypic profile, quiescent microglia were firstly thought to be “nonactivated” cells, though they are currently assumed to perform the brain surveillance. Indeed, M0 microglia are characterized by the expression of anti-inflammatory cytokines including IL-10 and IL-4, and molecules that are required for tissue repair such as resistin-like α (FIZZ1) and chitinase 3-like-3 lectin (Ym1, Chi3l3 in humans)
7 resembling alternative activated cells (Ponomarev et al. 2007). Moreover, M0 microglia are important for sustaining normal neuronal development and functions as they secrete transforming growth factor β (TGF-β), insulin growth factor 1 and brain-derived neurotrophic factor (BDNF) (Boche et al. 2013, Ponomarev et al. 2013) (Figure I.2).
After a brain injury, neurons secrete “help” signals that drive microglial migration towards damaged tissues, and “eat me” or “do not eat me” signals that respectively stimulate or inhibit neuronal phagocytosis. These signals are recognized through the respective receptors on microglial surface including cytokine/chemokine receptors and pattern recognition receptors such as toll-like receptors (TLRs), scavenger receptors, CD33 and TREM2 (Brown and Neher 2014, Kierdorf and Prinz 2013). These receptors are differently expressed in microglia in accordance to their phenotype upon activation, which in turn exhibit a wide spectrum of possibilities depending on stimuli.
2.2.
Microglial phenotypic diversity
Distinct molecules were shown to induce microglial polarization towards classic activated (M1), alternative activated (M2a), type II alternative activated (M2b) or acquired deactivated (M2c) phenotype, whereas microglial morphology is shifted from ramified to amoeboid(Ponomarev et al. 2013, Walker and Lue 2015) (Figure I.2).
Microglial classic activation associate with strong pro-inflammatory, cytotoxic and immune response to pathogen-associated molecular patterns or damage-associated molecular patterns. These agents are interferon γ (IFN-γ), GM-CSF, tumor necrosis factor α (TNF-α) and lipopolysaccharide (LPS). After these molecules bind to the respective receptors on microglial surface (IFNR, GM-CSFR, TNFR and TLR4) they promote the activation of signaling pathways including Janus kinase/signal transducer and activators of transcription 1-4 (JAK/STAT1-1-4), IFN regulatory factors (IRFs), p38 and JNK mitogen-activated protein kinase (MAPK) and nuclear factor κB (NF-κB) (Freilich et al. 2013). This leads to the transcription of pro-inflammatory genes such as cyclooxygenase 2, TNF-α, IL-1β, IL-6, IL-12 and IL-23. Functionally, classic activated microglia exhibit phagocytic ability due to the expression of the scavenger receptor CD68 and Fc receptors, which mediate phagocytosis of molecules that have been opsonized with antibodies. M1 microglia are fundamental in the innate immune response by typically expressing inducible nitric oxide synthase (iNOS), which metabolizes arginine towards NO. Together with the secretion of ROS, metalloproteinases and collagenases, the production of NO contributes for tissue degradation (Colton 2009). Besides participation in innate immunity, M1 microglia participate in the adaptive immune response by expressing CD40, CD45, CD80, CD86 which mediate T
I. Introduction
8
cell activation. Furthermore, they are often characterized by overexpression of MHC class II/human leucocyte antigen (HLA-DR), though some data report that MHC class II is not exclusively expressed by amoeboid/activated microglia (Michell-Robinson et al. 2015, Ponomarev et al. 2013, Walker and Lue 2015).
On the other hand, the presence of any of M2 microglial phenotypes correlates with anti-inflammatory events. Alternative microglial activation towards the M2a phenotype is induced by IL-4 and IL-13, and is particularly important for protecting the brain against parasites. IL-4-mediated activation of microglia involves several transcriptional networks such as STAT6. Concomitantly, IL-4 stimulation inhibits the expression of STAT1-4 and IRF3 genes involved in classic microglial activation (Freilich et al. 2013) and the M1-related markers CD45 and NO (Ponomarev et al. 2007). M2a microglia are particularly characterized by increased levels of IL-1 receptor antagonist and mannose receptor (CD206). Moreover, they overexpress arginase 1 (Arg1), which competes with iNOS for arginine required for the formation of collagen (Colton 2009). Along with FIZZ1 and Ym1, upregulation of Arg1 correlates with ECM reconstruction and protection. Moreover, M2a microglia secrete the anti-inflammatory cytokines TGF-β, IL-10, IL-4 and IL-13 which are important for immunosuppression (Cherry et al. 2014, Michell-Robinson et al. 2015, Ponomarev et al. 2013, Walker and Lue 2015).
