A aplicação de ferramentas proteómicas
na área da nefrologia
Dissertação de Mestrado em Genética Molecular Comparativa
e Tecnológica
José Eduardo Ferreira Araújo
Vila Real, 2013 Orientadores:
Professor Doutor Gilberto Igrejas Professor Doutor José Luís Capelo
A aplicação de ferramentas proteómicas
na área da nefrologia
Dissertação de Mestrado em Genética Molecular Comparativa
e Tecnológica
José Eduardo Ferreira Araújo
Composição do Júri: ___________________________________________ ___________________________________________ ___________________________________________ Vila Real, 2013 Orientadores:
Professor Doutor Gilberto Igrejas Professor Doutor José Luís Capelo
V
A elaboração deste trabalho é da exclusiva responsabilidade do autor. A reprodução de qualquer elemento textual, ou gráfico, não deve por isso ser efetuada sem o consentimento do mesmo. A autorização para a reprodução parcial de ideias apresentadas é portanto obtida, mediante declaração do interessado e só após a permissão do autor.
O autor:
José Eduardo Ferreira Araújo
VII
Orientador de estágio
Professor Doutor Gilberto Paulo Peixoto Igrejas
Departamento de Genética e Biotecnologia, UTAD
Co-orientador
Professor Doutor José Luís Capelo Martínez
Departamento de Química, FCT-UNL
IX
Este trabalho foi desenvolvido no âmbito da dissertação de 2º ciclo, para a obtenção do grau de mestre em Genética Molecular Comparativa e Tecnológica.
XI
Agradecimentos
No decorrer da realização deste trabalho, inúmeras pessoas e entidades intervieram, através da colaboração, disponibilidade, apoio e ensinamentos, quer diretamente como indiretamente. A todas essas pessoas presto os meus sinceros agradecimentos.
Ao Magnífico Reitor da Universidade de Trás-os-Montes e Alto Douro, Sr. Professor Carlos Alberto Sequeira, agradeço a possibilidade de ter realizado o 2º ciclo de estudos nesta instituição.
À atual Direção do Curso de Genética Molecular Comparativa e Tecnológica, Professoras Doutoras Paula Lopes e Raquel Chaves, assim como à anterior Direção, Professores Doutores Valdemar Pedrosa Carnide e Raquel Chaves, pela sua coordenação e disponibilidade.
Ao Complexo Hospitalar Universitário de Ourense, mais concretamente ao Serviço de Nefrologia e em especial ao Sr. Doutor Alfonso Otero González, por ter possibilitado a disponibilização das amostras para este estudo.
Ao meu Orientador, Professor Doutor Gilberto Paulo Peixoto Igrejas, pela orientação do meu estágio, por todos os conselhos dados, antes e no decorrer da dissertação, pela sua disponibilidade e apoio sempre que necessário, assim como a revisão crítica do texto que contribuíram para um maior rigor científico.
Ao meu Co-orientador, Professor Doutor José Luís Capelo Martínez, pela orientação do meu estágio, cedência de bibliografia, pelo voto de confiança, aceitando-me no seu grupo de investigação, por contribuir para a minha formação como investigador, por toda a dedicação, preocupação para que nada falte e amizade construída.
Ao Doutor Hugo Miguel Baptista Carreira dos Santos, por toda a sua disponibilidade e apoio laboratorial sempre que necessário mesmo quando tinha tanto a fazer, pelos ensinamentos transmitidos, cedência de bibliografia, revisão crítica, dedicação e preocupação, conselhos e amizade construída
Ao Professor Doutor Carlos Lodeiro Espiño pelo voto de confiança, simpatia e amizade.
A todos os meus colegas do grupo de investigação Bioscope, Doutor Mário Diniz, Diana Madeira, Carla Santos e Elisabete Oliveira. Agradeço também aos Amigos, Cristina Núñez e Júlio Miranda, pelo ânimo e palavra amiga dada no período de adaptação, os convívios extralaborais e companheirismo. Não poderia ainda deixar de agradecer ao meu
XII
Amigo de Berres, Javier Fernández-Lodeiro, por o tempo em que partilhamos casa, a confiança, as vivências e os dotes culinários que resultavam em tortilha a voar pela cozinha.
Aos meus companheiros de surf, Pedro Pinto e Vitaliy Sobchuk, por todos aqueles dias no fim do trabalho, em que fomos para o mar aproveitar os últimos raios de sol.
Aos colegas de Licenciatura em Genética e Biotecnologia, assim como os de Mestrado em Genética Molecular, Comparativa e Tecnológica.
Aos amigos que ficam desta vida académica, em especial ao meu grande Amigo Ricardo Monteiro, pela amizade, confiança, entreajuda e companheirismo.
À minha namorada Ana Laço, pelo carinho, estima, cumplicidade, palavra de incentivo e conselhos dados, um muito obrigado.
A todos os meus Familiares que sempre me apoiaram em tudo, aos meus queridos avós dos quais me orgulho, por toda a ligação e carinho.
Ao meu Primo e Irmão, por não estar tão presente, pelas visitas esporádicas que terminam sempre com a sensação de “pouco”.
Aos meus Pais, que tudo fazem por mim, pela educação que me deram, pela palavra amiga que sabem dar melhor do que ninguém, por todo o esforço para que nada falte e todo o apoio incondicional, o meu mais sincero Obrigado!
XIII
Resumo
Os equipamentos de espectrometria de massa dedicados à análise de biomoléculas são cada vez mais rápidos mais sensíveis e mais precisos, razões pelas quais, estas técnicas se aplicam ao estudo do proteoma de amostras tão complexas como líquido peritoneal e biópsias parafinadas. A proteómica surge assim como uma ferramenta útil na identificação de biomarcadores associados a doenças humanas, providenciando informações de diagnóstico que podem vir a ser essenciais para diagnóstico, prognóstico ou intervenções terapêuticas.
As biópsias parafinadas, existentes em bancos de tecidos hospitalares são um método recorrente de preservação e uma importante fonte de informação, no entanto problemas associados ao efeito cross-linkage da formalina em análise proteica constituem uma dificuldade inerente ao uso de tecidos parafinados. Para ultrapassar esta dificuldade diferentes abordagens foram estabelecidas e otimizadas com o intuito de reverter as modificações introduzidas pela fixação com formalina, com vista a maximizar a extração proteica e posterior análise proteómica. Este trabalho baseou-se portanto na elaboração de um desenho fatorial, assente em diferentes variáveis tanto para o constituinte do tampão, como para as condições de extração. Alguns dos fatores em estudo que se mostraram fundamentais foram: (i) a temperatura e o tempo de aquecimento, importante para reverter o efeito
cross-linkage, (ii) a sonicação, essencial para a eficiência da extração proteica e eficácia da
solubilização das amostras, (iii) o SDS, crítico para incrementar a extração proteica. Com a elaboração deste estudo foi possível concluir que a estratégia passa por usar o desenho fatorial para determinar a direção para investigações futuras e usar estes fatores na pesquisa desenvolvida.
Relativamente à análise do líquido peritoneal, esta também se torna um desafio, pois existem milhares de proteínas, lípidos e produtos metabólicos na amostra, enquanto os biomarcadores para a doença tendem a aparecer em concentrações muito baixas. O objetivo deste trabalho passou por
integrar a análise proteómica em líquido peritoneal, tendo como principal tarefa a identificação dos constituintes do proteoma desta amostra biológica, para possíveis previsões terapêuticas. No total foram identificadas 25 proteínas. O resultado de maior relevo neste estudo, foi a descoberta de que a perda das proteínas através do líquido peritoneal está associada ao metabolismo e regulação do cálcio no corpo humano. Estas descobertas, abrem assim novas perspetivas para o potencial uso do líquido peritoneal como amostra em diagnóstico e prognóstico de pacientes com falha renal.
Palavras-chave: Tecidos fixados com formalina; Líquido peritoneal; Desenho fatorial; Proteómica; Biomarcadores; Espectrometria de massa.
XV
Abstract
Mass spectrometry equipments dedicated to biomolecule analysis are increasingly more fast, sensitive and precise. Consequently, these techniques have been applied to the proteome study of complex samples like peritoneal liquid and paraffin embedded biopsies. Proteomics emerges as a useful tool in the identification of biomarkers associated with human diseases, providing diagnostic information that may be essential to diagnostic, prognostic or therapeutic interventions.
