iii
o júri
presidente Prof. Doutora Maria Inês Purcell de Portugal Branco
Professora auxiliar do Departamento de Química da Universidade de Aveiro
Prof. Doutora Maria Alice Zarur Coelho
Professora associada nível 1 da Escola de Química da Universidade Federal do Rio de Janeiro
Prof. Doutor Francisco Avelino da Silva Freitas
Professor auxiliar do Departamento de Química da Universidade de Aveiro
Prof. Doutor Carlos Manuel Santos Silva
iv
agradecimentos Gostaria de agradecer em primeiro lugar aos meus orientadores, Doutor Carlos
Manuel Silva e Doutor Avelino Silva, pelo seu incentivo na realização deste trabalho, pela sua orientação e disponibilidade e pela oportunidade que tive de evoluir, quer cientificamente quer como pessoa, ao longo deste último ano. Um agradecimento ainda aos meus pais e ao meu irmão pelo apoio, não só durante este último ano, mas em todo o meu percurso académico.
Queria agradecer à Helena, pelo incentivo e companheirismo ao longo dos últimos 3 anos, em que me transmitiu sempre confiança de que eu iria conseguir atingir os meus objectivos.
Por fim, um agradecimento ainda a todos os meus amigos, quer da residência quer do curso, em especial à Andreia, pela sua amizade e pela ajuda que sempre me deu.
v
palavras-chave Aminoácidos, Peptídeos, Equilíbrio, Coeficiente de Actividade, UNIFAC,
Debye-Hückel, Modelo
resumo O objectivo deste trabalho foi desenvolver um modelo para coeficientes de
actividade de aminoácidos/peptídeos em solução aquosa que exiba uma boa capacidade de correlação e de previsão.
O modelo proposto compreende quatro aspectos fundamentais: (i) calcula o equilíbrio químico do aminoácido (AA) em solução aquosa, que dá origem às espécies AA± (zeuterião), AA+, AA–, H+ e OH–; (ii) calcula os desvios à idealidade das interacções de curto alcance entre as espécies em solução, utilizando o método de contribuições de grupo de UNIFAC; (iii) as interacções de longo alcance, de natureza electrostática, são também quantificadas com a inclusão de um termo de Debye-Hückel (DH); (iv) o zeuterião é considerado uma molécula contendo dois grupos distintos carregados electricamente. Por este motivo também contribui para os desvios à idealidade calculados por DH. Com o objectivo de optimizar os parâmetros de interacção energética de UNIFAC foi compilada uma base de dados com 14 aminoácidos/peptídeos e desenvolvido um programa em Matlab. Foram também efectuadas previsões. A qualidade dos resultados foi medida pelo rmsd (root mean square deviation). De forma a interpretar correctamente os dados experimentais de aminoácidos de cadeia aberta contendo substituintes alquilo no carbono-
α
foi definido um grupo CH3 modificado (mCH3). Globalmente foram optimizados 17 parâmetros de interacção energética.O modelo apresentado correlacionou com precisão os dados experimentais de oito aminoácidos/peptídeos, fornecendo rmsdcorrel = 0,90%. A previsão de seis sistemas foi efectuada com um rmsdprev = 5,65%. Estes resultados são muito bons quando comparados com outros trabalhos baseados no método de UNIFAC, como o de Gupta e Heidemann (1990) (rmsdcorrel = 3,74% e rmsdprev = 14,94%) e o de Pinho et al. (1994) (rmsdcorrel = 0,80% e rmsdprev = 20,17%).
vi
keywords Amino Acids, Peptide, Equilibrium, Activity Coefficient, UNIFAC, Debye-Hückel.
Model
abstract The purpose of this work was the development of a model for the activity
coefficients of amino acids/peptides in aqueous solution with good correlation and prediction capabilities.
The proposed model comprises four aspects: (i) the chemical equilibrium calculation of the amino acid (AA) in aqueous solution, which originates species AA± (zwitterion), AA+, AA-, H+ and OH-, (ii) the deviations from the ideal solution behaviour due to the short-range interactions between species in solution are taken into account by the UNIFAC group contribution method, (iii) the long-range interactions, owing to electrostatic forces, are accounted for a Debye-Hückel (DH) term, (iv) the zwitterion is assumed to be a molecule with two distinct groups electrically charged, which contribute to additional deviations from ideal behaviour.
In order to optimize the interaction parameters of the UNIFAC model, a database containing 14 amino acids/peptides was compiled and a Matlab program was developed. Predictions were also accomplished for the activity coefficients. The accuracy of the results for both correlation and prediction was measured via rmsd (root mean square deviation). A mCH3 (modified CH3) group was introduced to interpret the experimental data of amino acids containing alkyl groups on its alpha-carbon. Altogether 17 interaction parameters were fitted.
