Adriana da Costa Meneses
Carcinomas do Epitélio Folicular da
Tiroide: Formas Familiares
2011/2012Adriana da Costa Meneses
Carcinomas do Epitélio Folicular da
Tiroide: Formas Familiares
Mestrado Integrado em Medicina
Área: Endocrinologia
Trabalho efetuado sob a Orientação de:
Dr.ª Elisabete Rodrigues
Trabalho organizado de acordo com as normas da revista:
Acta Médica Portuguesa
Projeto de Opção do 6º ano -
D
ECLARAÇÃO DEI
NTEGRIDADEEu, Adriana da Costa Meneses, abaixo assinado, nº mecanográfico 060801155, estudante do 6º ano do Mestrado Integrado em Medicina, na Faculdade de Medicina da Universidade do Porto, declaro ter atuado com absoluta integridade na elaboração deste projeto de opção.
Neste sentido, confirmo que NÃO incorri em plágio (ato pelo qual um indivíduo, mesmo por omissão, assume a autoria de um determinado trabalho intelectual, ou partes dele). Mais declaro que todas as frases que retirei de trabalhos anteriores pertencentes a outros autores, foram referenciadas, ou redigidas com novas palavras, tendo colocado, neste caso, a citação da fonte bibliográfica.
Faculdade de Medicina da Universidade do Porto, ___/___/______
Projeto de Opção do 6º ano – DECLARAÇÃO DE REPRODUÇÃO
Nome: Adriana da Costa Meneses
Endereço eletrónico: [email protected] Telefone ou Telemóvel: 912945008 Número do Bilhete de Identidade: 13363293
Título da Monografia: Carcinomas do Epitélio Folicular da Tiroide: Formas Familiares Orientador: Maria Elisabete Gonçalves Rodrigues
Ano de conclusão: 2012
Designação da área do projeto: Endocrinologia
É autorizada a reprodução integral desta Monografia para efeitos de investigação e de divulgação pedagógica, em programas e projetos coordenados pela FMUP.
Faculdade de Medicina da Universidade do Porto, ___/___/______
Título: Carcinomas do Epitélio Folicular da Tiroide: Formas Familiares Tipo de Artigo: Monografia
Contagem de Palavras: Artigo: 3427 Referências: 60 Resumo: 265 Abstract: 253 Figuras: 0 Tabelas: 5 Agradecimentos:
À Dr.ª Elisabete Rodrigues, pela disponibilidade, orientação dedicada e críticas pertinentes ao longo da execução da tese.
Ao Professor Davide Carvalho pela disponibilidade sempre demonstrada.
À minha família pelo apoio incondicional, paciência e carinho ao longo de todo este percurso. Aos meus amigos por me terem feito sempre sorrir ao longo do projeto.
Lista de Autores: Adriana, MENESES Serviço de Endocrinologia
Faculdade de Medicina da Universidade do Porto Alameda Professor Hêrnani Monteiro, 4200 Porto +351225513618
ÍNDICE
LISTA DE ABREVIATURAS E SIGLAS ... 4
RESUMO ... 5
ABSTRACT ... 7
INTRODUÇÃO ... 8
MÉTODOS ... 9
CARCINOMAS FAMILIARES DO EPITÉLIO FOLICULAR DA TIROIDE ... 10
FORMAS ASSOCIADAS A SÍNDROMES ... 10
Polipose Adenomatosa Familiar ... 10
Síndrome PTEN/Hamartoma ... 11 Complexo de Carney ... 13 Síndrome de Werner ... 13 FORMAS NÃO-SÍNDRÓMICAS ... 14 Critérios de Diagnóstico ... 14 Aspetos Genéticos ... 15 Aspetos Clínicos ... 16 Rastreio e Diagnóstico ... 18 Tratamento ... 19 CONCLUSÃO ... 19 REFERÊNCIAS ... 21 TABELAS... 25 ANEXOS ... 30
LISTA DE ABREVIATURAS
BMN Bócio multinodular
BAAF Biopsia aspirativa por agulha fina CFT Carcinoma Folicular da Tiroide CNMT Carcinoma não-medular da tiroide
CNMTf Forma familiar de carcinoma não-medular da tiroide CPT Carcinoma Papilar da Tiroide
CPTf Carcinoma papilar da tiroide familiar
HCEPR Hipertrofia congénita do epitélio pigmentado da retina NEM Neoplasia Endócrina Múltipla
PAF Polipose Adenomatosa Familiar
PRKAR1-α Subunidade reguladora da proteína quinase A tipo 1-α
CARCINOMAS DO EPITÉLIO FOLICULAR DA TIROIDE: FORMAS FAMILIARES
RESUMO
Os carcinomas da tiroide podem classificar-se em carcinomas medulares com origem nas células parafoliculares e em carcinomas não-medulares com origem nas células foliculares. Os carcinomas não-medulares são os mais frequentes sendo na sua maioria esporádicos. Todavia, cerca de 5% são considerados formas familiares.
As formas familiares dos carcinomas não-medulares da tiroide podem estar associados a síndromes ou serem não-sindrómicas.
O objetivo deste trabalho é fazer uma revisão do estado da arte acerca das formas familiares do carcinoma não-medular da tiroide, incluindo aspetos relacionados com as suas características clínicas, aspetos genéticos, formas de rastreio e diagnóstico bem como tratamento e prognóstico.
Nas formas associadas a síndromes existe um aumento da prevalência de carcinomas não-medulares da tiroide, no contexto de um síndrome neoplásico familiar, caracterizado pela predominância de tumores não tiroideus. Estes encontram-se bem caracterizados em termos clínicos e genéticos. Já o mesmo não acontece com as formas não-sindrómicas. Foram publicados critérios de diagnóstico para esta patologia, que mais tarde foram revistos, não existindo atualmente consenso. Este facto leva a que ainda não esteja bem estabelecido se há ou não uma maior agressividade clínica comparativamente às formas esporádicas. Continua a investigar-se a existência de um locus genético suscetível, não se tendo chegado a nenhuma conclusão definitiva, colocando-se a hipótese de poder tratar-se de um modelo poligénico. As formas de rastreio e tratamento também ainda não estão definidas de forma consensual.
São necessários mais estudos para se chegar a conclusões mais precisas e conseguir-se uma melhor gestão no reconhecimento de casos. Assim poder-se-ia instituir uma forma de rastreio eficaz e um potencial tratamento para indivíduos com risco aumentado.
ABSTRACT
Thyroid carcinomas can be classified as medullary carcinomas with origin in parafollicular cells and non-medullary carcinomas with origin in follicular cells, respectively. The non-medullary carcinomas are the most frequent and the majority of them are sporadic. However, around 5% are considered familiar forms.
The familiar forms of non-medullary thyroid carcinomas can be classified as being associated to syndromes or as being non-syndromic.
The purpose of this paper is to review the state of the art about, the familiar forms of non-medullary thyroid carcinomas, including aspects related to their clinical characteristics, genetics, screening, diagnosis as well as treatment and prognosis.
In the syndrome associated forms there is an increased prevalence of non-medullary thyroid carcinomas, within a familiar neoplastic syndrome, characterized by the predominance of non thyroid tumors. These are well classified both clinically and genetically. The same doesn’t happen with the non-syndromic forms. Diagnostic criteria were published for this pathology which were later revised, and currently there is no consensus. It is not yet well stablished whether or not there is greater clinical aggressiveness compared to the sporadic forms. The existence of a susceptible genetic locus is still being investigated without having reached any definitive conclusion. It also exists the possibility of this being a polygenic model. The screening and treatment form also are not well defined by consensus.
Further studies are needed to reach more accurate conclusions and to achieve a better case identification management. That way, there could be an effective screening procedure and a potential treatment for individuals with increased risk.
INTRODUÇÃO
A patologia nodular da tiroide é muito frequente na população geral, ainda mais nas mulheres que nos homens (razão de 3:1). A sua prevalência estimada por estudos ecográficos é de 20 a 76% o que é comparável com os dados das séries de autópsias1, 2. No entanto, apenas uma minoria é maligna, representando menos de 1% da totalidade dos carcinomas3, 4. Nos últimos anos tem havido um aumento da prevalência desta patologia maligna, sendo mesmo considerado um dos mais frequentes dentro do grupo das neoplasias malignas 3. Este aumento poderá, em parte, ser explicada pela maior acuidade e facilidade de acesso aos meios complementares de diagnóstico, o que leva à maior deteção de nódulos de pequenas dimensões, não palpáveis5.