When microglia are exposed to immune complexes and LPS, they acquire the immunoregulatory M2b (or type II) phenotype which represents a lower alternative activated state as a result of its mixed M1/M2a profile. M2b microglia are essential for clearing away ROS and NO released during M1 activation, protecting the brain against LPS insult. The polarization of myeloid cells towards the M2b phenotype requires two signals, firstly involving the binding of ligands through the Fcγ receptor and secondly through TLR4. The resulting M2b phenotype is characterized by the release of pro-inflammatory (IL-1β, TNF-α and IL-6) simultaneously with anti-inflammatory (IL-10 and IL-4) cytokines. However, M2b microglia secrete low levels of IL-12, which can be used to differentiate M1 and M2b microglia. In addition, M2b cells express MHC class II, CD80 and CD86, making them prone to participate in adaptive immunity (Cherry et al. 2014, Michell-Robinson et al. 2015, Ponomarev et al. 2013, Walker and Lue 2015).
Finally, stimulation with either glucocorticoids or TGF-β and IL-10 induces microglial deactivation towards the M2c phenotype. M2c microglia exhibit some markers of M2a and M2b cells including FIZZ1, TGF-β, IL-10, Arg1 and CD206, though they are particularly distinguished from them by the expression of the scavenger receptor CD163. Functionally, M2c microglia are mainly important for immunosuppression and debris scavenging (Cherry et al. 2014, Michell-Robinson et al. 2015, Ponomarev et al. 2013, Walker and Lue 2015).
Besides M0, M1, M2a, M2b and M2c microglial phenotypic markers and functions have been quite well described in the healthy brain, due to microglial plasticity some of their
9 characteristics might overlap, and novel phenotypes may arise making it difficult to assess microglia under pathological conditions (Walker and Lue 2015). Furthermore, once studying AD, additional age-related phenotypes should be taken into account.
2.3.
Microglia in the aged brain
It is accepted that aging induces marked changes in microglia culminating in overwhelming dysfunction, but it has not been established whether aged microglia become over-activated upon stimulation or degenerate and lack their ability to respond as a consequence of cellular senescence (Figure I.2).
Some reports demonstrated that a range of pro-inflammatory and immune markers are aberrantly expressed in the brain of healthy elderly (Cribbs et al. 2012, Schuitemaker et al. 2012). These observations support the hypothesis of several authors who argue that microglia are primed in the aged brain, thereby developing exacerbated and prolonged neuroinflammatory response after stimulation (Norden and Godbout 2013, Perry V. H. et al. 2010, van Gool et al. 2010). In elderly, microglial activation during sepsis was associated with enhanced detrimental behavioral outcome (Lemstra et al. 2007). After peripheral LPS administration in aged mice, microglia were shown to produce exaggerated amounts of pro-inflammatory and reactive species (Chen et al. 2008, Godbout et al. 2005). In similar study conditions, it was demonstrated that microglia lack their ability to respond to the M2a mediator IL-4, failing to recover from LPS stimulation due to lack of Arg1 (Fenn et al. 2012, Fenn et al. 2014). Failure in polarizing aged microglia towards the M2a phenotype might provide an explanation for the presence of reduced levels of IL-4 in the brain of aged mice which in turn impacts in neuronal synaptic function (Nolan et al. 2005). These data suggest that microglial dysfunction in the aged brain occurs as a consequence of their primed response upon pro-inflammatory stimulation concomitantly with irresponsiveness to anti-inflammatory stimulation, leading to the generation of uncontrolled anti-inflammatory and oxidative stress in the brain increasing the vulnerability to neurodegeneration.