The proposed model correlated accurately the experimental data of 8 amino acids/peptides, providing rmsdcorrel = 0.90%. The predictions achieved for the remaining 6 systems gave rise to rmsdpred = 5.65%. These results are very good in comparison with other UNIFAC based models, such as those by Gupta and Heidemann (1990) (rmsdcorrel = 3.74% and rmsdpred = 14.94%) and Pinho et al. (1994) (rmsdcorrel = 0.80% and rmsdpred = 20.17%).
Tabela 1.2: Lista de aminoácidos usados no estudo e algumas propriedades físicas. ... 9
Tabela 3.1: Grupos constituintes da espécie zeuteriónica de aminoácidos e peptídeos. .. 30
Tabela 3.2: Parâmetros para o cálculo de pK1 e pK2, para cada aminoácido ... 38
Tabela 3.3: Valores de pK1 e pK2 para alguns aminoácidos/peptídeos a 25ºC... 38
Tabela 3.4: Dados experimentais de coeficientes de actividade ... 39
Tabela 4.1: Principais parâmetros de interacção entre grupos m e n utilizados... 40
Tabela 4.2: Número de pontos experimentais, parâmetros de interacção usados na correlação e rmsd obtido para cada aminoácido/peptídeo. ... 41
Tabela 4.3: Comparação dos resultados obtidos neste trabalho com outros modelos publicados. ... 44
Tabela A.1: Conversão entre escalas de concentração... 48
Tabela A.2: Conversão entre coeficientes de actividade na convenção não simétrica ... 48
Tabela A.3: Parâmetros Rk e Qk do modelo UNIFAC ... 49
Figura 1.3: Reacção de desidratação de dois aminoácidos formando um dipeptídeo. ... 6
Figura 1.4: Mercado de aminoácidos – relação entre preço e consumo. ... 7
Figura 1.5: Esquema do processo de produção de um aminoácido, por fermentação ... 8
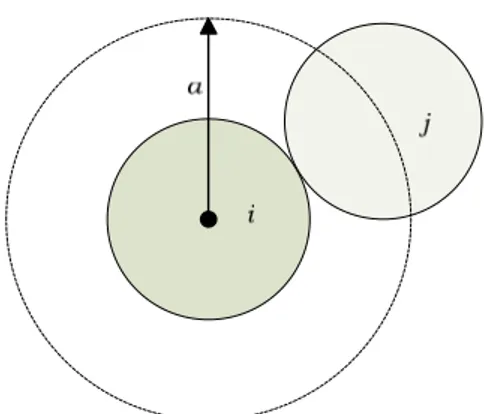
Figura 2.1: Representação esquemática da distância a, entre as espécies i e j... 22
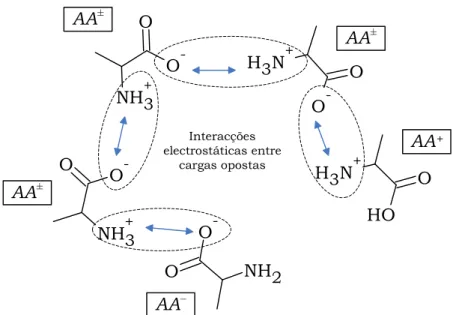
Figura 3.1: Interacções electrostáticas entre as espécies com carga. ... 29
Figura 3.2: Coeficientes de actividade experimentais em função da molalidade para 5 aminoácidos de cadeia lateral alifática. ... 31
Figura 3.3: Representação esquemática da estrutura do programa de cálculo... 33
Figura 3.4: Exemplo de ficheiro de dados da glicina. ... 34
Figura 3.5: Algoritmo de cálculo usado na optimização. ... 35
Figura 4.1: Coeficientes de actividade (convenção não simétrica e escala molal) de aminoácidos em água: dados experimentais e correlação obtida com o modelo deste trabalho. ... 41
Figura 4.2: Coeficientes de actividade (convenção não simétrica e escala molal) de aminoácidos/peptídeos em água: dados experimentais e correlação obtida com o modelo deste trabalho. ... 42
Figura 4.3: Coeficientes de actividade (convenção não simétrica e escala molal) de aminoácidos/peptídeos em água, experimentais e previstos pelo modelo. .... 43
Figura 4.4: Coeficientes de actividade (convenção não simétrica e escala molal) de aminoácidos/peptídeos em água, experimentais e previstos pelo modelo. .... 43
NOMENCLATURA
Símbolosa actividade; distância entre iões amn parâmetro interacção UNIFAC
A parâmetro Debye-Hückel AA aminoácido
B parâmetro Debye-Hückel c molaridade
f fugacidade
G energia livre de Gibbs H constante de Henry
I força iónica i soluto ou espécie j espécie
Ka constante de acidez
K1 constante de equilíbrio químico
K2 constante de equilíbrio químico
Kb constante de basicidade
KD constante de equilíbrio químico
KW produto iónico da água
M massa molar m molalidade
n número de moles Nsolu número de solutos