Os carcinomas da tiroide classificam-se em carcinomas não-medulares, com origem nas células do epitélio folicular (nestes incluem-se o carcinoma papilar da tiroide (CPT), o carcinoma folicular da tiroide (CFT), as suas variantes de Hurthle e ainda formas mais raras e indiferenciadas do carcinoma) e carcinomas medulares, com origem nas células parafoliculares6. Ambos podem apresentar-se como formas esporádicas ou familiares, as quais pressupõem um componente hereditário na forma de transmissão.
Os carcinomas não-medulares da tiroide (CNMT) , os mais frequentes, correspondem a cerca de 95%, sendo a maioria esporádicos3. O primeiro caso de forma familiar de carcinoma não medular da tiroide (CNMTf) foi descrito quando se publicou o caso de dois gémeos de 24 anos com o diagnóstico desta patologia7. Nos últimos anos tem-se verificado um aumento da descrição de casos de CNMT com predisposição familiar, com prevalências que variam, em diversos estudos, entre 3,5 a 15% dos cancros da tiroide3, 6, 8-15.
As formas familiares do CNMT são um grupo heterogéneo de doenças podendo classificar-se em dois grupos: associadas a síndromes e não sindrómicas. No primeiro grupo existe um aumento da prevalência de CNMT no contexto de um síndrome neoplásico familiar,
caracterizado por predominância de tumores não tiroideus. Nas formas não sindrómicas a neoplasia predominante é o CNMT, ainda que outras neoplasias possam ocorrer com maior frequência3, 4, 6. (Ver tabela 1)
Ao contrário do que acontece com outras patologias tiroideias, como é o caso do carcinoma medular da tiroide, na CNMTf, a relação entre mecanismos moleculares e características fenotípicas não é linear, não existindo consenso sobre as implicações clínicas e terapêuticas. Como tal, o objetivo deste trabalho é fazer uma revisão do estado da arte acerca das formas familiares de CNMT, incluindo aspetos relacionados com as suas características clínicas, aspetos genéticos, formas de rastreio e diagnóstico, bem como tratamento e prognóstico. O interesse do conhecimento dos genes responsáveis por estas formas familiares advêm da possibilidade de eles contribuírem, através de mutações somáticas, para o aparecimento de formas esporádicas de carcinoma da tiroide16.
MÉTODOS
Para a realização deste trabalho efetuaram-se duas pesquisa na Medline, com as seguintes questões: “Familial thyroid carcinoma AND non-medullary” e “Familial papillary thyroid carcinoma”, tendo-se obtido um total de 36 e 149 artigos, respetivamente.
Os critérios de inclusão usados foram: 1) artigos escritos em português, inglês, espanhol ou francês; 2) estudos realizados em humanos; 3) artigos de revisão, originais, metanálises e editoriais. Foram excluídos os artigos: 1) sem resumo; 2) a que não se teve acesso ao artigo integral; 3) casos clínicos e cartas ao editor.
Foram selecionados pelo título, de acordo com os critérios de inclusão, 23 e 77 artigos respetivamente. Após a leitura do resumo foram excluídos, de acordo com os critérios usados, 4 e 39 artigos, respetivamente, ficando 54 artigos para leitura integral (3 deles repetidos nas
estudo. Foram ainda incluídos 19 outros artigos obtidos da bibliografia já adquirida, considerados úteis.
No total foram então utilizados 58 artigos e ainda as recomendações mais recentes relativas à patologia nodular tiroideia e carcinomas da tiroide.
CARCINOMAS FAMILIARES DO EPITÉLIO FOLICULAR DA TIROIDE
1. Formas Associadas a Síndromes
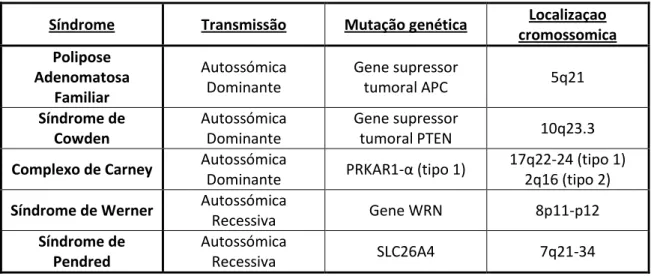
Os CNMT são encontrados com maior frequência em alguns síndromes caracterizados por outro tipo de tumores, como a Polipose Adenomatosa Familiar (FAP), Síndrome de Gardner, Síndrome de Cowden (SC), Complexo de Carney, Síndrome de Werner, Síndrome de Pendred e Neoplasias Endócrinas Multiplas (NEM) tipo II. Nestas síndromes as características anatomopatológicas dos tumores da tiroide são por vezes diferentes devendo alertar os anatomopatologistas para sugerir uma investigação mais cuidadosa do indivíduo afetado, bem como dos seus familiares3, 4, 6. Na tabela 2 e 3 encontra-se o resumo das alterações genéticas e manifestações clínicas das várias síndromes acima referidas.
Existem ainda outras síndromes onde foi diagnosticado CNMT, no entanto, a sua ligação ainda não está bem estabelecida. São exemplos a síndrome de McCune Albright, a síndrome de Peutz Jeghers e a síndrome da ataxia-telangiectasia17.
Polipose Adenomatosa Familiar
É uma síndrome com transmissão autossómica dominante, onde ocorre uma mutação germinativa no gene APC localizado no cromossoma 5q21. Caracteriza-se pela presença de centenas de pólipos adenomatosos no cólon que se desenvolvem nos adultos jovens, e se não forem retirados originam carcinoma do cólon em virtualmente 100% dos casos. As alterações extra-cólicas da PAF incluem osteomas, cistos epidérmicos, tumores desmóides, hamartomas
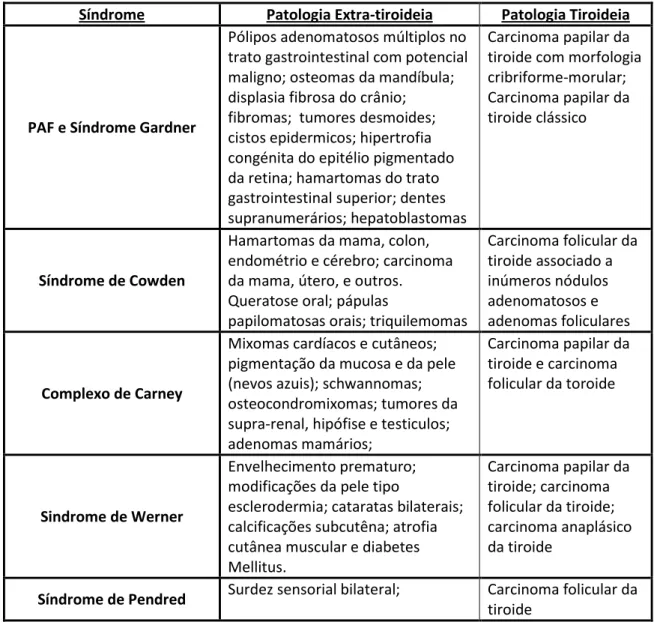
poliposos do trato gastrointestinal superior, hipertrofia congénita do epitélio pigmentado da retina (HCEPR), hepatoblastoma e tumores da tiroide3, 4, 6, 17, 18.
Os tumores da tiroide ocorrem, segundo diversos autores, numa frequência de 2 a 15%5, 6, 19, 20, tendo as mulheres jovens com PAF uma probabilidade 160 vezes maior de desenvolver carcinoma da tiroide que a população geral. Segundo Cetta e col., em 33% dos indivíduos diagnostica-se primeiro o carcinoma da tiroide19. Os carcinomas da tiroide associados à PAF são geralmente multifocais, bilaterais e histologicamente diferentes dos tumores esporádicos3, 17, 20, 21
. Em 90% dos casos a histologia revela morfologia cribriforme-morular, que é rara na forma esporádica (0,1 a 0,2%). Por isso, indivíduos que sejam diagnosticado com esta forma de CNMT devem ser investigados à procura de uma possível PAF17, 22-24. O prognóstico destes tumores é excelente (menos de 10% são agressivos)3, 17, 20, 21. Esta variante do CPT pode ser distinguida pela expressão aumentada de betacatenina17.
Foi descrita uma região onde as mutações ocorrem com maior frequência nos indivíduos com PAF e CPT. Cetta e col. concluíram que 87% dos doentes com estas duas entidades associadas tinham mutações germinativas, e 80% possuíam a mutação na região genómica associada à HCEPR, e na região cluster mutacional da região 5’ do exão 1519. Nos doentes com HCEPR e nos seus familiares deve realizar-se rastreio tiroideu a partir dos 15 anos5. Nos indivíduos com mutações noutras regiões do gene APC não está indicada a realização de rastreio de rotina17. Os doentes diagnosticados com CPT devem ser submetidos a tiroidectomia total20, 24.