The existing paraffin embedded biopsies in hospital tissue banks are a recurrent preservation methodology and an important source of information. However, associated problems with the formalin cross-linkage effect in protein analysis constitute one inherent difficult to the use of paraffin-embedded tissues. To overcome this difficulty different approach were established and optimized in order to reverse the changes introduced by fixation with formalin, aiming to maximize protein extraction and subsequent proteomic analysis. This work was based therefore on the development of a factorial design, based in different variables both for the constituent buffer as for the extraction conditions. Some of the factors in study that proved essential were: (i) the temperature and the heating time, important to reverse the cross-linkage effect, (ii) sonication, essential for protein extraction and solubilization efficiency of the samples, (iii) SDS, critical to increase the protein extraction. With the development of this study was possible to conclude that the strategy is to use de factorial design to determine a direction for future researches and to use these factors in research.
Relatively to peritoneal fluid analysis, this also became a challenge because there are thousands of proteins, lipids and metabolic products in the sample, as biomarkers for disease tend to appear in low concentrations. The objective of this work went through integrating the proteomic analysis in peritoneal fluid, with the main task to identify the proteome constituents of this biological sample for possible therapeutic predictions. Through this study was show that the proteome peritoneal fluid is far to being well known in its entirety. In total, 25 proteins were identified. The most relevant result in this study, was the finding that the loss of protein through the peritoneal fluid is associated to the metabolism and regulation of calcium in the human body. These findings open new perspectives for the potential use of peritoneal fluid sample in diagnostic and prognostic of patients with renal failure.
Keywords: Formalin embedded tissues, Peritoneal fluid, Factorial design, Proteomics, Biomarkers, Mass Spectrometry.
XVII
Índice Geral
I. INTRODUÇÃO... 1
1. Proteómica e o Proteoma ... 1
1.1. Importância da proteómica em investigação clínica ... 1
2. Biomarcadores ... 2
2.1. Biomarcadores associados ao diagnóstico terapêutico ... 2
3. Tecidos fixados com formalina ... 2
3.1. Importância dos tecidos FFPE e a desvantagem inerente ... 3
4. Líquido peritoneal ... 5
5. Diálise Peritoneal ... 5
6. Desenho Fatorial... 6
7. Métodos Analíticos... 6
7.1. Princípios da eletroforese ... 8
7.1.1. Eletroforese monodimensional (1-DE) ... 9
7.1.2. Eletroforese bidimensional (2-DE) ... 9
7.2. Ionização / Dessorção Assistida por Laser - Matrix-Assisted Laser Desorption Ionization (MALDI) . 10 8. Objetivos ... 15
II. MATERIAL E MÉTODOS ... 17
1. Desenvolvimento e otimização de metodologias de extração proteica em tecidos fixados com formalina ... 17
1.1. Material biológico ... 17
1.2. Reagentes... 17
1.2.1. Reagentes usados na fixação do material biológico e extração do fixador ... 17
1.2.2. Reagentes usados para os tampões de extração ... 17
1.2.3. Reagentes usados no tratamento dos géis excisados para a precedente identificação proteica . 18 1.3. Fixação das amostras ... 18
1.4. Preparação do tecido para posterior extração de proteína ... 18
1.5. Otimizações e Metodologias ... 18
1.5.1. Otimização da variável da indução da extração proteica por calor (Tempo/ ºC)... 18
1.5.1.1. Metodologia para a otimização da variável da indução da extração proteica por calor (Tempo/ ºC) ... 19
1.5.2. Escolha do detergente de extração: SDS 2 % / Triton X-100 1 % ... 19
1.5.2.1. Metodologia para otimização da variável SDS 2 % / Triton X-100 1 % ... 20
1.5.3. Otimização da variável da amplitude (%) de ultrassons (US) ... 20 1.5.3.1. Metodologia para a otimização da variável da amplitude (%) de ultrasons (US) . 21
XVIII
1.6. Desenho Fatorial: Otimização e metodologias ... 21
1.6.1. Metodologia para amostras sem extração por indução de calor (-) e ultrassons (-) .. 22
1.6.2. Metodologia para amostras com extração por indução de calor (+) e sem ultrassons (-) ... 23
1.6.3. Metodologia para amostras sem extração por indução de calor (-) e com ultrassons (+) ... 23
1.6.4. Metodologia para amostras com extração por indução de calor (+) e ultrassons (+) 24 1.6.5. Determinação da quantidade de proteína total após extração ... 24
1.7. Separação das proteínas extraídas por SDS-PAGE ... 24
1.8. Tratamento da amostra para identificação de proteínas ... 25
1.9. Análise por MALDI-TOF-MS ... 26
2. Análise do proteoma do efluente de diálise peritoneal ... 27
2.1. Material biológico ... 27
2.2. Reagentes... 27
2.3. Precipitação de proteína ... 29
2.4. Eletroforese bidimensional (2-DE) ... 29
2.5. SDS-PAGE ... 30
2.6. Coloração do gel e análise de imagem ... 30
III. RESULTADOS E DISCUSSÃO ... 31
1. Desenvolvimento e otimização de metodologias de extração proteica em tecidos fixados com formalina ... 31
1.1. Otimização da variável da indução da extração proteica por calor (Tempo/ ºC) ... 31
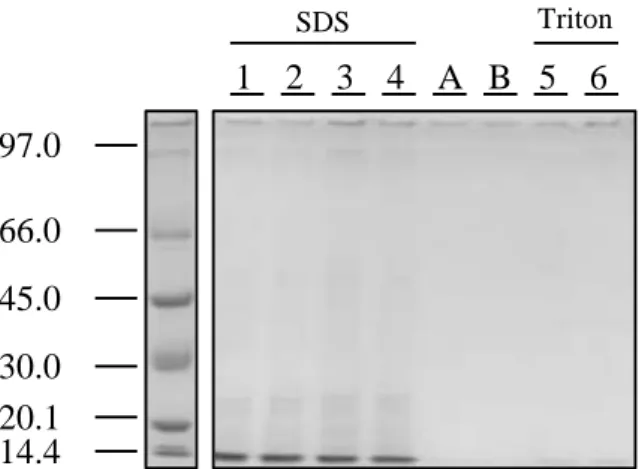
1.2. Seleção do detergente de extração: SDS 2 % ou Triton X-100 1 % ... 32
1.3. Otimização da variável amplitude de ultrasons (US) ... 34
1.4. Desenho Fatorial: Otimização e metodologias ... 36
1.4.1. Espectrometria de massa e interrogação das bases de dados ... 47
2. Análise do proteoma do efluente de diálise peritoneal ... 50
2.1. Análise individual das amostras de efluente de diálise peritoneal por 2-DE ... 50
2.2. Análise integrativa das proteínas identificadas ... 56
2.3. Comparação de literatura relativa ao efluente de diálise peritoneal ... 57
IV. Conclusões ... 59
1. Desenvolvimento e otimização de metodologias de extração proteica em tecidos fixados com formalina ... 59
2. Análise do proteoma do efluente de diálise peritoneal ... 60
V. Referências bibliográficas ... 61
VI. ANEXOS………...69
XIX
Índice Figuras
Figura 1- Organização estrutural e modificações prováveis que as proteínas sofrem sob condições de
FFPE. ... 3
Figura 2- Esquema representativo da formação hipotética de péptidos de tecidos FFPE e o sucesso da sua identificação.. ... 4
Figura 3- Figura representativa da diálise peritoneal e da forma como os produtos residuais atravessam a membrana para a cavidade peritoneal e são drenados juntamente com a solução de diálise.. ... 5
Figura 4- Identificação proteica com peptide mass fingerprinting. As massas de péptidos das proteínas digeridas são correspondidas com a lista da massa teórica de péptidos, que são matematicamente derivados da base de dados do genoma de certos organismos ... 8
Figura 5- Princípio da focagem isoelétrica. ... 10
Figura 6- Processo de ionização por MALDI. ... 12
Figura 7- Diferentes configurações do espectrómetro de massa/fonte iónica. ... 13
Figura 8- Representação esquemática dos espectros de massa, dos péptidos obtidos por MS e MS/MS. ... 14
Figura 9- Géis 1D da variável de indução da extração proteica por calor (Tempo/ ºC) ... 32
Figura 10- Géis 1D da variável do detergente SDS 2 %/ Triton X-100 1 % ... 33
Figura 11- Géis 1D da variável amplitude (%) de ultrasons (US) ... 35
Figura 12- Gráfico de barras representativo da quantidade de proteína extraída / mg de tecido em função da amplitude de ultrassons utilizada ... 35
Figura 13- Gel 1D relativo ao quadro de otimização ... 37
Figura 14- Quantidade de proteína extraída / mg de tecido para cada experiencia do quadro de desenho experimental 24. ... 39
Figura 15- Esquema representativo dos efeitos estimados e interações para cada uma das variáveis .. 46
Figura 16- Diagrama geométrico representativo da extração proteica. (A) extração aquecimento mínimo; (B) extração aquecimento máximo. ... 46