Nsolv número de solventes
P pressão
Ps pressão de vapor
q parâmetro de superfície de molécula Q parâmetro de superfície de grupo
R constante universal dos gases
r parâmetro de volume de van der Waals de molécula R parâmetro de volume de van der Waals de grupo S solubilidade
s solvente
T temperatura absoluta V volume
x fracção molar
y fracção molar na fase gasosa z carga do ião
Índices 1 AA± 2 AA+ 3 AA− 4 H+ 5 OH− i espécie 0 condição inicial c escala molaridade DATA dados experimentais
DH modelo de Debye-Hückel j reacção; espécie
k número grupos funcionais (UNIFAC) l soluto
m escala molalidade; grupo funcional (UNIFAC) n grupo funcional (UNIFAC)
s solvente
UNIFAC modelo UNIFAC w água
x escala fracção molar Expoentes
o estado padrão ‘ livre de soluto
• estado padrão para o solvente * convenção não simétrica id ideal
∇ convenção não simétrica para fugacidade
ξ
φ avanço de reacção intensivo
Φ fracção de volume de molécula
Θ fracção de área de molécula
θ fracção de área de grupo
Γ coeficiente de actividade de grupo
τ parâmetro UNIFAC
δ critério de paragem da função objectivo
ρ massa volúmica da solução
ρo massa volúmica do solvente
◊ convenção simétrica para fugacidade ∞ diluição infinita + catião - anião ± zeuterião E propriedade em excesso DH modelo de Debye-Hückel UNIFAC modelo UNIFAC
C termo combinatorial (modelo UNIFAC) R termo residual (modelo UNIFAC) calc valor calculado
exp valor experimental k iteração
1.1. Caracterização e classificação de aminoácidos... 2
1.2. Propriedades ácido-base dos aminoácidos ... 4
1.3. Reacções dos aminoácidos ... 5
1.4. Produção e mercado de aminoácidos ... 6
1.5. Estrutura dos aminoácidos/peptídeos estudados ... 9
2. FUNDAMENTOS TEÓRICOS ... 11
2.1. Termodinâmica de soluções de electrólitos ... 11
2.1.1. Escalas de concentração ... 11
2.1.2. Soluções de electrólitos... 13
2.2. Equilíbrio químico de aminoácidos em solução aquosa... 16
2.3. Modelos de coeficientes de actividade ... 18
2.3.1. Modelo UNIFAC ... 18
2.3.2. Modelo de Debye-Hückel ... 21
2.3.3. Revisão de modelos de coeficientes de actividade para aminoácidos ... 23
3. MODELO PROPOSTO PARA COEFICIENTES DE ACTIVIDADE DE AMINOÁCIDOS ... 25
3.1. Cálculo do equilíbrio químico ... 25
3.2. Descrição do modelo ... 28
3.3. Programa de cálculo... 32
3.4. Estratégia de cálculo... 36
3.5. Base de dados utilizada neste trabalho ... 36
4. RESULTADOS E DISCUSSÃO... 40
5. CONCLUSÕES ... 45
6. REFERÊNCIAS BIBLIOGRÁFICAS ... 46
– 1 –
1.
QUÍMICA DOS AMINOÁCIDOS
As proteínas são as macromoléculas mais abundantes nas células, constituindo mais de 50% do seu peso seco [1]. Já presentes nas mais antigas formas de vida na Terra, as proteínas são constituídas a partir do mesmo conjunto básico de blocos, os aminoácidos, unidos entre si por ligações covalentes. Entre as diversas funções desempenhadas pelas proteínas, há a destacar [2]:
– Funções estruturais em tecidos vivos, como por exemplo o colagénio; – Funções específicas sobre órgãos ou determinadas estruturas de um
organismo (como é o caso da insulina);
– Funções de defesa no que diz respeito ao reconhecimento e neutralização
de vírus, bactérias e outras substâncias estranhas, como é o caso dos anticorpos;
– Funções energéticas, a partir dos aminoácidos que as compõem; – Funções enzimáticas, com por exemplo as lipases;
– Funções de transporte de gases, como por exemplo a hemoglobina.