Streit and his collaborators identified, in brain samples of elderly, a population of microglia with specific characteristics which was reported as dystrophic/senescent microglia (Streit et al. 2004). Phenotypically, dystrophic microglia might be misinterpreted due to the expression of HLA-DR (Streit et al. 2004) as it is also expressed in functional human young microglia (Broderick et al. 2000, Melief et al. 2012). Morphologically, dystrophic microglia are distinct from ramified and amoeboid cells by exhibiting condensed nucleus, fragmented cytoplasmic processes (cytorrhexis), deramification and spheroidal/bulbous swellings. All these features have been attributed to progressive telomere shortening and decreased
I. Introduction
10
telomerase activity which can lead to replicative cellular senescence (Flanary and Streit 2004, 2005, Flanary et al. 2007), culminating in accidental microglial death manifested by remarkable cytorrhexis (Streit 2002, 2005, Streit and Xue 2009, Streit et al. 2004). Senescent microglia exhibit irreversible dysfunction that includes self-renewal inability, reduced vitality as well as motility and phagocytic impairment. This compromises the brain homeostasis predisposing to the development of age-related neurodegenerative disorders, particularly AD (Streit 2005, 2006).
Dystrophic microglia are thought to be spontaneously seen only in the aged human brain and not in rodents as a consequence of lifestyle and environmental factors that contribute to microglial senescence throughout life (Streit and Xue 2012, 2013, Streit et al. 2014). However, studies of primary murine microglia obtained from old mice (Njie et al. 2012, Sierra et al. 2007), or neonatal mice followed by prolonged in vitro culture as performed in our laboratory (Caldeira et al. 2014), demonstrated that aged microglia acquire properties of senescent cells including alterations in morphology, phenotypic profile and inability to respond appropriately to stimuli. Altogether, these findings support that a wide range of microglial phenotypes probably co-exist in the aged brain, playing different roles in neurodegeneration.
11 Figure I.2 | Microglial phenotypes in the healthy (M0, M1, M2a, M2b and M2c) and aged (dystrophic and primed) brain. In
the normal brain, neurons release several “resting” signals including CX3CL1 and CD200 inducing microglia to remain quiescent/vigilant (M0). In turn, M0 microglia excrete insulin growth factor 1 (IGF-1), brain-derived neurotrophic factor (BDNF) and transforming growth factor β (TGF-β) that are important for supporting normal neuronal development and functions. During brain surveillance, stimuli such as lipopolysaccharide (LPS) or interferon γ (IFN-γ) can induce microglial cytotoxic/classic activation (M1). In this state, microglia express increased levels of toll-like receptors (TLRs) and immune markers (e.g. major histocompatibility complex class II, MHC class II), while secrete pro-inflammatory mediators (e.g. interleukin (IL)-6, IL-1β, tumor necrosis factor α, TNF-α) and reactive species due to the enhanced expression of inducible nitric oxide synthase (iNOS). On the other hand, stimulation of microglia with IL-4/IL-13, LPS/immunocomplexes or IL-10/TGF-β/glucocorticoids trigger microglial alternative activation (M2a), less alternative activation (M2b) or acquired deactivation (M2c), respectively. M2a microglia are important for the generation of an anti-inflammatory environment by secreting anti-inflammatory cytokines (TGF-β), and for the extracellular matrix (ECM) repair by exhibiting increased levels of arginase 1 (Arg1). The expression of IL-1 receptor antagonist (IL-1Ra) is considered a key biomarker for M2a microglia, whereas they antagonize the synthesis of pro-inflammatory markers. M2b microglia are immunoregulatory cells by exhibiting a mixed M1/M2a phenotype characterized by pro-inflammatory (6, IL-1β, TNF-α) and anti-inflammatory (IL-10) markers. Finally, M2c microglia are important in the resolution phase of inflammation particularly for debris scavenging and immunoregulation, as they are characterized by enhanced levels of CD163, IL-10 and TGF-β. Additional microglial phenotypes have been proposed to populate the aged brain possibly playing a role in neurodegeneration: dystrophic/senescent microglia, which are characterized by condensed nucleus and swelling formation, concomitantly with irresponsiveness to stimuli and lack of neuronal supportive functions; and primed microglia, which are over-responsive to stimuli, especially systemic infection, leading to the generation of exacerbated and prolonged neuroinflammation.
2.4.