Figura 17- Gel 1D-SDS-PAGE representativo das condições experimentais óptimas. A numeração de 1 a 24 representa o número de bandas cortadas e analisada por espectrometria de massa com ionização MALDI... 47
Figura 18- Concentração de proteína em efluente de diálise peritoneal, determinada por Método de Bradford. ... 50
Figura 19- (A) - Géis-2D do efluente de diálise peritoneal de cada doente usado no estudo. (B) – Gel de referência com anotação de spots. ... 51
XXI
Índice de Quadros
Quadro 1- Matrizes de MALDI e as suas aplicações em proteómica ... 11
Quadro 2- Número experimental das amostras para cada variável da indução da extração proteica por calor e descrição da solução tampão. ... 19
Quadro 3- Número experimental das amostras para a variável de detergente SDS 2 % / Triton X-100 1 % e descrição da solução tampão. ... 20
Quadro 4- Descrição da solução tampão e das condições de extração ºC/ tempo e % de ultrasons/sem ultrasons. Número experimental das amostras para cada variável. ... 21
Quadro 5- Composição de cada uma das soluções tampão utilizadas no desenho experimental factorial 24. Os sinais (-) ou (+) representam os valores mínimos (-) ou máximos (+) para cada uma das variáveis. ... 22
Quadro 6- Representação esquemática da otimização, com a descrição do número experimental em relação às variáveis e à solução tampão correspondente. ... 22
Quadro 7- Informação clínica dos pacientes. ... 28
Quadro 8- Quantidade de proteína extraída / mg de tecido para cada uma das amplitudes de ultrassons testadas (30 %, 60 %, 100 % e sem US). ... 34
Quadro 9- Quantidade de proteína extraída normalizada pela massa de tecido utilizado. ... 38
Quadro 10- Variáveis e valores mínimos e máximos correspondentes. ... 40
Quadro 11- Matriz experimental do desenho fatorial 24. ... 40
Quadro 12- Sinais usados para calcular os efeitos no desenho fatorial 24. ... 42
Quadro 13- Principais efeitos de cada variável e as interações entre ambas. ... 43
Quadro 14- Quantidade média de proteína extraída normalizada pela massa de tecido utilizado, desvio padrão e a variância estimada utilizada para o cálculo dos desvios padrão dos efeitos ou interações. . 44
Quadro 15- Efeitos calculados de cada variável e as suas interações ... 45
Quadro 16- Proteínas identificadas em tecido FFPE de rim de vaca, para cada uma das condições ótimas (13, 14, 15 e 16). ... 48
XXIII
Abreviaturas e unidades
A
ACN Acetonitrilo
A1M Alfa-1-microglobulina
APS Persulfato de amónio A1AT Alfa 1- antitripsina B
BSA Albumina de soro bovino C
CBB Azul coomassie - coomassie blue R-250
CHAPS 3-[ (3-Colamidopropil) dimetilamónio-1-propanosulfato] CHUO Complexo Hospitalario Universitario de Ourense
CI Ionização química
CPD Diálise peritoneal crónica D
DOC Deoxicolato de sódio
DTT D,L-Ditiotreitol
E
ESI Ionização por electrospray
EI Impacto de eletrões
F
FFPE Fixado com formalina, embebido em parafina - Formalin-fixed,
paraffin-embedded
G
g Grama
×g Força centrífuga relativa g H h Hora HCl Ácido clorídrico HD Hemodiálise Hz Hertz I IAA Iodoacetamida
IEF Focagem isoelétrica
IPG Gradiente de pH imobilizado K
kV Kilovolt
L
LC Cromatografia líquida
LID Dissociação induzida por laser LIFT-TOF-TOF LIFT- Time-of-flight – time of flight
XXIV M
M Molar
mA Miliampére
MALDI Ionização / Dessorção Assistida por Laser
MALDI-TOF-MS Ionização / Dessorção Assistida por Laser – time-of-flight – mass
spectrometry
MALDI-MS Ionização / Dessorção Assistida por Laser – spectrometry
mg Miligrama mL Mililitro min Minuto mM Milimolar mg/mL Miligrama/mililitro MS Espectrometria de massa
MS/MS Espectrometria de massa/ Espectrometria de massa
MALDI-TOF/TOF Ionização / Dessorção Assistida por Laser – flight/
time-of-flight m/z Ratios massa-de-carga N nm Nanómetro ns Nanosegundo P PD Diálise peritoneal
PET Peritoneal equilibrium test
pH Potencial hidrogénio
PTH Hormona paratiróide
pI Ponto isoelétrico
PMF Peptide Mass Fingerprinting
PTM Modificação pós-tradução R
RET4 Proteina transportadora do retinol-4
rpm Rotações por minuto
RSD Desvio padrão relativo S
s Segundo
SDS Dodecil sulfato de sódio
SDS-PAGE Gel de poliacrilamida na presença de dodecil sulfato de sódio
SN Sobrenadante
T
TCA Ácido tricloroacético
TEMED (N, N, N’, N’,-tetrametileno-diamina) TFA Ácido Trifluoroacético
TOF Time-of-flight
TOF-TOF Time-of-flight – time of flight
TTHY Transtirretina
U
XXV UV Ultravioleta V v/v Volume/volume V Volts W w/v Massa/volume % Percentagem
1-DE Eletroforese monodimensional
1D-SDS-PAGE Electroforese monodimensional em gel de poliacrilamida na presença de dodecil sulfato de sódio
2-DE Eletroforese bidimensional
2D PAGE Eletroforese bidimensional em gel de poliacrilamida
µA/gel Microampére/gel
µg Micrograma
µg/µL Micrograma/microlitro
µm Micrometro
1
I.
INTRODUÇÃO
1.
Proteómica e o Proteoma
A Proteómica é uma área da biologia molecular em expansão, que se refere à análise sistemática e em larga escala das proteínas que compõem o proteoma. O termo “proteoma” deve a sua origem à fusão de duas palavras, proteína e genoma, e representa o conjunto de todas as proteínas codificadas pelo genoma de um organismo ou por uma dada célula sob determinadas condições. O proteoma é uma entidade dinâmica e complexa e, como tal, pode ser definido em termos da sequência, da estrutura, abundância, localização, modificações, interações e função bioquímica de cada uma das proteínas que o compõem1, 2. A análise de cada uma destas propriedades do proteoma requer a utilização de abordagens multidisciplinares na qual participam um grande número de técnicas desde a biologia molecular à química analítica e bioinformática, entre outras. As proteínas encontram-se envolvidas em quase todas as funções biológicas. Assim, a análise das proteínas na célula permite uma perspetiva global de como estas moléculas interatuam e cooperam de forma a criar e manter um sistema biológico funcional2. As proteínas com função alterada são por isso a causa principal de doenças, podendo servir como indicadores úteis no diagnóstico de certas doenças1.
1.1. Importância da proteómica em investigação clínica
Em investigação clínica a análise proteómica desenvolve-se, na maioria dos casos, com base na comparação de duas ou mais condições (doença vs. saudável, pacientes não tratados vs. doentes tratados com determinado fármaco, etc.) além disso, a identificação de modificações pós-traducionais (PTMs), a determinação da expressão diferencial, a quantificação de proteínas e a identificação de biomarcadores de diagnóstico são também objetivos do estudo do proteoma em investigação clínica 3.
Nos últimos anos a proteómica tem beneficiado cada vez mais dos avanços tecnológicos e instrumentais alcançados. Os equipamentos de espectrometria de massa dedicados à análise de biomoléculas são cada vez mais rápidos, mais sensíveis e mais precisos, razões pelas quais cada vez mais, estas técnicas, se aplicam ao estudo do proteoma de amostras tão complexas como tecidos biológicos ou fluídos corporais 4. A proteómica
2
surge assim como uma ferramenta útil na identificação de biomarcadores associados a doenças humanas, incluindo aqueles que resultam de mudanças subtis nas funções normais da célula e nas vias de sinalização5, 6. Assim, esta abordagem -ómica providencia informações de diagnóstico que podem vir a ser essenciais para novas previsões terapêuticas7, 8.
2. Biomarcadores
Um biomarcador é um analito bioquímico que pode ser identificado e/ou quantificado de modo a avaliar um processo biológico normal ou patológico, de resposta farmacológica ou de outra intervenção terapêutica 9-11. Definidos como alterações nos constituintes do tecido ou de fluídos corporais, estes marcadores constituem um modo de classificação homogéneo da doença e dos fatores de risco, podendo alargar a base informativa acerca da patogénese subjacente à doença12.