Dada a importância destas macromoléculas nos sistemas biológicos, torna-se fundamental aprofundar o conhecimento das propriedades dos seus blocos básicos. Para além de formarem as proteínas, os aminoácidos na sua forma livre estão presentes em alimentos e bebidas, em medicamentos e outros produtos de elevada importância económica. Quanto maior o conhecimento das propriedades destas pequenas moléculas, melhor será possível optimizar os seus processos de produção, em busca de produtos de melhor qualidade e, consequentemente, de maior valor comercial. Os aminoácidos são geralmente obtidos por síntese ou fermentação, processos estes que originam muitas vezes misturas complexas de biomoléculas e que requerem o recurso a processos de separação, de modo a extrair o produto desejado de subprodutos, excesso de reagentes ou impurezas, geralmente por processos de cristalização ou precipitação. Uma vez que os processos de separação podem representar cerca de 50% [2] do custo total de produção, é essencial conhecer o comportamento dos aminoácidos em solução, nomeadamente os seus coeficientes de actividade e a sua solubilidade.
– 2 –
Esta dissertação surge também da necessidade de encontrar um modelo preditivo que descreva o equilíbrio químico e físico de uma gama de aminoácidos/peptídeos, uma vez que apesar de já existirem alguns modelos que efectuam essa descrição, se continuar a verificar uma fraca capacidade preditiva.
1.1.
Caracterização e classificação de aminoácidos
Como já foi referido, os aminoácidos são uma classe de compostos orgânicos que podem ser encontrados em todos os organismos vivos, sendo os elementos básicos na formação das proteínas.
Desde o seu isolamento, efectuado pela primeira vez na segunda metade do século XIX, as suas propriedades físicas e químicas têm vindo a ser estudadas, não só pela sua importância em inúmeros processos fisiológicos e seu valor como elementos base de todas as formas de vida, mas também pela sua importância na indústria alimentar e farmacêutica, onde se pretende obter um produto com o máximo grau de pureza e com o mínimo custo possível.
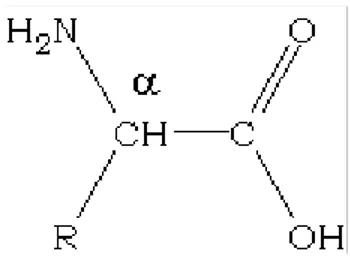
Quimicamente têm em comum a presença de um grupo carboxilo e um grupo amina ligados a um átomo de carbono central, denominado carbono-α, e diferem entre si pela cadeia lateral representada por R na Figura 1.1, variável em estrutura, tamanho, carga eléctrica e solubilidade em água [3].
Dos cerca de setecentos aminoácidos descobertos nos sistemas biológicos, vinte fazem parte de um grupo especial por serem utilizados pela natureza como blocos básicos na síntese de peptídeos e proteínas, sendo conhecidos como aminoácidos naturais [4].
Os aminoácidos mais comuns são os α−aminoácidos, onde o grupo amina está ligado ao carbono−α (ver Figura 1.1) [5].
– 3 – O mais simples destes é a glicina, de fórmula molecular C2H5NO2, e é o único
aminoácido que não é opticamente activo (não apresenta estereoisómeros), uma vez que não contém nenhum centro quiral. À excepção da glicina, todos os outros aminoácidos possuem um átomo de carbono assimétrico, o carbono−α, ao qual se encontram ligados quatro grupos constituintes diferentes como representado na Figura 1.1 [1]. Apresentam portanto actividade óptica, devido ao centro quiral no carbono−α, existindo os estereoisómeros D e L, predominando no entanto o isómero L [6].
Os aminoácidos podem classificar-se segundo o seu grupo R, distinguindo-se quatro classes [4]:
1. Grupo R não polar (hidrofóbico).
Os aminoácidos desta classe possuem grupos hidrocarbonados, apresentando menor solubilidade em água que os aminoácidos com grupo R polar. A esta classe pertencem, por exemplo, a leucina, a valina e a alanina, este último, o menos hidrofóbico desta classe [4].
2. Grupo R polar sem carga (hidrofílico).
Estes aminoácidos são relativamente mais solúveis em água do que os anteriores. Os seus grupos R são constituídos por grupos funcionais neutros polares, que podem formar pontes de hidrogénio com as moléculas de água. Como exemplo desta classe tem-se a serina, a treonina e a glicina [4].