Microglial deregulation in Alzheimer’s disease
As stated above, neuroinflammation is currently assumed to participate in AD pathogenesis by sustaining the accumulation of exacerbated amount of Aβ and NFTs in the AD brain. Since microglia play a pivotal role in the regulation of neuroinflammation, it is widely accepted that the whole neurodegenerative process might depend on microglial functionality, which can be affected by several environmental and genetic factors that ultimately can drive irreversible microglial impairment (Heneka et al. 2014, Heppner et al. 2015, Mosher and Wyss-Coray 2014, Prokop et al. 2013).
I. Introduction
12
Under normal conditions, quiescent microglia (M0) promptly recognize distinct stimuli that trigger microglial pro-inflammatory (M1) or anti-inflammatory (M2) response. Microglia are appropriately induced to proliferate, secrete cytokines, chemokines and oxidative species, undergo chemotaxis or phagocytosis to protect the healthy neurons against injurious agents. Following the resolution phase of inflammation, microglial cells return to their quiescent/vigilant state thereby restoring the brain homeostasis.
In AD, the presence of pathological protein aggregates particularly Aβ, alterations in the central nervous system (CNS) (e.g. trauma), the incidence of systemic or local inflammatory disorders and/or mutations in specific genes can support microglial deregulation by affecting their phagocytic ability and motility, as well as cytokine production (Heppner et al. 2015, Prokop et al. 2013). The M1 and M2a/M2b/M2c microglial activation states are quite well characterized based on their surface markers, products secreted and functions in normal conditions, though it has not been yet understood whether the presence of these microglial phenotypes individually affect the course of AD. Besides classic activated microglia typically correlate with cytotoxic features, numerous studies performed in transgenic AD mouse models demonstrate that the presence of microglia exhibiting M1-related markers might have beneficial effects by reducing Aβ pathology (Varnum and Ikezu 2012, Wilcock 2012). On the other hand, the presence of M2 microglia is not exclusively attributed to microglial protective functions in AD, as M2 microglial phenotypic markers are found in the brain both in early and late AD stages (Sudduth et al. 2013). Moreover, studies of transgenic AD mouse models provide controversial data regarding the role of M2a, M2b and M2c microglia controlling Aβ pathology (Varnum and Ikezu 2012, Wilcock 2012). For instance, it was recently demonstrated that the accumulation of IL-10, a major anti-inflammatory cytokine, might inhibits microglial ability to clear Aβ (Michaud and Rivest 2015). On the other hand, it has been highly discussed whether mutations in genes encoding TREM2 and CD33, which are considered markers of M2 microglia (Walker and Lue 2015), affect microglial ability to uptake Aβ in AD (Heppner et al. 2015, Prokop et al. 2013).
Besides it is still debatable whether M1 and M2 microglial phenotypes play detrimental or protective roles in AD, it seems clear that the use of inflammation modulator treatments, including non-steroidal anti-inflammatory drugs and glucocorticoids, should be carefully considered as they can deregulate microglial activation towards harmful phenotypes when microglia are still functional (Meraz-Rios et al. 2013). However, chronic exposure to Aβ, cytokines and chemokines can drive microglial dysfunction towards primed activation states, where microglia respond exaggeratedly to stimuli generating strong pro-inflammatory environment. As abovementioned, the presence of primed microglia in the aged brain is particularly deleterious in both humans and mice (Godbout et al. 2005, Lemstra et al. 2007), possibly predisposing to the development of AD. At this time, therapeutic interventions
13 should be aimed at the restoration of normal microglial function before microglia come in senescence, an irreversible state of microglial dysfunction/dystrophy. Streit and his colleagues have been proposing an alternative perspective on AD pathogenesis centered in microglial dysfunction, as they found that microglial dystrophy is enhanced in the brain of individuals with increased Aβ load compared with Aβ-free controls (Flanary et al. 2007). Moreover, they reported that dystrophic microglia co-localize with both Aβ deposits and tau-composing structures in the brain of AD patients (Streit et al. 2009), suggesting not only that Aβ induces microglial senescence rather than activation, but also that the presence of dystrophic microglia prompts tau pathology. On the other hand, they claim that some factors including lifestyle, diet, physical and mental activities, as well as exposure to drugs and pollutants are relevant for driving microglial senescence, supporting the idea that microglial dystrophy and AD can occur spontaneously only in humans (Streit and Xue 2012, 2013, Streit et al. 2014).