2.1. Biomarcadores associados ao diagnóstico terapêutico
Os biomarcadores são amplamente usados como ferramentas analíticas para aceder a parâmetros biológicos e a uma análise terapêutica rápida e abrangente9, 13, podendo ser
descobertos através de tecnologias de genómica, proteómica ou de imaging12. Além disso, a análise de biomarcadores pode promover o desenvolvimento e avaliação de novas terapias9 e refletir o espectro completo de uma doença, desde as primeiras manifestações até aos casos terminais12. Estes marcadores moleculares podem ser identificados/quantificados em amostras biológicas, como por exemplo o plasma, o soro, fluído cerebrospinal, urina, líquido peritoneal e biópsias ultracongeladas ou parafinadas11, 12, 14.
3. Tecidos fixados com formalina
Depois de removidas dos pacientes, as amostras de tecido (biópsias) são, geralmente, conservadas por fixação em soluções contendo aldeídos (e. g. 10 % formalina) ou alternativamente, ultracongelados em azoto líquido e, posteriormente armazenadas a -80 °C15. O efeito de redução forte do formaldeído inibe as proteases, evitando a degradação dos componentes celulares e fixando as proteínas através da formação de pontes de metileno entre os grupos amino das diferentes cadeias de polipéptidos16. Assim, o método de armazenamento usualmente utilizado para biópsias em bancos de tecidos hospitalares, consiste na fixação da amostra de tecido em formalina, sendo depois embebida em parafina (formalin-fixed,
3
paraffin-embedded - FFPE). Este método é recorrente e amplamente utilizado em anatomia
patológica a nível mundial, no entanto não é o método ideal de preservação quando o objetivo é a análise do proteoma ou o estudo funcional de algumas proteínas/enzimas 17.
3.1. Importância dos tecidos FFPE e a desvantagem inerente
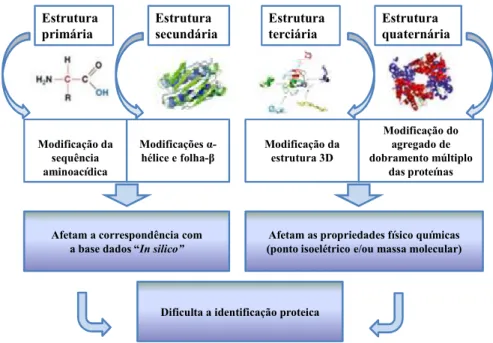
Quando comparados com tecidos frescos ou ultracongelados é evidente que os tecidos FFPE apresentam inúmeras vantagens. São estáveis por exemplo à temperatura ambiente, podem ser facilmente armazenados e não necessitam de condições de refrigeração especializada, providenciando também a arquitetura e estabilidade de detalhes celulares16. Os tecidos FFPE armazenados representam portanto, um valioso recurso de estudo, como alternativa às biópsias de tecidos frescos que são difíceis de obter e necessárias para as fases de deteção/validação na investigação de biomarcadores e para diagnóstico18. As amostras armazenadas de tecidos parafinados e o seu diagnóstico associado representam, uma fonte de informação proteómica em doenças onde os resultados dos pacientes já são conhecidos19. No entanto, o elevado grau de covalência cross-linked das proteínas em tecidos FFPE impede geralmente a extração eficiente destas, limitando assim a exploração bioanalítica da informação com potencial que se encontra disponível nos bancos de tecidos20. Em análise proteómica de tecidos FFPE o maior desafio é, talvez, definir as modificações exatas que afetam as cadeias polipeptídicas. Estas modificações podem originar redes de cross-link intra e/ou intercelulares16 (Figura 1).
Figura 1 - Organização estrutural e modificações prováveis que as proteínas sofrem sob condições de FFPE16.
Estrutura primária Estrutura secundária Estrutura terciária Estrutura quaternária Modificação da sequência aminoacídica Modificações α-hélice e folha-β Modificação da estrutura 3D Modificação do agregado de dobramento múltiplo das proteínas
Afetam a correspondência com a base dados “In silico”
Afetam as propriedades físico químicas (ponto isoelétrico e/ou massa molecular)
4
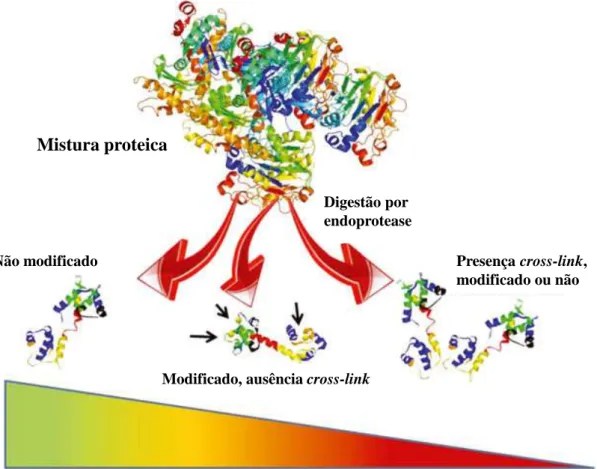
Apesar disto, tem sido feito um esforço significativo, necessário para ultrapassar o problema que se encontra associado ao efeito cross-linkage da formalina em análise proteómica. Resultados ambíguos são frequentemente obtidos devido à falha de correspondência entre os péptidos experimentalmente identificados com os teóricos que se encontram in silico. Dependendo do grau de modificação e do cross-link a possibilidade de identificar péptidos pode ser categorizada em alta, média ou baixa16, 18 (Figura 2).
Figura 2 - Esquema representativo da formação hipotética de péptidos de tecidos FFPE e o sucesso da sua identificação. Após digestão por endoproteases, três tipos de péptidos são produzidos com diferente sucesso de identificação: não modificados, que são os que apresentam um sucesso de identificação maior; modificados, ausência cross-link, em que o sucesso da sua identificação baseia-se principalmente na configuração variável do motor de pesquisa; presença cross-link, modificado ou não, que exibem uma forma complexa com as propriedades físico-químicas alteradas, tornando a correspondência In silico difícil e com baixo sucesso16.
Assim, para aumentar as probabilidades de deteção e identificação é necessário minimizar o efeito cross-link. Para isso é importante a otimização de protocolos capazes de garantir uma extração máxima de proteína, bem como capazes de eliminar as modificações causadas pela fixação com formalina.
Mistura proteica
Não modificado
Modificado, ausência cross-link
Presença cross-link, modificado ou não Digestão por
endoprotease
5
4. Líquido peritoneal
Os fluídos corporais humanos são matrizes biológicas complexas, constituídos por inúmeros tipos de proteínas6. O líquido peritoneal é um fluído seroso encontrado na cavidade peritoneal, que permite a livre movimentação dos orgãos abdominais sem fricção. Este fluído é produzido como um ultrafiltrado de plasma dependente da permeabilidade vascular e da força hidrostática e oncótica21,22. Encontrar biomarcadores em líquido peritoneal torna-se um desafio, pois existe um grande número de proteínas, lípidos e produtos metabólicos na amostra, enquanto os biomarcadores para a doença tendem a aparecer em concentrações muito baixas6. As proteínas alteradas no peritónio podem funcionar como biomarcadores na monitorização ou deteção de lesões resultantes da diálise peritoneal14.
5. Diálise Peritoneal
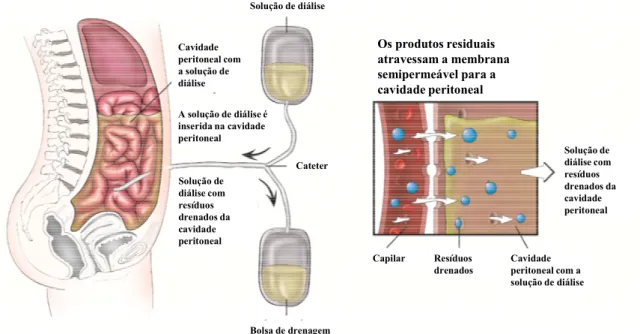
A diálise peritoneal (PD), é um método terapêutico usado como forma de regularização dos fluídos corporais no organismo para níveis normais23. O processo consiste na utilização do peritónio ou o celoma do paciente, como membrana através da qual fluídos e substâncias dissolvidas (eletrólitos, ureia, glucose, proteínas e outras moléculas pequenas) são trocadas do sangue, dialisado peritoneal e circulação6(Figura 3).
Figura 3- Figura representativa da diálise peritoneal e da forma como os produtos residuais atravessam a membrana para a cavidade peritoneal e são drenados juntamente com a solução de diálise. (Figura adaptada de [1]).