3. Grupo R carregado negativamente (acídico).
Os dois aminoácidos cujos grupos R possuem uma carga negativa a pH=7,0 são o ácido aspártico e o ácido glutâmico, cada um deles com um segundo grupo carboxilo [1].
4. Grupo R carregado positivamente (básico).
São aminoácidos básicos em que os grupos R apresentam uma carga positiva em pH=7,0. Como exemplo tem-se a lisina e a arginina [1].
– 4 –
1.2.
Propriedades ácido-base dos aminoácidos
O conhecimento do comportamento ácido-base dos aminoácidos é fundamental para se entender algumas das suas propriedades físico-químicas. Embora sejam representados como contendo um grupo amina e um grupo ácido carboxílico, os aminoácidos apresentam algumas propriedades que não condizem com esta estrutura [7].
– Ao contrário das aminas e dos ácidos carboxílicos, os aminoácidos são sólidos cristalinos, não voláteis, que fundem, com decomposição, a temperaturas bastante elevadas (tipicamente superiores a 200ºC).
– São solúveis em solventes apolares e são apreciavelmente solúveis em água.
– As respectivas soluções aquosas comportam-se como soluções de
substâncias de elevado momento dipolar.
– As constantes de acidez e basicidade são anormalmente pequenas, para grupos como COOH e NH2.
Estas características são as esperadas para sais onde a rede cristalina é estabilizada por forças electrostáticas de atracção entre grupos com cargas opostas, como o cloreto de sódio. Se os aminoácidos cristalizassem numa forma não iónica seriam estabilizados por forças de van der Waals, bastante mais fracas que as forças electrostáticas, e apresentariam pontos de fusão abaixo do que apresentam [4].
Todas as propriedades acima referidas estão perfeitamente concordantes com uma estrutura iónica dipolar dos aminoácidos, uma estrutura do tipo:
N H3 +
— CHR — COO−
Propriedades físicas como a solubilidade, ponto de fusão e momento dipolar elevado correspondem ao que é expectável de um sal com estrutura semelhante à acima representada [7].
– 5 – Ponto isoeléctrico
Quando um aminoácido é dissolvido em água, forma-se um ião dipolar, o zeuterião, e este pode agir como dador e receptor de protões. É portanto uma substância anfotérica, uma vez que pode participar em reacções quer como ácido quer como base, dependendo da espécie com a qual reage.
A Figura 1.2 mostra a curva de titulação da alanina, onde se observam duas fases distintas na zona dos pontos a e b. Nesta zona, apesar de incrementos progressivos de OH-, há uma menor alteração do pH. No ponto a, a concentração da espécie catiónica, dadora de protões e representada por 1, é igual à concentração da espécie zeuteriónica, receptora de protões e representada por 2. No ponto b, a espécie 2 é agora dadora de protões e de concentração igual a 3, receptora de protões. Em pH=6,01 existe um ponto de inflexão, chamado ponto isoeléctrico, cujo valor é calculado pela média aritmética de pKa e pKb, isto é,
(
pKa pKb)
pI = 12 + [4].
Na secção 3.3.1 apresentam-se todos os parâmetros necessários para calcular os valores de pKa e pKb dos aminoácidos em estudo.
Figura 1.2: Curva de titulação da alanina por adição de uma base. Adaptado de [8].
1.3.
Reacções dos aminoácidos
As reacções dos aminoácidos, como em todos os compostos orgânicos, são as reacções características dos seus grupos funcionais, os grupos amina e ácido
– 6 –
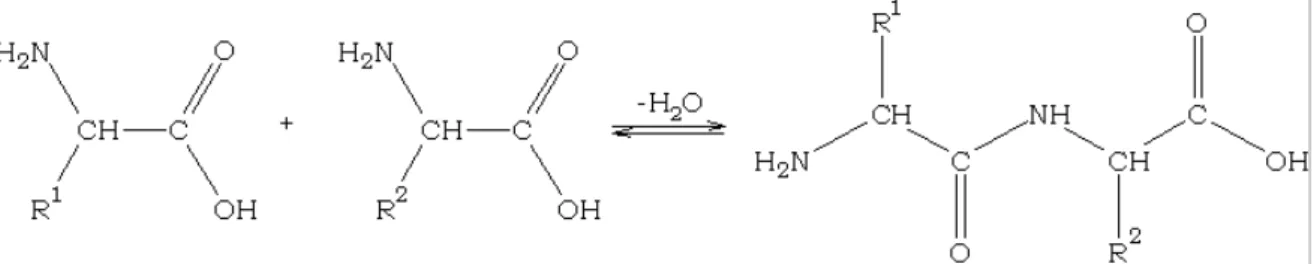
carboxílico. Não sendo objectivo deste trabalho analisar as reacções dos aminoácidos, importa referir a formação dos peptídeos. Através de uma reacção de desidratação, dois aminoácidos podem ligar-se covalentemente por uma ligação peptídica formando um dipeptídeo – Figura 1.3. Uma sucessão destas reacções pode levar à formação de uma proteína, sendo os grupos R responsáveis pela grande variedade de proteínas que podem ser formadas.