In summary, it seems clear that microglia are deregulated in AD, and that distinct functional and/or dysfunctional phenotypes may co-exist playing different roles in the AD brain. However, more attention should be focused on identifying relationships between microglial phenotypes and events in AD, particularly using brain tissues and cells of human origin.
3. MicroRNAs: biogenesis and functions
MicroRNAs (miRNAs or miRs) are small single-stranded RNAs that are endogenously expressed in diverse species and cells. Besides being non-coding, miRNAs regulate the expression of specific genes at the post-transcriptional level blocking protein synthesis.
MiRNAs can be originated through two different pathways depending on the primary genomic loci: the canonical and non-canonical pathways. In the canonical pathway, the RNA polymerase II mediates miRNA gene transcription to pri-miRNA, a large stem-loop hairpin structure. Then, the pri-miRNA is asymmetrically cleaved in the nucleus towards the generation of a pre-miRNA, mediated by an enzymatic complex containing Drosha, an RNAse III endonuclease, and DiGeorge syndrome critical region gene 8. In the non-canonical pathway, the miRNA precursor, named mirtrons, are directly spliced from the intronic sequences of transcribed genes forming a lariat structure which is de-branched to form the pre-miRNA hairpin structure. The processed pre-miRNA either obtained through the canonical or non-canonical pathways is then exported to the cytoplasm by Exportin-5/RanGTP where it is cleaved by another RNAse III endonuclease (Dicer), losing the stem-loop of the precursor to produce a double-stranding miRNA molecule. Then, the functional
I. Introduction
14
strand associates with Argonaute proteins 1-4 in order to originate a RNA-induced silencing complex, while the complementary strand, usually denoted as miRNA*, is rapidly degraded. Finally, RISC can recognize and bind to complementary seed sequences in the 3’ untranslated region of target mRNAs, resulting in their degradation and consequent repression of the translation process (Bartel 2004, Winter et al. 2009).
Several miRNAs have been implicated in many important biological processes including cell development, proliferation, differentiation, inflammation and immunity. Hence, understanding the role of miRNAs in the regulation of genes involved in microglial neuroinflammatory response is fundamental to assess microglial profile and functions in pathological conditions.
3.1.
Inflammation-related microRNAs in microglia
Several miRNAs have been so far identified to be likely involved in the regulation of microglial functions (Michell-Robinson et al. 2015). With regards to neuroinflammation, numerous studies support that the miR-124, miR-155 and miR-146a play a pivotal role in the regulation of microglial phenotype by promoting microglial quiescence (miR-124), or by driving microglial inflammatory and immune response (miR-155 and miR-146a) (Ponomarev et al. 2013).
3.1.1. MiR-124
The miR-124 is a brain-enriched miRNA, whereas it is particularly expressed in neurons (Jovicic et al. 2013). During the CNS development, the expression of miR-124 is important for supporting neuronal differentiation and maturation (Makeyev et al. 2007, Visvanathan et al. 2007) and regulate axonal and dendritic branching (Franke et al. 2012). Moreover, it can also disinhibit neurite outgrowth in an inflammatory environment (Hartmann et al. 2015).
In microglia, C/EBP-α is established as one of the main targets of miR-124. C/EBP-α was reported to be expressed in low levels in quiescent microglia, though it is upregulated upon microglial activation (Walton et al. 1998). Furthermore, C/EBP-α and its downstream target PU.1 play an important role driving the development of myeloid cells both in the first and second waves of hematopoiesis. Upon miR-124 inhibition of C/EBP-α/PU.1, differentiation of macrophages to adult microglia was shown to be favored in detriment of monocytes proliferation (Ponomarev et al. 2011).