Síndrome Tumor PTEN-Hamartoma
Esta síndrome é um complexo provocado por uma mutação inativante germinativa do gene supressor tumoral PTEN, que se localiza no cromossoma 10q23.3 e inclui a Síndrome de Cowden, Síndrome Bannayan-Riley-Ruvalcaba, Síndrome Proteus e Síndrome Proteus like. Apenas 10 a 50% dos doentes têm familiares afetados com a doença3, 4.
No SC ,as mutações do gene PTEN foram encontradas em 80 a 85% dos doentes afetados. As manifestações fenotípicas desenvolvem-se, em mais de 90% dos indivíduos, por volta dos 20 anos. Aos 30 anos quase todos já desenvolveram lesões mucocutâneas que são patognomónicas. O SC é diagnosticado em média por volta da quarta década de vida3. Ao contrário do que acontece com a PAF, aqui os hamartomas não são considerados pré-malignos18. Os indivíduos afetados irão desenvolver tumores benignos e malignos num variado número de tecidos como mama, útero e tiroide. Muitos doentes também têm lesões mucocutâneas como queratose acral, pápulas papilomatosas orais e triquilemomas5, 17, 18. Os critérios de diagnóstico foram definidos pelo International Cowden’s Consortium CD diagnosis criteria/National Cancer Network25
Os achados anatomopatológicos da tiroide são relativamente frequentes nesta síndrome (presentes em 60% dos indivíduos) e afetam principalmente as células foliculares17. Nesta síndrome, o CFT é geralmente multicêntrico e origina-se a partir de um adenoma folicular pré-existente, surge com uma frequência de 5 a 10% e é considerado um critério de diagnóstico major. O bócio multinodular, nódulos adenomatosos múltiplos e adenomas foliculares são critérios de diagnóstico minor, ocorrendo com uma frequência de 50 a 67%. Na SC os nódulos adenomatosos são caracteristicamente múltiplos, podendo exceder os 100 nódulos e variam entre 0,1 a 0,8 cm. Os adenomas foliculares são também muito comuns e geralmente múltiplos. O CPT raramente está associado a esta síndrome. Nalguns estudos identificou-se hiperplasia das células C, ainda que o carcinoma medular não faça parte do espectro da SC3, 26. O diagnóstico das lesões tiroideias geralmente precede o diagnóstico da SC em muitos anos17
A grande incidência de patologia tiroideia em doentes com SC leva a que alguns autores recomendem a realização de ecografia cervical de rotina5, 17, não existindo no entanto recomendações específicas que o aconselhem18. Nos casos onde a biópsia aspirativa por agulha fina (BAAF) é duvidosa, alguns autores recomendam a realização de tiroidectomia17.
Complexo de Carney
É uma síndrome de transmissão autossómica dominante caracterizada pela presença de pigmentação da pele, hiperatividade endócrina e mixomas (podem estar presentes no coração, pele (lentigos), tecido conjuntivo, canal auditivo externo e mama)17. É definido pela associação de múltiplas neoplasias endócrinas e outras manifestações como acromegalia, hiperplasia micronodular primária pigmentada do córtex suprarrenal, mixomas cardíacos e cutâneos, lentiginose, nevos azuis múltiplos, adenomas mamários, tumores testiculares, cistos ováricos, schwanomas, osteocondromixomas ou tumores tiroideus. Esta síndrome é diagnosticada aquando da presença de duas ou mais destas manifestações ou se o paciente tiver a mutação germinativa da subunidade reguladora da proteína quinase A tipo 1-alfa (PRKAR1-α) e/ou se um parente de primeiro grau estiver afetado com uma única manifestação3, 17. Esta síndrome é classificada como tipo 1 quando há uma mutação na PRKAR1-α, ou tipo 2 quando a mutação se encontra no cromossoma 2q1627.
Nestes doentes a tiroide é normalmente nodular, com nódulos adenomatosos bilaterais, adenomas foliculares, CPT e CFT. Estas características estão presentes em cerca de 11-15% dos doentes. Ainda que a probabilidade de carcinoma da tiroide seja baixa, a prevalência de nódulos tiroideus é bastante elevada4-6, 28.
Doentes com Complexo de Carney ou predisposição genética para o mesmo devem fazer rastreio anual das manifestações da doença17.
Síndrome de Werner
A síndrome de Werner é uma doença rara do tecido conjuntivo caracterizada por transmissão autossómica recessiva, que tipicamente se manifesta na terceira década de vida. É caracterizada por envelhecimento precoce, cataratas bilaterais, cabelo cinzento e atrofia
cutânea e muscular. Os indivíduos afetados com esta síndrome têm risco aumentado de desenvolver inúmeras neoplasias, entre as quais, tiroideias29.
Esta síndrome é provocada por uma mutação no gene WRN, que codifica uma proteína importante na reparação e replicação do DNA, que está presente no cromossoma 8p11-p12. Suspeita-se deste diagnóstico pela apresentação clínica, sendo depois confirmado com estudos moleculares5, 17, 29.
As neoplasias da tiroide nestes indivíduos ocorrem numa idade inferior à população geral, com uma razão mulher homem menor (2:1). Existe uma probabilidade três vezes maior de desenvolver CFT e seis vezes superior de desenvolver carcinoma anaplásico. Devem ser feitos exames de rastreio nestes doentes3-5, 17.
2. Formas Não Sindrómicas
Fazem parte deste grupo as formas familiares do carcinoma papilar, o carcinoma papilar da tiroide com ou sem oxifilia, o carcinoma papilar da tiroide associado a carcinoma papilar das células renais e o carcinoma papilar da tiroide com bócio multinodular6.
Critérios de Diagnóstico
Após a identificação do primeiro caso de carcinoma papilar da tiroide familiar (CPTf) em 1955 foram surgindo cada vez mais descrições de CPTf sem qualquer associação a outras síndromes clínicas ou exposição ambiental, como é o exemplo da publicação de Lote e col. em 198030. Perante esta heterogeneidade de publicações, sem que existissem critérios rigorosos, e para se conseguir realizar estudos epidemiológicos, genéticos e clinicopatológicos precisos, tornou-se fundamental a criação de critérios de diagnóstico para esta entidade clínica31, 32. Assim, Musholt e col., em 2000, realizaram uma meta-análise de toda a literatura inglesa de CPTf, conseguindo estabelecer os critérios de identificação para esta patologia (tabela 4). Estes
critérios, apesar de uteis pelo seu carater objetivo, não foram considerados absolutos, admitindo os próprios autores a possibilidade de exceções31.
No entanto, Charkes e col. demonstraram que devido à grande prevalência de cancro da tiroide na população em geral, se numa família existirem apenas dois membros com CNMT a probabilidade de ser esporádico é muito elevada (62-69%). Todavia se existirem três ou mais familiares de primeiro grau com CNMT a possibilidade ser esporádico é inferior a 6%33. Mais recentemente, outros autores consideram como definição da CNMTf a existência de três ou mais familiares de primeiro grau com CNMT, na ausência de outro síndrome familiar3, 4, 6, 34. Dotto e col. afirmam que famílias com 3 ou mais membros afetados e a presença de CPT em homens e crianças é mais sugestivo de predisposição familiar4.
Aspetos Genéticos
Vários autores sugerem que a CNMTf tem transmissão autossómica dominante com penetrância incompleta35. Todavia, há 10 anos que se tenta encontrar, sem sucesso, um locus genético suscetível. Até à data já se excluíram alguns genes importantes (RET, TRKA, JUNB, APC, PTEN e TSHR, MET, BRAF, PAX8 e NIS)6, 16, 36-41, existindo 6 regiões potenciais de localização genética (MNG1, TCO, fPTC/PRN, NMTC1, FTEN e complexo telomerase-telomerase)8, 16, 35, 38, 42-44.
Os genes anteriormente referidos foram encontrados em famílias isoladas, cujos resultados nem sempre são confirmados por outros autores11, 36-38, 42-49. A tabela 5 apresenta um resumo dos vários estudos a favor e contra estes genes específicos.