Solução de diálise com resíduos drenados da cavidade peritoneal Solução de diálise Bolsa de drenagem Cateter Cavidade peritoneal com a solução de diálise A solução de diálise é inserida na cavidade peritoneal Solução de diálise com resíduos drenados da cavidade peritoneal Capilar Cavidade peritoneal com a solução de diálise Resíduos drenados Os produtos residuais atravessam a membrana semipermeável para a cavidade peritoneal
6
Aproximadamente 12 % de pacientes em todo mundo são sujeitos à PD, em alternativa à hemodiálise (HD)23. Assim, este método de terapia é indiscutivelmente o primeiro a ser usado em casos de falha renal, encontrando-se associado a uma melhor preservação da função renal residual, em relação à observada em hemodiálise24, 25. A taxa de transporte de solutos através da membrana peritoneal é determinante para a prescrição da diálise e também o principal fator para predição da sobrevivência do paciente23. No entanto, a taxa de transporte de soluto e de ultrafiltração, varia muito em pacientes com doença renal em estado terminal, que são tratados com diálise peritoneal crónica (CPD). Este facto deve-se à diferença de capacidade da membrana peritoneal para transportar ou remover solutos e água, variando consoante o paciente26. Apesar da diálise peritoneal substituir a função renal, frequentemente gera lesões no peritónio que não são fáceis de identificar sem recorrer a técnicas invasivas. No entanto, alterações entre o dialisado peritoneal e o fluído peritoneal normal, podem ser essenciais à compreensão dos mecanismos de lesão peritoneal causada pela PD14.
6. Desenho Fatorial
Para se realizar um desenho fatorial padrão é necessário selecionar um número fixo de “níveis” (ou versões) para cada uma, de um conjunto de variáveis (fatores), fazendo-se depois experiências com cada uma das combinações possíveis27. O desenho fatorial de quatro fatores, cada um com dois níveis (desenho 24) é um exemplo, existindo 16 combinações possíveis. No entanto é possível prosseguir o estudo com metade das combinações (com replicação apropriada) e mesmo assim estimar o efeito de cada fator e a interação entre cada par de fatores28, 29. Através disto é possível descobrir que fatores influenciam o resultado da experiência e que níveis destes fatores originam uma investigação com uma maior sensibilidade30. Estes desenhos requerem relativamente poucas experiências por fator e apesar de não permitirem explorar exaustivamente uma ampla região do espaço de fatores, podem indicar tendências e assim determinar uma direção para investigações futuras31. Assim, a estratégia passa por usar o desenho fatorial para determinar os fatores e níveis mais importantes para o resultado e usar posteriormente estes fatores na investigação30.
7. Métodos Analíticos
Em espectrometria de massa, a amostra biológica é ionizada e os iões são posteriormente acelerados em direção ao analisador de massa. Assim, é possível determinar a
7
existência de iões (ratios massa-de-carga, m/z) específicos para cada amostra 32. No entanto, a espetrometria de massa (MS) está intimamente ligada e dependente de tecnologias de separação como a eletroforese bidimensional em gel de poliacrilamida (2D PAGE), que simplificam a complexidade das amostras biológicas antes de proceder-se à análise de massa33. Existem duas abordagens gerais na proteómica para a identificação e caracterização de proteínas por MS: a abordagem top-down que consiste na análise de proteína intacta e a abordagem bottom-up, baseada na análise de péptidos obtidos enzimaticamente ou quimicamente. Explicada mais detalhadamente, a abordagem top-down envolve uma fase de ionização da proteína intacta e a subsequente medição da relação massa-carga, seguindo-se a sua fragmentação direta dentro do espectrómetro de massa, sem ocorrer uma digestão antes da análise34, 35, 36.
Relativamente à estratégia bottom-up, esta permite inferir a presença de uma proteína através da deteção de péptidos, sendo usada para análises de larga escala e de alto rendimento em amostras complexas35, 37. A estratégia bottom-up pode ser realizada através do método
Sort-then-break ou Break-then-sort. O primeiro consiste na separação proteica antes da
digestão e precedente análise dos péptidos por peptide mass fingerprinting (PMF) (Figura 4) ou da separação adicional dos péptidos por cromatografia líquida (LC) acoplada ao espectrómetro de massa. O método Break-then-sort consiste na digestão proteica sem ocorrer nenhum passo de separação prévio, sendo os péptidos separados por LC acoplada ao espectrómetro de massa34, 35, 36.
8
Figura 4- Identificação proteica com peptide mass fingerprinting. As massas de péptidos das proteínas digeridas são correspondidas com a lista da massa teórica de péptidos, que são matematicamente derivados da base de dados do genoma de certos organismos3.
7.1. Princípios da eletroforese
A eletroforese é o processo pelo qual moléculas carregadas em solução se movimentam por ação de um campo elétrico. Quando submetidas a um campo elétrico as moléculas movimentam-se com uma velocidade dependente da sua carga, forma e tamanho. A técnica de eletroforese é amplamente utilizada na separação e purificação de biomoléculas como as proteínas ou ácidos nucleicos1.
As proteínas são geralmente separadas numa matriz sólida de gel de poliacrilamida1. A matriz é necessária porque a corrente elétrica ao passar através da solução de eletroforese gera calor, causando a difusão e convecção da mistura de bandas na ausência de um meio estabilizador. Os géis de poliacrilamida e de agarose são os mais usados como meio estabilizador e na separação de proteínas, atuando a matriz como uma malha seletiva de tamanho38.
Prática Experimental Pesquisa de dados
Excisão de spots do gel 2-D Digestão in vitro, eluição dos péptidos com tripsina Espectro de massa dos péptidos
Massa dos péptidos:
735.2258 657.7893 534.5399 383.9141 275.2567 Correspondem ? ! Tradução in silico Digestão in silico
Produto teórico do gene: Sequência de aminoácidos Péptidos trípticos teóricos Base dados genómica: Sequência de DNA Massa teórica: 735.2256 657.7896 593.9785 534.5397 395.6702 383.9147 275.2561
9
7.1.1. Eletroforese monodimensional (1-DE)
A electroforese monodimensional (1-DE) é uma técnica de separação na qual se utiliza apenas uma das propriedades da proteína para resolver uma mistura de proteínas. Quando se utiliza a electroforese monodimensional podem utilizar-se diversos meios de separação, sendo os mais comuns o gel nativo (condições não desnaturantes) e o gel de poliacrilamida com dodecil sulfato sódio (SDS-PAGE). No entanto o gel de SDS-PAGE é o mais amplamente utilizado em proteómica, devido à sua capacidade de separar grandes quantidades de proteínas, incluindo fragmentos39. Quando se utiliza a técnica de SDS-PAGE, a migração e consequente separação das proteínas é determinada não pela carga elétrica intrínseca das cadeias dos polipéptidos, mas pelo peso molecular. O SDS adicionado quer à matriz de gel quer à amostra proteica, é um detergente aniónico que provoca a desnaturação das proteínas, uma vez que quebra as pontes de hidrogénio, elimina as interações hidrofóbicas eliminando a estrutura terciária e secundária das proteínas. O SDS liga-se à maioria das proteínas numa proporção de 1,4g de SDS por 1g de proteína, conferindo uma carga total negativa à proteína proporcional ao seu tamanho. Ou seja, como o SDS possui carga negativa, na sua presença todas as proteínas na mistura ficam igualmente carregadas, migrando de acordo com o seu peso molecular e não pela sua carga1, 40. Aplicada antes da análise por espectrometria de massa, esta técnica amplia a dinâmica de análise e facilita a identificação de proteínas pouco abundantes39. A desvantagem desta técnica é o limite de resolução, associada ao facto de ser de natureza monodimensional. A sobreposição de bandas pode ser de difícil interpretação, por isso, esta técnica é mais eficaz na análise de um pequeno número de proteínas41.
7.1.2. Eletroforese bidimensional (2-DE)
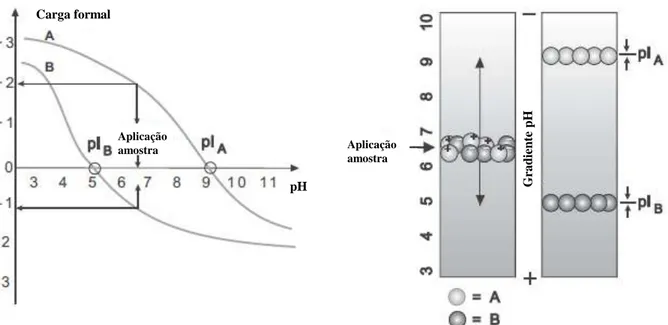
Originalmente desenvolvida em 1975, a eletroforese bidimensional é um método analítico que separa as proteínas de uma amostra biológica pelo seu ponto isoelétrico (primeira dimensão) e massa molecular relativa (segunda dimensão). O ponto isoelétrico corresponde ao pH no qual a carga formal de uma dada proteína é zero3,32. A 2-DE de proteínas é realizada sob condições desnaturantes. Relativamente à primeira dimensão as proteínas são solubilizadas na presença de ureia, causando a disrupção das ligações de hidrogénio. No entanto não afeta a carga intrínseca das proteínas o que permite separar as proteínas apenas pela sua carga (Figura 5).