The miR-124 was found to be crucial for maintaining microglial quiescence, as miR-124 inhibition of C/EBP-α reduces the expression of CD45, MHC class II and F4/80, iNOS and
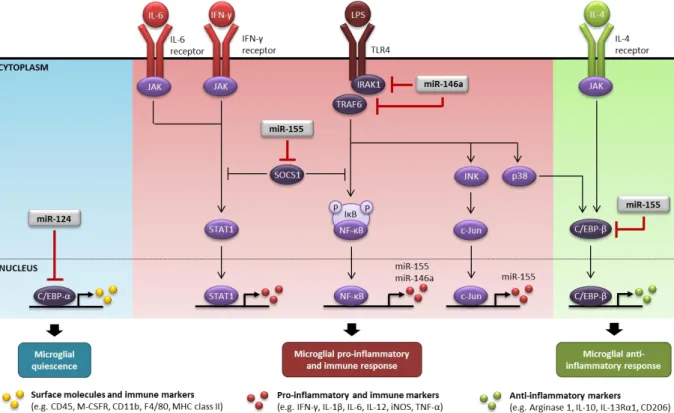
15 TNF-α, all involved in microglial pro-inflammatory and immune response, while enhances the expression of the anti-inflammatory agents FIZZ1, Arg1 and TGF-β (Ponomarev et al. 2011)
(Figure I.3). In a mice model of chronic pain, intrathecal administration of miR-124 was
shown to normalize the M1/M2 phenotypic markers ratio after an inflammatory insult, which might has beneficial effects in disease (Willemen et al. 2012). On the other hand, during experimental autoimmune encephalomyelitis, peripheral administration of miR-124 was shown to promote systemic deactivation of macrophages (Ponomarev et al. 2011). These data suggest that miR-124 not only contributes to maintain the MHC class IIlow/CD45low profile of quiescent microglia, but might also has an immunoregulatory role by promoting cell deactivation while enhance the expression of anti-inflammatory agents. When macrophages reach the brain, they are possibly deactivated due to direct transfer of 124 from miR-124+ neurons through exosomal shuttle vesicles (Ponomarev et al. 2013). Since microglial expression of miR-124 does not require neither IL-4/IL-13 receptors, typically activated when microglia are polarized towards the M2a phenotype, nor STAT6 signaling (Veremeyko et al. 2013), it is also reasonable that miR-124+ neurons, rather than IL-4, regulate the levels of miR-124 in quiescent microglia, besides IL-4 is highly expressed in the normal CNS (Ponomarev et al. 2007). On the other hand, when quiescent microglia are stimulated with IL-4 towards the alternative activated state, the levels of miR-12IL-4 decay (Freilich et al. 2013).
Recently, it was demonstrated that the TNF receptor-associated factor 6 (TRAF6) is a direct target of miR-124 (Qiu et al. 2015) providing alternative mechanisms for miR-124-mediated immunosuppression in microglia.
3.1.2. MiR-155
The miR-155 is considered a pro-inflammatory miRNA, as its expression is upregulated in response to LPS, TNF-α or IFN but downregulated in response to anti-inflammatory cytokines such as IL-10 and TGF-β in myeloid cells (McCoy et al. 2010, O'Connell et al. 2007, Tili et al. 2007).
In murine microglia, LPS was shown to induce miR-155 upregulation as a result of the activation of many inflammatory transcription factors including NF-κB, c-Jun, STAT1-4, and IRF1, IRF3, IRF7 and IRF8 (Freilich et al. 2013). In murine N9 microglial cell line, miR-155 overexpression was reported to directly inhibit the expression of suppressor of cytokine signaling 1 (SOCS1), a key repressor of NF-κB and JAK/STAT1 signaling pathways. Targeting SOCS1 induces the upregulation of several M1 markers including IL-1β, IFN-γ, iNOS, IL-6 and TNF-α, potentiating pro-inflammatory and immune microglial response (Cardoso et al. 2012) which can have detrimental impact on neurogenesis (Woodbury et al.
I. Introduction
16
2015) (Figure I.3). On the other hand, the expression of SOCS1 can be restored upon miR-155 suppression (Kim et al. 2014).
Besides supporting classic microglial activation, upregulation of miR-155 was shown to inhibit the STAT6 anti-inflammatory signaling pathway in microglia (Freilich et al. 2013) and macrophages (Martinez-Nunez et al. 2011). Additionally, miR-155 was reported to inhibit microglial alternative activation by repressing c-Maf, a transcription factor that regulates the anti-inflammatory response in myeloid cells (Su et al. 2014). In murine and human myeloid cells, miR-155 was also shown to directly repress C/EBP-β (He et al. 2009, Worm et al. 2009), a transcription factor that regulates the expression of several anti-inflammatory genes including IL-10, Arg1, IL-13 receptor α1 and CD206 (Ruffell et al. 2009). C/EBP-β can be positively regulated by the transcriptional activity of cyclic AMP response element-binding (CREB) via TLR4-p38 MAPK (Ananieva et al. 2008, El Kasmi et al. 2008), or STAT6 via IL-4 receptor-JAK (Albina et al. 2005), and it is particularly important for sustaining immunosuppression and tissue repair (Figure I.3).