Os casos descritos sugerem que o gene MNG1 pode estar envolvido apenas num pequeno número de casos com bócio multinodular (BMN)16. No que diz respeito ao gene TCO pensa-se que pode estar associado a uma minoria de casos de tumores com oxifilia celular38. O gene
estar associado especificamente à síndrome CPT/CPR16, 42, 43. O gene NMTC1 além de ter sido reportado como sendo um locus suscetível na CNMTf, nas variantes foliculares do CPT foi ainda sugerido posteriormente, que pode haver um aumento do risco da CNMTf em indivíduos que tenham os loci NMTC1 e TCO44. Em relação ao complexo telomerase-telomerase, Capezzone e col. sugeriram que o aumento da atividade desta enzima é provocado por uma mutação genética que regula de forma positiva a expressão do gene da telomerase16.
É importante notar que apesar de existirem estudos com boa evidência para determinados genes suscetíveis, análises subsequentes contradizem esses achados. Isto pode ser, em parte, explicado pelas variações existentes nos diferentes desenhos de estudo. Alguns incluem indivíduos com doença tiroideia benigna, que nem sempre progride para carcinoma podendo estar a diluir os resultados. Por outro lado, na maioria não é usado um grupo controlo com indivíduos com carcinoma esporádico. Por fim, o facto de serem usadas famílias isoladas também contribui para esta heterogeneidade de resultados10, 16.
Percebe-se então que existem alguns fenótipos que estão associados a loci particulares e que devido à raridade das doenças documentadas não é possível realizar-se uma subcaracterização da doença. Estes achados levam a ponderar a possibilidade de estarmos perante um modelo de doença poligénico16.
Continua a ser fundamental a identificação de marcadores moleculares já que isso permitirá uma melhor gestão no reconhecimento de casos prováveis e um potencial tratamento em indivíduos com um aumento de risco de desenvolver a doença16.
Aspetos Clínicos
Após a definição da CNMTf como uma entidade clínica distinta, e da criação de critérios precisos de diagnóstico tornou-se importante definir quais as diferenças clínicas entre formas familiares e esporádicas para se conseguir implementar eventuais formas de atuação distintas.
Apesar dos esforços realizados na tentativa de esclarecer esta questão, ainda não se conseguiu reunir consenso. Alguns estudos sugerem que não existem diferenças clinico-patológicas entre a forma familiar e esporádica dos CNMT12, 14, 50-52. Outros mais recentes, sugerem que as formas familiares são mais frequentemente multifocais, têm maior número de metástases ganglionares, maior percentagem de recidiva e são diagnosticadas mais cedo em comparação com as formas esporádicas9, 13, 32, 49, 53-58. Foi também sugerido que a agressividade da doença poderá ser mais comum no caso índice e nas famílias com três ou mais membros afetados13, 32. No entanto, Capezzone e col. demonstraram que indivíduos de segunda geração são diagnosticados em idades mais jovens e têm doença mais agressiva em comparação com os familiares afetados, o que sugere a existência de um fenómeno de antecipação49.
Por outro lado, ainda que em alguns estudos se tenha provado que há uma maior agressividade na forma familiar em comparação com as formas esporádicas, no que diz respeito à sobrevivência, na maioria não foram reportadas diferenças estatisticamente significativas13, 53, 54, 57. Isto pode ser explicado pelo facto do carcinoma da tiroide ter uma mortalidade muito baixa. Não é por isso, previsível que venha a existir um estudo coorte com uma catamnese suficientemente longa para se conseguir detetar pequenas alterações na sobrevida10, 58. Apesar disso, alguns estudos reportaram um menor tempo livre de doença entre os indivíduos com CNMTf, principalmente aqueles com três ou mais membros da família afetados 13, 53, 58.
Foi também evidenciado em indivíduos com CNMTf que os seus familiares têm uma maior incidência de bócio multinodular e hipotiroidismo. Já a frequência de anticorpos não se revelou aumentada8, 31, 42.
A diversidade de resultados clinicopatológicos encontrados pode, em parte, também ser explicada pela variedade de desenhos de estudo e diferentes critérios de inclusão usados pelos
parentesco em primeiro grau afetados como critério para definir a presença da CNMTf. Os resultados podem, por isso, estar a ser desviados pela introdução de casos esporádicos como sendo familiares 33.
Rastreio e Diagnóstico
Atualmente colocam-se várias questões relativamente à forma de atuação perante uma família com CNMTf. Deve fazer-se rastreio aos membros de familiares afetados? Com que idade e grau de parentesco?
Todos os indivíduos com patologia tiroideia devem ser sujeitos a uma anamnese minuciosa na tentativa de identificar história familiar. Sendo a patologia da tiroide muito frequente na população geral a distinção entre benignidade e malignidade torna-se essencial e a recolha da história clínica pode ser um fator importante na decisão clínica a tomar13, 59, 60.
Ainda não estão definidos de forma rigorosa quais os procedimentos a seguir perante uma família com CNMTf. Alguns autores consideram que deve ser realizada ecografia cervical a todos os indivíduos com familiares de primeiro grau afetados, com o objetivo de detetar a doença num estádio mais precoce13, 56. Uchino e col. concluíram que pelo menos 52% de familiares de doentes com CNMTf têm um nódulo tiroideu, e em 10% dos casos é diagnosticado carcinoma, um valor superior ao da população geral. A idade para iniciar este rastreio não é consensual mas, estes autores sugerem que antes dos 20 anos não traz benefícios56. Os principais problemas associados a esta metodologia prendem-se com a relação custo-benefício, bem como a ansiedade gerada nos indivíduos implicados.
De seguida é necessário definir qual a atitude a tomar com os doentes em que sejam detetados nódulos. As mais recentes recomendações aconselham que, ainda que se trate de um nódulo infra-centimétrico, pelo maior risco que estes indivíduos têm de carcinoma, e porque a probabilidade de malignidade não varia com o tamanho do nódulo, os membros de
famílias com CNMTf deviam ser sujeitos a BAAF1, 2. Apesar destas indicações alguns autores mostram-se reticentes relativamente a este procedimento devido à maior percentagem de falsos negativos nestes casos10, 13, 58, 60. Caso o resultado da biópsia seja negativo, sugere-se, ainda que não seja consensual, que o procedimento deve ser repetido31.
Tratamento
Caso seja diagnosticado carcinoma da tiroide a abordagem terapêutica preconizada pelas últimas recomendações da American Thyroid Association é a tiroidectomia total2. Este procedimento é vantajoso pois permite a posterior realização de terapêutica ablativa com I-131 radioativo, e o controlo da persistência/recorrência da doença através do doseamento da tiroglobulina9, 10, 13, 60. Apesar de não ser consensual, vários estudos defendem que a tiroidectomia total deve ser acompanhada de dissecção profilática do grupo ganglionar central. Caso haja metastização nos gânglios da cadeia ganglionar lateral diagnosticada em exames pré-operatórios, deve realizar-se exérese da cadeia ganglionar afetada10, 58. A ablação com iodo radioativo é também aconselhada nestes estudos15, 54, 60. Maxwell e col., pelo contrário, consideram que não deve haver uma atuação diferente nestes casos em relação aos esporádicos14.
CONCLUSÃO
Os CNMTf podem ocorrer como formas isoladas ou associadas a síndromes. Apesar de ser atualmente reconhecido como uma entidade clinica distinta ainda gera alguma controvérsia no que diz respeito aos critérios de diagnóstico, à sua maior agressividade relativamente à forma esporádica, alterações genéticas associadas, bem como formas de rastreio e tratamento que devem ser instituídos.
desta patologia. Assim, seria possível definir de forma mais precisa qual o tipo de tratamento e rastreio mais adequado para os doentes afetados e seus familiares, respetivamente.
A continuidade da investigação por um locus genético suscetível é fundamental já que permitirá uma melhor gestão no reconhecimento de casos e o desenvolvimento de um potencial tratamento para indivíduos com risco aumentado.
REFERÊNCIAS
1. Gharib H, Papini E, Paschke R, Duick DS, Valcavi R, Hegedus L, et al. American Association of Clinical Endocrinologists, Associazione Medici Endocrinologi, and EuropeanThyroid Association Medical Guidelines for Clinical Practice for the Diagnosis and Management of Thyroid Nodules. Endocr Pract. 2010;16 Suppl 1:1-43.
2. Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167-214.
3. Nose V. Familial non-medullary thyroid carcinoma: an update. Endocr Pathol. 2008;19(4):226-40.
4. Dotto J, Nose V. Familial thyroid carcinoma: a diagnostic algorithm. Adv Anat Pathol. 2008;15(6):332-49.
5. Richards ML. Familial syndromes associated with thyroid cancer in the era of personalized medicine. Thyroid. 2010;20(7):707-13.