Dos 20 aminoácidos naturais, o organismo humano apenas tem capacidade de produzir 11 destes, pelo que os restantes 9 devem fazer parte da alimentação, sendo conhecidos como aminoácidos essenciais. Os 20 aminoácidos naturais são: valina, leucina, isoleucina, lisina, treonina, metionina, histidina, fenilalanina, triptofano, alanina, arginina, glutamina, ácido aspártico, ácido glutâmico, prolina, cisteína, tirosina, asparagina, glicina e serina, sendo que os primeiros 9 aqui enunciados são os aminoácidos essenciais.
Figura 1.3: Reacção de desidratação de dois aminoácidos formando um dipeptídeo.
1.4.
Produção e mercado de aminoácidos
A produção de aminoácidos teve a sua origem no Japão, após trabalhos de investigação onde se descobriu que o glutamato monossódico, obtido a partir do ácido glutâmico, que acentuava e melhorava o sabor de alguns alimentos [9]. Iniciou-se então o utilização do glutamato monossódico na alimentação, sendo ainda hoje adicionado a uma grande variedade de alimentos.
Devido à sua vasta gama de aplicações, como a alimentação, a cosmética, a indústria farmacêutica, entre outras, a procura de aminoácidos cresceu rapidamente e foi acompanhada pelo desenvolvimento de tecnologias de produção. Nas últimas três décadas, o aumento da procura originou um crescimento de mercado de cerca de 5 a 10% ao ano [10]. Na Tabela 1.1 e Figura 1.4 podem ver-se, respectivamente, a quantidade de alguns aminoácidos produzidos, em toneladas por ano, e a relação entre o preço e o consumo:
– 7 – Tabela 1.1: Produção de alguns aminoácidos, em toneladas por ano. Adaptado de [10].
Aminoácido Produção
(ton/ano)
Método de
produção Aplicação
Ácido Glutâmico 1200000 Fermentação Aditivo alimentar Lisina 600000 Fermentação Aditivo alimentar
Metionina 550000 Síntese Aditivo alimentar
Treonina 40000 Fermentação Aditivo alimentar Glicina 16000 Síntese Aditivo alimentar, edulcorante Aspartato 14000 Catálise enzimática Aspartamo, polímeros
Fenilalanina 13000 Fermentação Aspartamo
Císteina 4500 Fermentação Aditivo alimentar, indústria farmacêutica
Cistina 3500 Extracção,
fermentação Indústria farmacêutica
Alanina 1500 Extracção,
fermentação Edulcorante
Leucina 1200 Extracção,
fermentação Indústria farmacêutica
Valina 1000 Extracção,
Fermentação
Indústria farmacêutica, Pesticidas
Isoleucina 500 Extracção,
fermentação Indústria farmacêutica
Figura 1.4: Mercado de aminoácidos (publicado em 2006) – relação entre preço e
consumo. Adaptado de [10]. P re ç o ( U S $ / k g ) Consumo (ton/ano)
– 8 –
Na Figura 1.4 observa-se que a quantidade produzida é inversamente proporcional ao preço de mercado, sendo o glutamato monossódico o aminoácido mais produzido e o mais barato.
Como se observa na Tabela 1.1, os principais métodos de obtenção de aminoácidos à escala industrial são a fermentação, a extracção após hidrólise de proteínas e síntese química [11].
A fermentação é um método que utiliza microrganismos que, através do seu metabolismo, convertem nutrientes que lhes são fornecidos em produtos, neste caso um ou vários aminoácidos. Na fermentação, açúcares (fonte de carbono) são adicionados ao meio de cultura onde os microrganismos se encontram, e o seu metabolismo, por acção de enzimas, pode produzir vários aminoácidos através de sucessivas reacções. Na Figura 1.5 pode observar-se uma representação esquemática da produção do glutamato monossódico.
Não sendo um objectivo deste trabalho, mais detalhes acerca do processo de fermentação para produção de aminoácidos podem ser encontrados nas referências [9] e [10].
– 9 –
1.5.