Overexpression of miR-155 was also reported to occur in brain sections and microglia of aged mice compared with samples of adult counterparts (Fenn et al. 2013), suggesting that deregulated expression of miR-155 in elderly potentially provide favorable conditions to the development of inflammation-related neurodegenerative disorders.
3.1.3. MiR-146a
Like miR-124, the miR-146a is also considered a brain-enriched miRNA though it is rather expressed in microglia than in neurons (Jovicic et al. 2013).
The miR-146a is upregulated in myeloid cells in response to TNF-α, IL-1β or LPS, through the activation of NF-κB (Li Y. Y. et al. 2011c, Lukiw 2012, Perry M. M. et al. 2008). However, upregulation of miR-146a negatively regulates NF-κB by targeting two components of the TLR signaling pathway, the IL-1R-associated kinase 1 (IRAK1) and TRAF6 (Taganov et al. 2006). Inhibition of IRAK1 and TRAF6 blocks TLR signaling by reducing phosphorylation and degradation of the inhibitor of κB, which consequently blocks the translocation of the NF-κB to the nucleus. As a consequence, miR-146a promotes abrogation of NF-κB-mediated transcription of several pro-inflammatory genes such as IL-1β, IFN-γ, iNOS, IL-6 and TNF-α, attenuating both the immune and inflammatory responses. Due to the inhibition of NF-κB transcriptional activity, miR-146a also generates a negative feedback on its own expression, as well as on the expression of miR-155 (Rusca and Monticelli 2011)
(Figure I.3). In primary young murine macrophages, lack of miR-146a expression was shown
17 LPS, generating harmful uncontrolled chronic inflammation (Boldin et al. 2011). Altogether, these data suggest that miR-146a overexpression acts like an inflammatory break while triggers the resolution of inflammation.
However, some studies evoked that miR-146a overexpression in the brain potentiates inflammation rather than constraints. Like other NF-κB-sensitive miRNAs, upregulation of miR-146a was found to inversely correlate with the expression of the complement factor H (CFH), an important repressor of the innate immune response and inflammatory signaling (Lukiw et al. 2008). These findings were validated in human neuron-glial (HNG) co-cultures, human astroglial (HAG) and human microglial (HMG) cells, whereas TNF-α-induced miR-146a coupled with CFH and IRAK1 repression (Li Y. Y. et al. 2011b). In HAG cells, it was also shown that miR-146a inhibition of IRAK1 resulted in compensatory NF-κB-mediated upregulation of IRAK2 (Cui et al. 2010), suggesting that IRAK2 can drive an alternative mechanism for NF-κB activation after miR-146a overexpression. Nevertheless, IRAK2 was reported to be repressed by miR-146a in murine macrophages (Hou et al. 2009), as well as in human astrocytes (Iyer et al. 2012) and in CHME3 microglia (Sharma et al. 2015), suppressing NF-κB activation and inflammation. These data support the controversy regarding the role of miR-146a in the regulation of inflammation and immunity, especially concerning the brain.
On the other hand, miR-146a is considered a marker of cellular senescence (Olivieri et al. 2013a, Olivieri et al. 2013b). A recent study performed in our laboratory reported that, when primary neonatal murine microglia are cultured in vitro for long time, they acquire a miR-146a-enriched profile and decreased levels of miR-124 and miR-155, which together with other markers is indicative of microglial senescence and loss of function (Caldeira et al. 2014). Interestingly however, studies performed in primary cells and brain tissues obtained from aged mice demonstrated that miR-146a is overexpressed in macrophages and brain tissues but not in microglia (Fenn et al. 2013, Jiang et al. 2012, Li N. et al. 2011a). Age-related NF-κB activation can be one of the reasons for the upregulation of miR-146a in elderly (Ye and Johnson 2001), which in turn might limit senescence-associated inflammation by inhibiting the expression of pro-inflammatory cytokines (Bhaumik et al. 2009, Jiang et al. 2012).