10
Figura 5- Princípio da focagem isoelétrica. Esquerda: Curva da carga formal de duas proteínas modelo, A e B. No ponto de aplicação, a curva A tem duas cargas positivas e a curva B tem uma carga negativa. Direita: Migração de A e B para os seus pIs no gradiente de pH do gel de focagem isoelétrica3.
Quanto à segunda dimensão as proteínas são separadas na presença de SDS, este liga-se as proteínas carregando-as negativamente e tornando-as idênticas em termos de densidade de carga, assim as proteínas migram estritamente de acordo com o seu tamanho42, 43. Resumidamente, esta técnica consiste na focagem isoelétrica (IEF) num gradiente de pH, seguida da separação em SDS em gel de poliacrilamida (SDS-PAGE). Este método permite assim, separar, exibir e armazenar milhares de proteínas num só gel32, 43.
7.2. Ionização / Dessorção Assistida por Laser - Matrix-Assisted Laser Desorption
Ionization (MALDI)
A base fundamental da técnica de espectrometria de massa (MS) consiste na ionização de uma ou mais moléculas, e posterior deteção destes iões em fase gasosa. Os métodos clássicos para a ionização de moléculas são o impacto de eletrões (EI) e a ionização química (CI). Até ao desenvolvimento de técnicas de ionização suaves, como a ionização por
electrospray (ESI) e da ionização por MALDI, as ferramentas de espectrometria de massa não
eram consideradas para aplicação em ciências biológicas44-46. Hoje em dia a ionização por MALDI é essencial em espectrometria de massa aplicada à proteómica, devido à sua extrema sensibilidade, facilidade de aplicação e tolerância a contaminantes, além destas características a técnica de MALDI-TOF-MS é relativamente rápida permitindo a aquisição de uma grande quantidade de dados em pouco tempo47. Resumidamente, no MALDI-MS, a amostra é
Aplicação amostra Aplicação amostra G ra d ien te p H Carga formal pH
11
embebida numa estrutura cristalina de pequenos compostos orgânicos (matriz) e colocada num suporte condutivo para amostras. O sucesso das análises por MALDI-MS são fortemente dependentes da matriz, grande parte das matrizes bem-sucedidas e que ainda hoje são usadas, foram descobertas empiricamente devido à falta de diretrizes para escolher e desenhar um sistema de matriz ótimo para um dado analito. O Quadro 1 apresenta algumas das matrizes usadas em proteómica48.
Quadro 1- Matrizes de MALDI e as suas aplicações em proteómica 48.
Nome Aplicação Comprimento
de onda (nm) Ácido 2, 5-Dihidroxibenzóico Análise comum a proteínas e péptidos; proteínas e
péptidos modificados pós-transducionalmente
337
Ácido α-Ciano-4-hidroxicinamico Análise de péptidos 337, 355
2, 6-Dihidroxiacetofenona Proteínas e péptidos modificados pós-transducionalmente
337
2, 4, 6-Trihidroxiacetofenona Proteínas e péptidos modificados pós-transducionalmente
337
Ácido Sinapínico Proteínas 337
Ácido Sucínico Análise comum a proteínas e péptidos; proteínas e péptidos modificados pós-transducionalmente
2940
A matriz do MALDI absorve energia laser e transfere-a para o analito acidificado, enquanto o rápido aquecimento a laser provoca a dessorção da matriz e dos iões [M+H]+ do analito para a fase gasosa (Figura 6). A ionização por MALDI requer várias centenas de disparos laser para alcançar um ratio de signal-to-noise aceitável33, 44, 46.
12
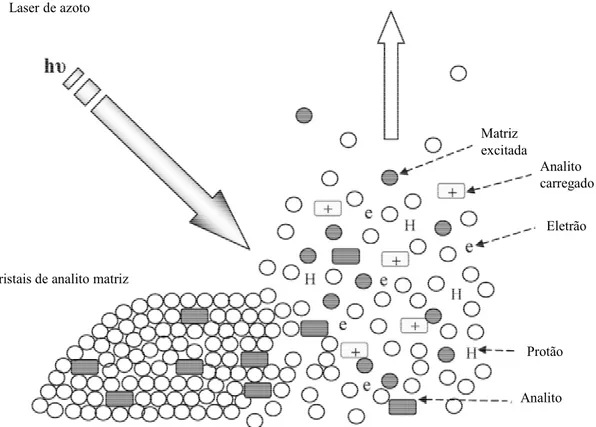
Figura 6- Processo de ionização por MALDI. Os cristais de analito matriz são bombardeados com laser UV excitando a matriz, esta por sua vez transfere a energia para os analitos. Isto resulta na ionização e dessorção dos analitos, carregados sobretudo de forma isolada44.
Para melhorar a medição da massa molecular para níveis de partes por milhão (ppm) é acoplado ao MALDI um reflectrão, um espelho de iões e um time of flight (TOF)41. As massas dos iões são calculadas pela medição TOF que se torna mais longo para moléculas maiores em relação a menores. Encontrando-se as massas disponíveis como dados numéricos para o processamento direto e análise subsequente46. Os iões gerados pelo MALDI são geralmente carregados de forma isolada, isto torna o MALDI aplicável a análises top-down de proteínas de alto peso molecular com instrumentos de análise pulsada. As desvantagens são a baixa reprodutibilidade shot-to-shot e a forte dependência de métodos de preparação de amostra33. Relativamente aos mecanismos dos aparelhos de espectrometria de massa existem diferentes configurações. Na Figura 7 estão representadas três configurações, sendo a última (C) a correspondente ao MALDI usado na análise proteómica dos tecidos fixados com formalina e de líquido peritoneal.
Laser de azoto
Cristais de analito matriz
Matriz excitada Analito carregado Eletrão Protão Analito
13
Figura 7-Diferentes configurações (A, B e C) do espectrómetro de massa/fonte iónica. A- na configuração reflector time-of-flight (TOF) os iões são acelerados a uma energia cinética alta e separados ao longo do tubo de voo devido às suas diferentes velocidades. Os iões são refletidos no refletor, compensando assim pequenas diferenças de energia cinética, estes colidem depois no detetor que amplifica e realiza a sua contagem à chegada. B- na configuração TOF-TOF é incorporada uma célula de colisão entre duas secções de TOF. Na primeira secção TOF1 os iões de um certo ratio massa-de-carga (m/z) são selecionados, a fragmentação ocorre na célula de colisão, sendo depois os fragmentos obtidos separados na segunda secção TOF2. C- na configuração LIFT-TOF/TOF, a secção TOF1 abrange da fonte iónica do MALDI até à célula LIFT. A secção TOF2 abrange do segundo acelerador, na célula LIFT, até ao refletor. Adaptado de Aebersold & Mann49e Suckau et al.50.
Quando se parte de uma amostra de proteína ou de uma mistura complexa de proteínas, uma enzima, geralmente a tripsina, digere as proteínas em péptidos (digestão tríptica). Obtidos os péptidos trípticos recorre-se à identificação e caracterização proteica por
A - Reflector time-of-flight (TOF)
B - Time-of-flight
time-of -flight (TOF- TOF)
Laser pulsado
Placa com amostra
Refletor TOF TOF2 TOF1 Célula de colisão TOF1 TOF2 Detetor Detetor Fonte iónica Placa com amostra Analisador Refletor Célula de colisão Timed Ion Selector (TIS) Fonte 2 (LIFT) Post LIFT Metastable Suppressor (PLMS) C- LIFT Time-of-flight
time-of -flight (LIFT-TOF/TOF)
14
espectrometria de massa, os péptidos são selecionados um a um, através do primeiro estado de análise de massa (MS1). Cada péptido isolado é então induzido à fragmentação e o segundo estado de análise de massa é usado para capturar um espectro MS/MS37(Figura 8).
Figura 8- Em (1) é representado esquematicamente o espectro de massa dos péptidos obtidos (espectro MS1 ou “espectro de massa normal”), o computador gera então uma lista de péptidos que serão fragmentados (516.27 m/Z), obtendo-se uma série de espectros MS/MS (2). Esta análise consiste no isolamento de um ião peptídico específico, na fragmentação por colisão energética com gás e a obtenção dos espectros MS/MS. Os espectros MS e MS/MS são obtidos a cada segundo e gravados para fazer a comparação com bases de dados, resultando assim na identificação dos péptidos e por conseguinte da proteína. (Adaptado de Aebersold & Mann49).