6. Nose V. Familial thyroid cancer: a review. Mod Pathol. 2011;24 Suppl 2:S19-33.
7. Robinson DW, Orr TG. Carcinoma of the thyroid and other diseases of the thyroid in identical twins. AMA Arch Surg. 1955;70(6):923-8.
8. Cross G, Pitoia F, Suarez H, Kral M, Manavela M, Morando D, et al. High prevalence of thyroid disorders in relatives of patients with familial papillary thyroid cancer. Medicina (B Aires). 2010;70(2):139-42.
9. Ito Y, Kakudo K, Hirokawa M, Fukushima M, Yabuta T, Tomoda C, et al. Biological behavior and prognosis of familial papillary thyroid carcinoma. Surgery. 2009;145(1):100-5. 10. Kebebew E. Hereditary non-medullary thyroid cancer. World J Surg. 2008;32(5):678-82. 11. Prazeres HJ, Rodrigues F, Soares P, Naidenov P, Figueiredo P, Campos B, et al. Loss of heterozygosity at 19p13.2 and 2q21 in tumours from familial clusters of non-medullary thyroid carcinoma. Fam Cancer. 2008;7(2):141-9.
12. Moses W, Weng J, Kebebew E. Prevalence, clinicopathologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid. 2011;21(4):367-71.
13. Triponez F, Wong M, Sturgeon C, Caron N, Ginzinger DG, Segal MR, et al. Does familial non-medullary thyroid cancer adversely affect survival? World J Surg. 2006;30(5):787-93. 14. Maxwell EL, Hall FT, Freeman JL. Familial non-medullary thyroid cancer: a matched-case control study. Laryngoscope. 2004;114(12):2182-6.
15. Marchesi M, Biffoni M, Biancari F, Faloci C, Cresti R, Mariotti F, et al. Familial papillary carcinoma of the thyroid: a report of nine first-degree relatives of four families. Eur J Surg Oncol. 2000;26(8):789-91.
16. Khan A, Smellie J, Nutting C, Harrington K, Newbold K. Familial nonmedullary thyroid cancer: a review of the genetics. Thyroid. 2010;20(7):795-801.
17. Nose V. Thyroid cancer of follicular cell origin in inherited tumor syndromes. Adv Anat Pathol. 2010;17(6):428-36.
18. Harb WJ, Sturgis EM. Differentiated thyroid cancer associated with intestinal polyposis syndromes: a review. Head Neck. 2009;31(11):1511-9.
19. Cetta F, Montalto G, Gori M, Curia MC, Cama A, Olschwang S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: results from a European cooperative study. J Clin Endocrinol Metab. 2000;85(1):286-92. 20. Herraiz M, Barbesino G, Faquin W, Chan-Smutko G, Patel D, Shannon KM, et al. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(3):367-73.
inactivation of the adenomatous polyposis coli gene. J Clin Endocrinol Metab. 2001;86(1):427-32.
22. Donnellan KA, Bigler SA, Wein RO. Papillary thyroid carcinoma and familial adenomatous polyposis of the colon. Am J Otolaryngol. 2009;30(1):58-60.
23. Lee S, Hong SW, Shin SJ, Kim YM, Rhee Y, Jeon BI, et al. Papillary thyroid carcinoma associated with familial adenomatous polyposis: molecular analysis of pathogenesis in a family and review of the literature. Endocr J. 2004;51(3):317-23.
24. Ito Y, Miyauchi A, Ishikawa H, Hirokawa M, Kudo T, Tomoda C, et al. Our experience of treatment of cribriform morular variant of papillary thyroid carcinoma; difference in clinicopathological features of FAP-associated and sporadic patients. Endocr J. 2011;58(8):685-9.
25. Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8(8):1461-72.
26. Harach HR, Soubeyran I, Brown A, Bonneau D, Longy M. Thyroid pathologic findings in patients with Cowden disease. Ann Diagn Pathol. 1999;3(6):331-40.
27. Horvath A, Bertherat J, Groussin L, Guillaud-Bataille M, Tsang K, Cazabat L, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat. 2010;31(4):369-79.
28. Stratakis CA, Courcoutsakis NA, Abati A, Filie A, Doppman JL, Carney JA, et al. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J Clin Endocrinol Metab. 1997;82(7):2037-43.
29. Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. 2008;124(4):369-77.
30. Lote K, Andersen K, Nordal E, Brennhovd IO. Familial occurrence of papillary thyroid carcinoma. Cancer. 1980;46(5):1291-7.
31. Musholt TJ, Musholt PB, Petrich T, Oetting G, Knapp WH, Klempnauer J. Familial papillary thyroid carcinoma: genetics, criteria for diagnosis, clinical features, and surgical treatment. World J Surg. 2000;24(11):1409-17.
32. Sturgeon C, Clark OH. Familial nonmedullary thyroid cancer. Thyroid. 2005;15(6):588-93.
33. Charkes ND. On the prevalence of familial nonmedullary thyroid cancer in multiply affected kindreds. Thyroid. 2006;16(2):181-6.
34. Nose V. Familial follicular cell tumors: classification and morphological characteristics. Endocr Pathol. 2010;21(4):219-26.
35. Malchoff CD, Malchoff DM. Familial nonmedullary thyroid carcinoma. Cancer Control. 2006;13(2):106-10.
36. McKay JD, Williamson J, Lesueur F, Stark M, Duffield A, Canzian F, et al. At least three genes account for familial papillary thyroid carcinoma: TCO and MNG1 excluded as susceptibility loci from a large Tasmanian family. Eur J Endocrinol. 1999;141(2):122-5.
37. Malchoff CD, Sarfarazi M, Tendler B, Forouhar F, Whalen G, Joshi V, et al. Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab. 2000;85(5):1758-64.
38. Canzian F, Amati P, Harach HR, Kraimps JL, Lesueur F, Barbier J, et al. A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am J Hum Genet. 1998;63(6):1743-8.
39. Corvi R, Lesueur F, Martinez-Alfaro M, Zini M, Decaussin M, Murat A, et al. RET rearrangements in familial papillary thyroid carcinomas. Cancer Lett. 2001;170(2):191-8.
40. Kraimps JL, Canzian F, Jost C, Menet E, Amati P, Levillian P, et al. Mapping of a gene predisposing to familial thyroid tumors with cell oxyphilia to chromosome 19 and exclusion of JUN B as a candidate gene. Surgery. 1999;126(6):1188-94.
41. Bakhsh A, Kirov G, Gregory JW, Williams ED, Ludgate M. A new form of familial multi-nodular goitre with progression to differentiated thyroid cancer. Endocr Relat Cancer. 2006;13(2):475-83.
42. Cavaco BM, Batista PF, Martins C, Banito A, do Rosario F, Limbert E, et al. Familial non-medullary thyroid carcinoma (FNMTC): analysis of fPTC/PRN, NMTC1, MNG1 and TCO susceptibility loci and identification of somatic BRAF and RAS mutations. Endocr Relat Cancer. 2008;15(1):207-15.
43. Bevan S, Pal T, Greenberg CR, Green H, Wixey J, Bignell G, et al. A comprehensive analysis of MNG1, TCO1, fPTC, PTEN, TSHR, and TRKA in familial nonmedullary thyroid cancer: confirmation of linkage to TCO1. J Clin Endocrinol Metab. 2001;86(8):3701-4.
44. McKay JD, Thompson D, Lesueur F, Stankov K, Pastore A, Watfah C, et al. Evidence for interaction between the TCO and NMTC1 loci in familial non-medullary thyroid cancer. J Med Genet. 2004;41(6):407-12.
45. Bignell GR, Canzian F, Shayeghi M, Stark M, Shugart YY, Biggs P, et al. Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am J Hum Genet. 1997;61(5):1123-30.
46. Lesueur F, Stark M, Tocco T, Ayadi H, Delisle MJ, Goldgar DE, et al. Genetic heterogeneity in familial nonmedullary thyroid carcinoma: exclusion of linkage to RET, MNG1, and TCO in 56 families. NMTC Consortium. J Clin Endocrinol Metab. 1999;84(6):2157-62. 47. McKay JD, Lesueur F, Jonard L, Pastore A, Williamson J, Hoffman L, et al. Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am J Hum Genet. 2001;69(2):440-6.
48. Stankov K, Pastore A, Toschi L, McKay J, Lesueur F, Kraimps JL, et al. Allelic loss on chromosomes 2q21 and 19p 13.2 in oxyphilic thyroid tumors. Int J Cancer. 2004;111(3):463-7. 49. Capezzone M, Marchisotta S, Cantara S, Busonero G, Brilli L, Pazaitou-Panayiotou K, et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr Relat Cancer. 2008;15(4):1075-81.