Estrutura dos aminoácidos/peptídeos estudados
Na Tabela 1.2 são apresentadas as estruturas de todos os 14 aminoácidos/peptídeos estudados neste trabalho, bem como a sua temperatura de fusão, massa molar e solubilidade em água a 25ºC.
Tabela 1.2: Lista de aminoácidos usados no estudo e algumas propriedades físicas.
Adaptado de [13].
Aminoácido Estrutura Tfusão
(ºC) M (kg/mol) Solubilidade em água a 25ºC (g/kgágua) Glicina 290 75,07 250,9 Alanina 297 89,10 165,0 Ác. Amino Butírico 304 103,12 210,0 Valina 315 117,15 88,5 Ác. Amino Valérico n.d. 117,15 n.d. Hidroxiprolina 274 131,13 361,0 Prolina 221 115,14 1623,0 Serina 228 105,10 421,7
– 10 – Treonina 256 119,12 98,1 Glicilglicina 263 132,12 n.d. Glicilalanina n.d. 146,15 n.d. Alanilglicina n.d. 146,15 n.d. Alanilalanina n.d. 160,17 n.d. Triglicina n.d. 189,17 n.d.
– 11 –
2.
FUNDAMENTOS TEÓRICOS
Apresentam-se neste capítulo os conceitos fundamentais necessários à modelação dos coeficientes de actividade de aminoácidos e peptídeos em solução aquosa de acordo com a abordagem adoptada nesta dissertação. Assim, começa-se pelas ferramentas termodinâmicas utilizadas para descrever soluções de electrólitos e o equilíbrio químico e aminoácidos em solução aquosa, faz-se uma breve apresentação dos modelos de UNIFAC e Debye-Hückel, concluindo-se com uma revisão dos principais modelos existentes na literatura para calcular os coeficientes de actividade destas moléculas.
2.1.
Termodinâmica de soluções de electrólitos
A termodinâmica de soluções de electrólitos difere da termodinâmica de soluções de não electrólitos, devido à presença de cargas eléctricas nas espécies em solução e que dão origem a interacções electrostáticas que não são de todo desprezáveis. O comportamento da fase líquida é mais difícil de descrever, onde as interacções electrostáticas de longo alcance são responsáveis por fortes desvios à idealidade mesmo em soluções diluídas. Também outros aspectos são diferentes e devem ser tidos em conta, como as escalas de concentração usadas e a definição de estados padrão [14].
2.1.1.
Escalas de concentração
As escalas de concentração geralmente usadas para exprimir a composição de uma solução de electrólitos são: molalidade, molaridade e fracção molar.
Escala de molalidade
A molalidade, mi, é definida como o número de moles (ni) de soluto i por kg de
solvente s:
∑
= = solv N s s s i i M n n m 1 (1)– 12 –
em que ns é o número de moles do solvente s, Ms a sua massa molecular em
kg/mol e Nsolv é o número total de solventes em solução.
Escala de molaridade
A molaridade, ci, é definida como o número de moles de soluto i por dm3 de
solução:
V n
c i
i = (2)
em que V é o volume do sistema.
Escala de fracção molar
É a escala mais usada para sistemas de não electrólitos; a fracção molar da espécie i, xi, é definida como:
∑
= = espécies N i i i i n n x 1 (3)em que ni é o número de moles da espécie i e Nespécies é o número de espécies,
solutos e solventes em solução. A relação entre fracção molar e a molalidade é dada por:
∑
∑
= = + = solv solu 1 ' 1 1 N s s s N l l i i M x m m x (4)Considerando o caso particular de um sal, a molalidade e a fracção molar são dadas por, respectivamente:
∑
= s s s sal M n n msal (5)∑
+ = s sal s sal sal n n n x (6)– 13 – em que nsal e ns são o número de moles do sal e do solvente respectivamente e Ms
a massa molar do solvente, em kg/mol. As equações (5) e (6) estão relacionadas por:
∑
−
=
s s sM
x
x
x
m
'
1
1
sal sal sal (7)Na equação (7), x's representa a fracção de solvente em base livre de soluto. Mais conversões entre cada uma das escalas de concentração podem ser obtidas, sendo apresentadas em apêndice na Tabela A.1.
2.1.2.
Soluções de electrólitos
É importante analisar a termodinâmica de soluções contendo um soluto involátil num solvente, antes de introduzir o efeito de ionização dos aminoácidos em solução. Esta discussão torna-se importante na medida em que os aminoácidos se comportam como sólidos não voláteis à temperatura ambiente.