15
8. Objetivos
Com a realização deste trabalho no âmbito da dissertação de mestrado em Genética Molecular Comparativa e Tecnológica pretende-se:
- Desenvolver e otimizar um conjunto de metodologias necessárias à extração proteica em tecidos fixados com formalina;
- Integrar a análise proteómica em líquido peritoneal, tendo como principal tarefa a identificação dos constituintes do proteoma desta amostra biológica, para possíveis previsões terapêuticas.
17
II.
MATERIAL
E
MÉTODOS
Este capítulo encontra-se dividido em dois subcapítulos inerentes a cada um dos principais objetivos referidos no capítulo anterior. No subcapítulo 1. será abordado o objetivo referente ao desenvolvimento e otimização de metodologias necessárias ao incremento da extração proteica em tecidos fixados com formaldeído.
1. Desenvolvimento e otimização de metodologias de extração proteica em
tecidos fixados com formalina
Diferentes abordagens foram estabelecidas e otimizadas com o intuito de reverter as modificações introduzidas pela fixação com formalina com vista a maximizar a extração proteica e posterior análise proteómica.
1.1. Material biológico
O material biológico usado para a otimização foram rins de vaca. A escolha deste material deveu-se ao facto de poder ser usado sem qualquer restrição de quantidade. Os rins foram obtidos no matadouro e colocados em gelo, de forma a evitar e minimizar o tempo de exposição a condições propícias para que ocorresse proteólise dos tecidos.
1.2. Reagentes
1.2.1. Reagentes usados na fixação do material biológico e extração do fixador
Os reagentes usados para a fixação e posterior extração foram o formaldeído (37 %), ácido acético e o Etanol absoluto (CH3CH2OH) (100 %), adquiridos da Panreac.
1.2.2. Reagentes usados para as soluções tampão de extração
Os seguintes reagentes foram utilizados como constituintes das soluções tampão de extração, designadamente: β- mercaptoetanol (C2H6OS) (≥ 99 %) da Merck; Sal de Dodecil
Sulfato Sódio (SDS) (C12H25NaO4S) da Panreac; Tris(hidroximetil)aminometano
(NH2C(CH2OH)3) (≥ 99,8 %), Glicerol (HOCH2CH(OH)CH2OH) (≥ 99 %) e o Triton 100-X
18
1.2.3. Reagentes usados no tratamento dos géis excisados para a precedente identificação proteica
Os seguintes reagentes foram utilizados para o tratamento dos géis excisados: Bicarbonato de amónio, Acetonitrilo (ACN), D,L-Ditiotreitol (DTT), Iodoacetamida (IAA), Tripsina, Ácido Fórmico e Ácido Trifluoroacético (TFA) foram adquiridos da Sigma-Aldrich.
1.3. Fixação das amostras
As amostras biológicas foram cortadas em pequenas porções (0,1-0,5 g), e incubadas numa solução de fixador Bouin (etanol 50 % v/v; formaldeído 10 % v/v; ácido acético 7 % v/v) durante 48 h de modo a induzir o cross-link entre as proteínas. Após fixação as amostras foram lavadas 3 vezes com etanol 90 % (v/v) de modo a remover o excesso de fixador.
Terminado o procedimento de fixação as amostras foram congeladas com azoto líquido e maceradas num almofariz arrefecido com azoto líquido até à obtenção de um pó fino e homogéneo. Este pó foi posteriormente guardado a -60 °C.
1.4. Preparação do tecido para posterior extração de proteína
Em microtubos de 1,5 mL, foram colocadas aproximadamente 16 mg de amostra de tecido, e o tecido foi hidratado com uma série decrescente de álcoois (etanol a 100 %, 96 % e 70 %), sendo adicionado por série 200 µL em cada microtubo, durante 10 min. Entre cada série e no fim destas, foi realizada uma centrifugação a 9000 rpm (ELMI skyline), durante 5 min e posterior remoção do etanol. Os microtubos foram deixados abertos para evaporar o etanol, durante 20 min.
1.5. Otimizações e Metodologias
1.5.1. Otimização da variável da indução da extração proteica por calor (Tempo/ °C)
Para esta otimização a solução tampão usada tinha como constituintes os seguintes reagentes: Tris-HCl 20 mM a pH 9; Glicerol a 10 % (p/v); SDS a 2 % (p/v); β-mercaptoetanol a 6 % (v/v). As variáveis de temperatura (°C) e tempo testadas no bloco de aquecimento, foram duas: 20 min a 100 °C seguido de 2 h a 60 °C e 2 h a 70 °C (Quadro 2). Foram realizadas duas réplicas experimentais (em dias diferentes) com 3 amostras para cada uma das variáveis temperatura (°C) e Tempo, sendo portanto 12, o número total de amostras.
19
Quadro 2- Número experimental das amostras para cada variável da indução da extração proteica por calor e descrição da solução tampão.
1.5.1.1. Metodologia para a otimização da variável da indução da extração proteica por calor (Tempo/ °C)
Em microtubos de 1,5 mL e após hidratação do tecido, adicionou-se a cada amostra 100 µL de solução tampão descrita no Quadro 2 (6 µL β-mercaptoetanol + 94 µL de solução tampão). O β-mercaptoetanol só deve ser adicionado à solução tampão no momento da utilização. As amostras foram incubadas em gelo durante 15 min. Em seguida dos 6 microtubos com as amostras (1.ª réplica), 3 delas foram sujeitas a extração proteica com a solução tampão a 100 °C no bloco de aquecimento, durante 20 min e a 60 °C durante 2 h. As outras 3 foram sujeitas a extração proteica com a solução tampão a 70 °C no Bloco aquecimento, durante 2 h, sendo as amostras agitadas a cada 30 min. Após arrefecerem, foram centrifugadas a 14000 ×g, durante 15 min a 4 °C. O sobrenadante foi recuperado para outro microtubo, adicionou-se 50 µL de solução tampão às amostras e centrifugou-se a 9000 rpm (ELMI skyline), durante 5 min, sendo o sobrenadante transferido para o microtubo com o sobrenadante anterior.
1.5.2. Escolha do detergente de extração: SDS 2 % / Triton X-100 1 %
Para esta otimização uma das soluções tampão usada tinha como constituintes os seguintes reagentes: Tris-HCl 20 mM a pH 9; Glicerol a 10 % (p/v); SDS a 2 % (p/v); β-mercaptoetanol a 6 % (v/v) (solução tampão A). A segunda solução tampão tinha como reagentes constituintes: Tris-HCl 20 mM a pH 9; Glicerol a 10 % (p/v); Triton X-100 a 1 % (p/v); β-mercaptoetanol a 6 % (v/v); (solução tampão B, Quadro 3). Foi realizada uma réplica com 4 amostras para a variável de solução tampão A e 2 amostras para a de solução tampão B.
Solução tampão Temperatura (°C) / Tempo
Tris-HCl 20 mM 20 min a 100 °C + 2 h a 60 °C # Experimental (1, 2, 3) pH 9 Glicerol 10 % 2 h a 70º C # Experimental (4, 5, 6) SDS 2 %
20
Quadro 3- Número experimental das amostras para a variável de detergente SDS 2 %/ Triton X-100 1 % e descrição da solução tampão.
Solução tampão A Solução tampão B
Tris-HCl 20 mM Tris-HCl 20 mM
pH 9 pH 9
Glicerol 10 % Glicerol 10 %
SDS 2 % Triton X-100 1 %
# Experimental (1, 2, 3 e 4) # Experimental (5 e 6)
1.5.2.1. Metodologia para otimização da variável SDS 2 %/ Triton X-100 1 %
Em microtubos de 1,5 mL e após a hidratação do tecido, adicionou-se a cada amostra 100 µL de solução tampão (6 µL mercaptoetanol + 94 µL de solução tampão). O β-mercaptoetanol só deve ser adicionado à solução tampão no momento da utilização. As amostras foram incubadas em gelo durante 15 min. Em seguida dos 6 microtubos com as amostras, 4 delas foram sujeitas a extração proteica com a solução tampão A a 70 °C no Bloco de aquecimento, durante 2 h. As outras 2 foram sujeitas a extração proteica com a solução tampão B a 70 °C no Bloco aquecimento, durante 2 h. Sendo todas as amostras agitadas a cada 30 min. Após arrefecerem, foram centrifugadas a 14000 ×g, durante 15 min a 4 °C. O sobrenadante foi recuperado para outro microtubo, adicionou-se 50 µL de solução tampão às amostras e centrifugou-se a 9000 rpm (ELMI skyline), durante 5 min, sendo o sobrenadante transferido para o microtubo com o sobrenadante anterior.