50. Loh KC. Familial nonmedullary thyroid carcinoma: a meta-review of case series. Thyroid. 1997;7(1):107-13.
51. Leprat F, Bonichon F, Guyot M, Trouette H, Trojani M, Vergnot V, et al. Familial non-medullary thyroid carcinoma: pathology review in 27 affected cases from 13 French families. Clin Endocrinol (Oxf). 1999;50(5):589-94.
52. Mosso L, Velasco S, Salazar I, Solar A, Gonzalez H, Cardona B, et al. [Clinical features of 17 patients with familial non medullary thyroid carcinoma]. Rev Med Chil. 2007;135(6):718-24. Carcinoma familiar del tiroides no medular (CFTNM): caracteristicas de presentacion en 17 casos.
53. Alsanea O, Wada N, Ain K, Wong M, Taylor K, Ituarte PH, et al. Is familial non-medullary thyroid carcinoma more aggressive than sporadic thyroid cancer? A multicenter series. Surgery. 2000;128(6):1043-50;discussion 50-1.
54. Lupoli G, Vitale G, Caraglia M, Fittipaldi MR, Abbruzzese A, Tagliaferri P, et al. Familial papillary thyroid microcarcinoma: a new clinical entity. Lancet. 1999;353(9153):637-9.
55. Uchino S, Noguchi S, Kawamoto H, Yamashita H, Watanabe S, Shuto S. Familial nonmedullary thyroid carcinoma characterized by multifocality and a high recurrence rate in a large study population. World J Surg. 2002;26(8):897-902.
56. Uchino S, Noguchi S, Yamashita H, Murakami T, Watanabe S, Ogawa T, et al. Detection of asymptomatic differentiated thyroid carcinoma by neck ultrasonographic screening for familial nonmedullary thyroid carcinoma. World J Surg. 2004;28(11):1099-102.
57. Pitoia F, Cross G, Salvai ME, Abelleira E, Niepomniszcze H. Patients with familial non-medullary thyroid cancer have an outcome similar to that of patients with sporadic papillary thyroid tumors. Arq Bras Endocrinol Metabol. 2011;55(3):219-23.
58. Vriens MR, Suh I, Moses W, Kebebew E. Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid. 2009;19(12):1343-9.
59. Tsilchorozidou T, Vafiadou E, Yovos JG, Romeo G, McKay J, Lesueur F, et al. A Greek family with a follicular variant of familial papillary thyroid carcinoma: TCO, MNG1, fPTC/PRN, and NMTC1 excluded as susceptibility loci. Thyroid. 2005;15(12):1349-54.
60. Alsanea O. Familial nonmedullary thyroid cancer. Curr Treat Options Oncol. 2000;1(4):345-51.
TABELAS
Tabela 1 Classificação das formas associadas a síndromes e não sindrómicas das formas familiares dos carcinomas não medulares.
Formas Associadas a Síndromes
Formas Não-Sindrómicas
Polipose Adenomatosa Familiar Síndrome de Gardner
Síndrome de Cowden Síndrome Peutz-Jeghers Síndrome de Carney Síndrome de Werner
Carcinoma papilar da tiroide familiar (CPTf) CPTf associado a neoplasia papilar renal
Carcinoma não-medular da tiroide familiar tipo 1 Bócio Multinodular Familiar
Tabela 2 Alterações genéticas presentes nas formas associadas a síndromes
Síndrome Transmissão Mutação genética Localizaçao cromossomica Polipose Adenomatosa Familiar Autossómica Dominante Gene supressor tumoral APC 5q21 Síndrome de Cowden Autossómica Dominante Gene supressor tumoral PTEN 10q23.3
Complexo de Carney Autossómica
Dominante PRKAR1-α (tipo 1)
17q22-24 (tipo 1) 2q16 (tipo 2)
Síndrome de Werner Autossómica
Recessiva Gene WRN 8p11-p12
Síndrome de Pendred
Autossómica
Tabela 3 Manifestações clínicas das formas associadas a síndromes17
Síndrome Patologia Extra-tiroideia Patologia Tiroideia
PAF e Síndrome Gardner
Pólipos adenomatosos múltiplos no trato gastrointestinal com potencial maligno; osteomas da mandíbula; displasia fibrosa do crânio; fibromas; tumores desmoides; cistos epidermicos; hipertrofia congénita do epitélio pigmentado da retina; hamartomas do trato gastrointestinal superior; dentes supranumerários; hepatoblastomas
Carcinoma papilar da tiroide com morfologia cribriforme-morular; Carcinoma papilar da tiroide clássico
Síndrome de Cowden
Hamartomas da mama, colon, endométrio e cérebro; carcinoma da mama, útero, e outros. Queratose oral; pápulas
papilomatosas orais; triquilemomas
Carcinoma folicular da tiroide associado a inúmeros nódulos adenomatosos e adenomas foliculares Complexo de Carney
Mixomas cardíacos e cutâneos; pigmentação da mucosa e da pele (nevos azuis); schwannomas; osteocondromixomas; tumores da supra-renal, hipófise e testiculos; adenomas mamários; Carcinoma papilar da tiroide e carcinoma folicular da toroide Sindrome de Werner Envelhecimento prematuro; modificações da pele tipo
esclerodermia; cataratas bilaterais; calcificações subcutêna; atrofia cutânea muscular e diabetes Mellitus. Carcinoma papilar da tiroide; carcinoma folicular da tiroide; carcinoma anaplásico da tiroide
Síndrome de Pendred Surdez sensorial bilateral; Carcinoma folicular da
Tabela 4 Critérios para as formas familiares do carcinoma não-medular da tiroide31
Critérios Primários
Carcinoma Papilar da tiroide em dois ou mais familiares de primeiro grau
Bócio multinodular em pelo menos 3 familiares de primeiro ou segundo grau de um doente com carcinoma papilar da tiroide
Critérios Secundários
Diagnóstico de carcinoma papilar da tiroide em doente com idade inferior a 33 anos
Carcinoma papilar da tiroide multifocal ou biltateral
Invasão local do tumor (pT4)
Metastização ganglionar (pN1) ou metastização à distância (M1)
Acumulação familiar de doenças tiroideias com origem na adolescência
Considera-se haver predisposição familiar para CPT quando estão reunidos:
Dois critérios primários ou
Um critério primário e três secundários
Critérios de Exclusão:
Exposição prévia a radiações
Identificação de síndromes neoplásicos associados (nomeadamente acumulação de carcinomas colorrectais, ováricos ou mamários no aglomerado familiar)
Identificação de alterações somáticas no DNA tumoral (nomeadamente rearranjos RET/PTC)
Tabela 5 Estudos a favor e contra as alterações genéticas identificadas nas formas não sindrómicas
Região potencialmente
candidata
Evidências a favor da associação Evidências Contra a associação Autor
Ano Características da Família Ref
Autor Ano Características da família Ref. MNG1 (14q31)
Bignell e col., 1997 1 Fiatria (18 BMN, 2 CPT) 42 Bignell e col., 1997 37 Genogramas, 122 CNMTf/doença benigna
42 McKay e col. , 1999 1 Fiatria (9 PTC, 11 BMN) 34
Lesueur e col., 1999 56 Genogramas 43
Malchoff e col., 2000 1 Fiatria (5 PTC, 2 CPR) 35
Bevan e col., 2001 22 Genogramas 44
Cavaco e col., 2008 7 Familias 40
TCO
(19p132)
Canzian e col., 1998 1 Fiatria (6 BMN, 3 PTC) 36 McKay e col., 1999 1 Fiatria (9 CPR, 11 BMN)
34
Bevan e col., 2001 1 Familia 44 Lesueur e col., 1999 56 Genogramas 43
McKay e col., 2004 10 Familia (35 CNMT, 18 benignos)
45 Malchoff e col., 2000 1 Fiatria (5 PTC, 2 CPR) 35
Prazeres e col., 2008 9 Famílias (14 CNMT) 11 Bevan e col., 2001 21 Familias 44
Cavaco e col., 2008 7 Famílias 40
fPTC/PRN
(1q21)
Malchoff e col., 2000 1 Fiatria (5 PTC, 2 CPR) 35 Bevan e col., 2001 22 Famílias 44
Cavaco e col., 2008 7 Famílias 40
NMTC1
(2q21)
McKay e col., 2001 1 Fiatria ( 80 pedigrees) 46 Stankov e col., 2004 1 Fiatria (6 BMN, 3 CPT) 47 McKay e col., 2004 10 Famílias (35 CNMT) 45
Prazeres e col., 2008 9 Famílias (14 CNMT) 11 Cavaco e col., 2008 7 Famílias 40
Complexo telomerase-telomerase Capezzone e col., 2008 47 CPTf 48 FTEN (8p23.1-p22)
Cavaco e col., 2008 1 Fiatria (11 benignos, 5 CNMT)
ANEXOS
INSTRUÇÕES A AUTORES
Edição
Acta Med Port 2011Normas de publicação acta médica
portuguesa
Os artigos propostos não podem ter sido objecto de qualquer outro tipo de publicação. As opiniões expressas são da inteira res-ponsabilidade dos autores. Os artigos publicados ficarão propriedade conjunta da AMP e dos autores.
A AMP reserva-se o direito de comercialização do artigo enquanto parte integrante da revista (na elaboração de separatas, por exemplo). O autor deverá enviar com a carta de submissão a declaração de cedência de direitos de autor para fins comerciais. Relativamente à utilização por terceiros a AMP rege-se pelos termos da licença Creative commons ‘Atribuição – Uso Não--Comercial – Proibição de Realização de Obras Derivadas (by-nc-nd)’.
A Acta Médica Portuguesa segue um rigoroso processo de revisão por pares (externos à revista). Os manuscritos recebidos serão enviados a peritos das diversas áreas, os quais deverão fazer os seus comentários, incluindo a sugestão de aceitação, aceitação condicionada a modificações ou rejeição.
Estipula-se para esse processo o seguinte plano temporal:
• Após a recepção do artigo, o Editor-Chefe, ou um dos Editores Associados, enviará o manuscrito a, no mínimo, dois revisores.
• No prazo de um mês, o revisor deverá responder ao editor indicando os seus comentários relativos ao manuscrito sujeito a revisão, e a sua sugestão de quanto à aceitação ou rejeição do trabalho.
• O Conselho Editorial tomará, num prazo de 15 dias, uma primeira decisão que poderá incluir a aceitação do artigo sem modificações, o envio dos comentários do(s) revisor(es) para que os Autores procedam de acordo com o indicado, ou a rejeição do artigo.
• Os Autores dispõem de um mês para submeter a nova versão revista do manuscrito, contemplando as modificações recomendadas pelos peritos e pelo Conselho Editorial.
• O Editor-Chefe ou um dos Editores Associados, dispõe de 15 dias para tomar a decisão de rejeitar o artigo na sua nova versão, aceitar o artigo na nova versão, ou submeter essa nova versão a um ou mais revisores externos, que poderão, ou não, coincidir com os que já fizeram a primeira revisão.
• Caso o manuscrito seja reenviado para revisão externa, os peritos dispõem de um mês para o envio dos seus comentários e da sua sugestão quanto à aceitação ou recusa para publicação do manuscrito.
Atendendo às sugestões dos revisores, o Editor Chefe poderá aceitar o artigo nesta nova versão, rejeitá-lo ou voltar a solicitar modificações. Neste último caso, os Autores dispõem de um mês para submeter uma versão revista, a qual poderá, caso o Editor Chefe assim o determine, voltar a passar por um processo de revisão por peritos externos.
No caso da aceitação, em qualquer das fases anteriores, a mesma será comunicada ao Autor principal. Num prazo inferior a um mês, o Conselho Editorial enviará o artigo para revisão dos Autores já com a formatação final, mas sem a numeração definitiva. Os Autores dispõem de 5 dias para a revisão do texto e comunicação de quaisquer erros tipográficos. Nesta fase, os Autores não podem fazer qualquer modificação de fundo ao artigo, fora das correcções de erros. Não são permitidas, nomeadamente, alterações a dados de tabelas ou gráficos, alterações de texto, etc.
Após a resposta dos Autores, ou na ausência de resposta, após o decurso dos 5 dias, o artigo considera-se concluído, e será dis-ponibilizado como [ahead of print] no site da Acta Médica Portuguesa.
Quando recepcionarem a comunicação de aceitação, têm os autores que remeter de imediato, por correio o formulário de cedência de direitos que se encontra no site da AMP, devidamente preenchido e assinado por todos os autores.
1. iNtrodução
Normas de publicação Acta Médica Portuguesa , Acta Med Port. 2011
A submissão de qualquer tipo de artigos à AMP deve ser feita exclusivamente por correio electrónico, seguindo com atenção as normas indicadas de seguida.
Deverão ser enviados num único correio electrónico apenas os seguintes ficheiros, utilizando estas designações no nome do ficheiro: • Submissao
• Texto_principal
• Figura (tantos ficheiros quantas as figuras)
No e-mail deverão os autores indicar caso não desejem ser incluídos na base de dados de revisores da AMP para futuros contactos. Normas gerais
a) submissão
O ficheiro «Submissao» tem que ser remetido através do preenchimento do formulário que se encontra disponível no site e que inclui o seguinte conteúdo:
- Folha de título - lista de autores - check list - Folha de título
A Folha de Título deve indicar o tipo de artigo e a razão da submissão (a mais-valia resultante da respectiva publicação). O envio da folha de título implica a Declaração de Responsabilidade que certifica que o artigo não foi submetido a outra entidade e que todos os autores contribuíram de forma significativa para a sua elaboração. A Folha de Título confirma de forma inequívoca que todos os autores têm conhecimento da presente submissão e com ela concordam.
A Folha de Título contém o título do artigo, o tipo de artigo (ver os tipos de artigo permitidos pela AMP e respectivas normas), identificação do autor que ficará responsável pelo contacto com a revista e prestação de informações aos co-autores; deverá igualmente indicar e referir o número de palavras do artigo, o número de palavras do resumo, o número de referências, de tabelas e de figuras. Estas informações, incluindo a autoria, não podem ser referidas em mais nenhum local do artigo.
Título: o título do artigo (independentemente da sua tipologia) deve ser conciso e não deverá exceder os 120 caracteres. Não se aconselha a utilização de subtítulos. Deverá ser claramente identificativo do conteúdo do texto e não deverão utilizar-se títulos alegóricos ou metafóricos.
Agradecimentos: os agradecimentos deverão ser colocados apenas na folha de título. Caso a pesquisa tenha usufruído de pa-trocínios externos, este facto deverá ser referido nos agradecimentos. Caso tenha sido recebido financiamento público, deverá indicar-se a referência completa do projecto financiado.
Conflito de interesses: os autores deverão comunicar na folha de título a existência ou inexistência de laços financeiros/conflitos de interesse com a instituição que patrocinou a pesquisa. Caso não existam quaisquer conflitos, deverão incluir a seguinte afir-mação: Os autores declaram a inexistência de conflitos de interesse.
-lista de autores
Inclui o nome e filiação profissional de todos os autores. A normalização dos nomes é essencial para a indexação nas bases de dados, especialmente nas estrangeiras.
Os autores deverão identificar-se sempre: com um nome (apenas um e apresentado em minúsculas), um segundo nome (opcional, mas apenas um e que deverá igualmente ser apresentado em minúsculas) e um apelido (que deverá ser escrito em maiúsculas). Exemplo: João A. COSTA ou João António COSTA
Se o autor quiser utilizar dois apelidos (não se aceitarão mais do que dois apelidos), deverá colocar os dois em maiúsculas. Exemplo: João A. COSTA SILVA ou João António COSTA SILVA
O uso de partículas no apelido (de, da, e) não é aconselhado. No entanto, se algum autor desejar utilizá-las, deverá considerá-las como parte do apelido e escrevê-las em maiúsculas.
Exemplo 1: João A. COSTA DA SILVA Exemplo 2: João DA COSTA
Normas de publicação Acta Médica Portuguesa , Acta Med Port. 2011
- check list
Deve preencher sempre a check list e submeter o artigo apenas quando cumpra todos os requisitos constantes da mesma. b) texto principal
Num ficheiro, chamado ‘texto_principal’, que começa com o título do artigo (o mesmo título em português e em inglês), deverá ser enviado o resumo em português (máximo: 350 palavras), e a sua versão em inglês (tem que corresponder a uma tradução fidedigna do resumo em português), e o texto do artigo, sem figuras (que são enviadas à parte) mas incluindo, no final, as tabelas. A AMP não usa palavras-chave.
Referências bibliográficas: de acordo com as Normas para uniformização dos Manuscritos submetidos a Revistas Biomédicas do ICMJE, e seguindo o estilo da U.S. National Library of Medicine. As referências deverão numerar-se pela ordem de apare-cimento no texto, e devem ser colocadas no fim do ficheiro texto_principal pela mesma ordem da numeração. Não deverão ser incluídos na lista de referências quaisquer artigos ainda em preparação ou observações não publicadas, comunicações pessoais, etc., tais inclusões só são permitidas no corpo do artigo (ex: P. Andrade, comunicação pessoal).