1.5.3. Otimização da variável da amplitude (%) de ultrassons (US)
Para esta otimização a solução tampão usada tinha como constituintes os seguintes reagentes: Tris-HCl 20 mM a pH 9; Glicerol a 1 % (p/v); SDS a 2 % (p/v); β-mercaptoetanol a 6 % (v/v). As variáveis amplitude (%) de ultrassons (US) testadas com a sonda “Dr.
HielscherGmbH”, foram quatro, designadamente: 4×10s a 30 % de amplitude de US; 4×10s a 60
% de amplitude de US; 4×10s a 100 % de amplitude de US; Sem ultrasons (Quadro 4). Todas as amostras foram sujeitas a 100 °C no bloco de aquecimento, durante 20 min, seguidas de 2 h a 60 °C, tendo sido realizadas réplicas técnicas para cada uma das variáveis amplitude (%) de ultrassons (US). O número total de amostras foi portanto de 12.
21
Quadro 4-Descrição da solução tampão e das condições de extração °C/ tempo e % de ultrasons/sem ultrasons. Número experimental das amostras para cada variável.
Solução tampão 20 min a 100 °C + 2 h a 60 °C
Tris-HCl 20 mM 4×10s a 30 % de amplitude de US, #Experimental (A1, A2 e A3) pH 9 4×10s a 60 % de amplitude de US, #Experimental (A4, A5 e A6) Glicerol 1 % 4×10s a 100 % de amplitude de US, #Experimental (A7, A8 e A9)
SDS 2 % Sem ultrasons, #Experimental (A10, A11 e A12)
1.5.3.1. Metodologia para a otimização da variável da amplitude (%) de ultrasons (US)
Em microtubos de 1,5 mL e após a extração do fixador, adicionou-se a cada amostra 100 µL de solução tampão (6 µL mercaptoetanol + 94 µL de solução tampão). O β-mercaptoetanol só deve ser adicionado à solução tampão no momento da utilização. As amostras foram incubadas em gelo durante 15 min. Todas as amostras foram sujeitas a extração proteica com a solução tampão a 100 °C no Bloco aquecimento, durante 20 min, seguidas de 2 h a 60 °C, sendo as amostras agitadas a cada 30 min. Sonicar com a sonda “Dr.
HielscherGmbH”as amostras correspondentes a cada % de amplitude (30 %, 60 %, 100 %) (4 ×
10s), exceto as amostras sem US. Em seguida as amostras foram centrifugadas a 14000 ×g, durante 15 min a 4º C. O sobrenadante foi recuperado para outro microtubo, adicionou-se 50 µL de solução tampão às amostras e centrifugou-se a 9000 rpm (ELMI skyline), durante 5 min, sendo o sobrenadante transferido para o microtubo com o sobrenadante anterior.
1.6. Desenho Fatorial: Otimização e metodologias
No decorrer deste desenho fatorial foram usadas diferentes soluções tampão e condições de extração. As soluções tampão usadas tinham como constituintes os seguintes reagentes: solução tampão I, Tris-HCl 20 mM a pH 7,4, Glicerol a 1 % (p/v), SDS a 2 % (p/v); solução tampão II, Tris-HCl 20 mM a pH 9, Glicerol a 1 % (p/v), SDS a 2 % (p/v); solução tampão III, Tris-HCl 20 mM a pH 7,4, Glicerol a 10 % (p/v), SDS a 2 % (p/v); solução tampão IV, Tris-HCl 20 mM a pH 9, Glicerol a 10 % (p/v), SDS a 2 % (p/v). Antes da utilização de cada solução tampão, foi adicionado 6 % (v/v) de β- mercaptoetanol (6 µL de β- mercaptoetanol + 94 µL de solução tampão). Quanto às condições de extração estas foram também quatro, nomeadamente: sem temperatura, 20 min a 100 °C e 2 h a 60 °C no Bloco aquecimento, sem ultrassons e por fim, 4×10s de US a uma amplitude de 100 % com a sonda
22
“Dr. HielscherGmbH” (Quadro 5). Foram realizadas três réplicas experimentais (em dias
diferentes) para cada um dos 16 números experimentais (Quadro 6), sendo portanto 48, o número total de amostras.
Quadro 5- Composição de cada uma das soluções tampão utilizadas no desenho experimental factorial 24. Os sinais (-) ou (+) representam os valores mínimos (-) ou máximos (+) para cada uma das variáveis.
Quadro 6- Representação esquemática da otimização, com a descrição do número experimental em relação às variáveis e à solução tampão correspondente.
Número Experimental pH % Glicerol Temperatura Ultrasons (US) Solução tampão 1 − − − − I 2 + − − − II 3 − + − − III 4 + + − − IV 5 − − + − I 6 + − + − II 7 − + + − III 8 + + + − IV 9 − − − + I 10 + − − + II 11 − + − + III 12 + + − + IV 13 − − + + I 14 + − + + II 15 − + + + III 16 + + + + IV
1.6.1. Metodologia para amostras sem extração por indução de calor (-) e ultrassons (-)
Em microtubos de 1,5 mL e após a hidratação do tecido, adicionou-se por cada amostra com variáveis diferentes para o pH e % de glicerol, 100 µL da solução tampão correspondente (6 µL β-mercaptoetanol + 94 µL de solução tampão). O β-mercaptoetanol foi adicionado à solução tampão no momento da utilização. As amostras foram incubadas em
Solução tampão I Solução tampão II Solução tampão III Solução tampão IV Tris-HCl 20 mM Tris-HCl 20 mM Tris-HCl 20 mM Tris-HCl 20 mM
pH 7,4 pH 9 pH 7,4 pH 9
Glicerol 1 % Glicerol 1 % Glicerol 10 % Glicerol 10 %
SDS 2 % SDS 2 % SDS 2 % SDS 2 %
pH % Glicerol Temperatura Ultrasons (US)
7,4 (-) 1 % (-) Sem temperatura (-) Sem US (-)
23
gelo durante 15 min. Em seguida as amostras foram centrifugadas a 14000 ×g, durante 15 min a 4 °C. O sobrenadante (SN1) foi recuperado para outro microtubo, adicionando-se 50 µL de solução tampão correspondente em cada uma das amostras. Agitaram-se durante 30s. Centrifugaram-se novamente a 14000 ×g durante 15 min a 4 °C, recuperando o sobrenadante e juntando com SN1. Por fim, o SN1 foi guardado a -20 °C.
1.6.2. Metodologia para amostras com extração por indução de calor (+) e sem ultrassons (-)
Em microtubos de 1,5 mL e após a hidratação do tecido, adicionou-se por cada amostra com variáveis diferentes para o pH e % de glicerol, 100 µL de solução tampão correspondente (6 µL β-mercaptoetanol + 94 µL de solução tampão). O β-mercaptoetanol foi adicionado à solução tampão no momento da utilização. As amostras foram incubadas em gelo durante 15 min. Colocaram-se os microtubos no Bloco aquecimento a 100 °C durante 20 min, em seguida incubaram-se a 60 °C, durante 2 h. As amostras foram depois centrifugadas a 14000 ×g, durante 15 min a 4 °C. O sobrenadante (SN1) foi recuperado para outro microtubo, adicionando-se 50 µL de solução tampão correspondente em cada uma das amostras. Agitaram-se durante 30s. Centrifugaram-se novamente a 14000 ×g durante 15 min a 4 °C, recuperando o sobrenadante e juntando com SN1. Por fim, o SN1 foi guardado a -20 °C.
1.6.3. Metodologia para amostras sem extração por indução de calor (-) e com ultrassons (+)
Em microtubos de 1,5 mL e após a hidratação do tecido, adicionou-se por cada amostra com variáveis diferentes para o pH e % de glicerol, 100 µL de solução tampão correspondente (6 µL β-mercaptoetanol + 94 µL de solução tampão). O β-mercaptoetanol foi adicionado à solução tampão no momento da utilização. As amostras foram incubadas em gelo durante 15 min. Sendo depois sonicadas com a sonda “Dr. HielscherGmbH” a 100 %
amplitude (4 × 10s) e em seguida centrifugadas a 14000 ×g, durante 15 min a 4 °C. O sobrenadante (SN1) foi recuperado para outro microtubo, adicionando-se 50 µL de solução tampão correspondente em cada uma das amostras. Agitaram-se durante 30s. Centrifugaram-se novamente a 14000 ×g durante 15 min a 4 °C, recuperando o sobrenadante e juntando com SN1. Por fim, o SN1 foi guardado a -20 °C.