Legendas das figuras: Após as referências bibliográficas, ainda no ficheiro «texto_principal», envie uma legenda detalhada (sem abreviaturas) para cada figura, referencie a figura no texto e indique a sua localização aproximada no corpo do texto com o comentário “Inserir Figura nº 1… aqui”.
Tabelas: É obrigatório o envio das tabelas a preto e branco no final do ficheiro «Texto_principal». As tabelas devem ser elaboradas e submetidas em documento word, em formato de tabela simples (simple grid), sem utilização de tabuladores, nem modificações tipográficas. Todas as tabelas devem ser mencionadas no texto do artigo e devem ser numeradas pela ordem que surgem no texto. Indique a sua localização aproximada no corpo do texto com o comentário “Inserir Tabela nº 1… aqui”. Neste caso os autores autorizam uma reorganização das tabelas caso seja necessário.
Abreviaturas: não é permitido o uso de abreviaturas idealizadas pelos autores, limitando-se o seu uso às abreviaturas comum-mente aceites na literatura biomédica (SIDA, OMS, etc..) As abreviaturas utilizadas devem ser objecto de especificação anterior. Símbolos e unidades de medida: deverão utilizar-se as unidades incluídas no Sistema Internacional de Unidades (S.I.Units, the SI for Health Professions, WHO, 1977). Os números de um a dez devem ser escritos por extenso, excepto quando têm decimais ou se usam para unidades de medida. Números superiores a dez são escritas em algarismo, salvo no início de uma frase. c) Figuras
Os ficheiros «figura» podem ser tantos quantas imagens tiver o artigo. Cada um destes elementos deverá ser submetido em ficheiro separado, obrigatoriamente em versão electrónica, pronto para publicação. As figuras (fotografias, desenhos e gráficos) não são aceites em ficheiros word.
As legendas têm que ser colocadas no ficheiro «texto_principal».
Caso a figura esteja sujeita a direitos de autor, é responsabilidade dos autores do artigo adquirir esses direitos antes do envio do ficheiro à AMP.
Só são aceites imagens de doentes quando necessárias para a compreensão do artigo. Se for usada uma figura em que o doente seja identificável deve ser obtida e remetida à AMP a devida autorização.
• Fotografias
Devem ter uma das seguintes extensões: tiff, jpeg, psd. O tamanho dos ficheiros terá de ser no mínimo de 300 dpi’s ao tamanho real da publicação (mínimo 80mm de largura – correspondente ao espaço de uma coluna).
• Desenhos e gráficos
Os desenhos e gráficos devem ser enviados com uma resolução mínima de 600 dpi. Estas figuras deverão ser enviadas prefe-rencialmente numa das seguintes extensões: AI (adobe ilustrator), EPS, CDR (Corel Draw). As fontes devem ser transformadas em curvas ou enviadas à parte.
Permite-se o envio de desenhos e gráficos com extensão fotográfica (tiff, jpeg, psd). Neste tipo de ficheiro o tamanho terá de ser no mínimo 300 dpi ao tamanho real da publicação (largura mínima: 80 mm, correspondente a uma coluna), ou em PDF (de alta qualidade com as fontes embebidas ou convertidas em curvas).
4. tipos de artigo e reQuisitos editorial
Artigo elaborado pelo Conselho Editorial da revista ou a convite do mesmo, sobre tema específico; Deve conter 1200 – 1500 palavras e um máximo de 15 - 20 referências bibliográficas e só pode conter 1 tabela ou 1 figura. Um Autor que pretenda submeter para publicação um editorial não solicitado deve entrar em contacto previamente com o Editor-Chefe.
perspectiva
Artigos elaborados a convite do Conselho Editorial que podem cobrir grande diversidade de temas com interesse nos cuidados de saúde, problemas actuais ou emergentes, gestão e política de saúde, história da medicina, ligação à sociedade, etc. Um Autor que deseje propor um artigo desta categoria deverá remeter previamente ao Editor-Chefe o respectivo resumo, indicação dos autores e título do artigo para análise.
Deve conter no máximo 1200 palavras e um máximo de 10 referências bibliográficas e só pode conter 1 tabela ou 1 figura. revisão
Os artigos de revisão são elaborados a convite do Conselho Editorial. Um Autor que deseje propor a publicação de uma revisão não solicitada deverá remeter previamente à AMP o respectivo resumo, indicação dos autores e título para análise.
Os artigos de revisão seguem os mesmos processos editoriais e de peer-review que os artigos originais.
Uma revisão não pode exceder as 3500 palavras e não tem limite do número de referências, com um máximo de 5 tabelas ou figuras (total).
original
Artigos originais não podem exceder as 4000 palavras, excluindo o resumo, um total máximo de 6 figuras ou tabelas, e até 60 referências.
Deve ser sempre subdividido em 5 secções:
introdução, material e métodos, resultados, discussão, e conclusão.
A introdução deve conter uma revisão do estado da arte que ajude a compreensão do estudo. No final da introdução, deverão ser declarados com clareza os objectivos do estudo.
Métodos: devem ser descritos de modo a que o leitor entenda como foi realizada a pesquisa. Em pesquisas com seres humanos, é necessário informar a existência de consentimento informado, e da submissão à Comissão de Ética para a investigação ou à Comissão de investigação da Instituição dos Autores.
Resultados: devem ser apresentados de maneira coerente e estar ligados aos objectivos e métodos anteriormente descritos. Discussão: deve reiterar os principais resultados do trabalho, comentar aspectos negativos do mesmo, discutir e comparar a importância e implicações dos resultados e referir as limitações ao estudo encontradas pelos autores.
Conclusão: o autor deve abster-se de deduções ou infe¬rências não baseadas nos resultados de seu estudo. caso clínico
Breves relatórios que apresentam uma avaliação crítica de determinado percurso clínico nos quais se pretende realçar alguns elementos específicos como associações clínicas, relatórios de reacções adversas ou outras associações relevantes.
Os casos clínicos não podem ter mais de 3 autores. O texto não pode exceder as 750 palavras, ter um máximo de 1 tabela ou 1 figura e até 5 referências.
imagens em medicina
Imagens de condições médicas. Estes artigos pretendem capturar a noção de potencial diagnóstico visual e de diversidade que os médicos experienciam na sua prática clínica.
Só são aceites fotografias originais, de alta qualidade, sem prévia publicação. Devem ser enviados dois ficheiros: um com a qualidade exigida para a publicação de imagens e outra que serve apenas para referência em que o topo da fotografia deve vir indicado com uma seta.
Deve incluir um título com um máximo de 8 palavras e um texto com um máximo de 150 palavras onde se dê informação clínica relevante, incluindo um breve resumo do historial do doente, dados laboratoriais relevantes, terapêutica, e condição actual. Não pode ter mais do que 3 autores e 5 referências.
Para informação sobre o envio de imagens digitais consultar as «Normas técnicas para a submissão de figuras, quadros ou fotografias»
guidelines / Normas de orientação
As sociedades médicas ou os colégios das especialidades que desejem publicar na AMP recomendações de prática clínica, deverão contactar previamente o Conselho Editorial e submeter o texto completo e a versão para ser publicada. O Editor-Chefe poderá colocar como exigência a publicação exclusiva das recomendações na AMP.
Poderá ser acordada a publicação de uma versão resumida na edição impressa cumulativamente a à publicação da versão com-pleta no site da AMP.
cartas ao editor
Apresentação de comentários críticos sobre artigos publicados na AMP. Neste caso a carta só é aceite se enviada ao Editor em tempo de ser publicada numa das duas edições seguintes à da publicação do artigo e não pode exceder as 200 palavras. Outros temas de investigação com interesse na área da medicina. Neste caso o texto não ultrapassará as 400 palavras. Em qualquer dos casos, a contagem de palavras exclui o título, bibliografia, assinatura dos autores, tabela ou figura.
As cartas só poderão ter um máximo de 5 referências bibliográficas e uma tabela ou uma figura e só poderão ser assinadas por um máximo de 3 autores. Caso seja aplicável, as respostas dos autores devem ter as mesmas características.
errata
Após a publicação dos artigos (seja online, seja na versão impressa), apenas se efectuam alterações sob a forma de Errata, que incluirá indicação do URL do artigo.
Todos os tipos de artigo devem ser preparados de acordo com as normas internacionais do ICMJE. Artigos que não cumpram as normas editoriais serão recusados liminarmente pela redacção e não serão enviados para análise dos revisores.