Quando se fala em soluções ideais de não-electrólitos, estas seguem a Lei de Raoult, dada pela equação (8):
σ i i iP x P
y = (8)
em que xi e yi são as fracções molares do componente i nas fases líquida e
gasosa, respectivamente, P é a pressão total e Piσ a pressão de vapor do
componente i. Quando se lida com soluções reais, o comportamento do solvente segue a Lei de Raoult quando a concentração dos solutos tende para zero. Uma solução diluída tem comportamento ideal quando o solvente segue a Lei de Raoult e o soluto segue a Lei de Henry, equação (9) [15]:
i i iP x H
y = (9)
No entanto, em soluções de electrólitos, os iões interagem fortemente entre si e com o solvente, ainda mais no caso de solventes polares como a água, originando fortes desvios à idealidade, mesmo a baixas concentrações.
– 14 –
Soluções reais de electrólitos podem ser descritas termodinamicamente em termos das suas propriedades em excesso, calculadas a partir dos potenciais químicos de cada espécie, µi.
Da termodinâmica de não electrólitos, para um componente i a uma dada temperatura, pressão e composição, o potencial químico µi relaciona-se com a
actividade ai por: i o i i =
µ
+RT lnaµ
(10)onde
µ
io é o potencial químico de i num estado padrão convenientemente definido, que, para misturas de não electrólitos voláteis, corresponde a líquido puro à temperatura e pressão do sistema [16].Actividade para solvente
Para uma mistura contendo um soluto involátil num solvente, aplica-se directamente a equação (10) para o solvente s, ou seja:
s o
s
s =
µ
+RT lnaµ
(11)onde a actividade do solvente, as, se calcula por: s x s
s
x
a
=
γ
, (12)em que
γ
s,x é o coeficiente de actividade para o solvente, escrito na convenção simétrica e em fracção molar. Numa solução real, os coeficientes de actividade são normalizados de acordo com a convenção simétrica em que γs,x → 1 quandoxs → 1.
Actividade para soluto
No entanto, para um soluto involátil não é conveniente escolher para estado padrão o líquido puro à temperatura e pressão do sistema, na medida em que nessas condições o soluto não é líquido [14].
Escrevemos então o potencial químico como:
(
i i)
i
i
µ
RT
γ
ε
µ
=
+
ln
,ε– 15 – em que
µ
i+ é o potencial químico padrão da espécie i (a uma dada composiçãofixa, mas dependente da temperatura, pressão e natureza do soluto e solvente),
ε
ié a escala de concentração adoptada (mi, ci ou xi) e
γ
i,ε é o coeficiente de actividade do soluto nesta escala de concentração e em convenção simétrica. No caso de involáteis selecciona-se comummente a convenção não simétrica, segundo a qual os coeficientes de actividade,γ
i*, são unitários a diluição infinita.Assim, a actividade de um componente i pode calcular-se de várias formas:
i x i x i
x
a
,=
γ
*, (14) i m i m im
a
,=
γ
*, (15) i c i c ic
a
.=
γ
*, (16)Onde, por definição, os coeficientes de actividade assimétricos escritos em fracção molar (
γ
i ,∗x), molalidade (∗ m i ,
γ
) e molaridade (γ
i ,∗c) satisfazem as condições* ,x i
γ
→ 1 ,γ
i*,m → 1 eγ
i*, c → 1, quando∑
= solu 1 N i iε
→ 0.Os valores tabelados de coeficientes de actividade surgem normalmente em convenção não simétrica e escala molal, enquanto os calculados por modelos termodinâmicos surgem maioritariamente em convenção simétrica e escala de fracção molar. Tal como para as escalas de concentração, também é possível relacionar essas duas convenções. Partindo então da definição de coeficiente de actividade e usando fracções molares tem-se que:
◊ ∧ ∧ ◊ ∧ = ≡ i i i id i i x i f x f f f , ,
γ
(17) ∇ ∧ ∧ ∇ ∧ = ≡ i i i id i i x i f x f f f , * ,γ
(18) em que ∧ if é a fugacidade do componente i na solução à temperatura T e pressão
– 16 –
convenções simétrica e não simétrica, respectivamente. Igualando a fugacidade calculada pelas duas equações anteriores, vem:
∇ ◊
=
i i x i i i x ix
f
x
f
* , ,γ
γ
(19)No limite, quando xi → 0 verifica-se que
γ
i*,x→ 1 eγ
i → ∞ iγ
. Logo: ∇ ◊ ∞ = i i i f fγ
(20)em que
γ
i∞é o coeficiente de actividade do componente i a diluição infinita.Combinando as equações (19) e (20), obtém-se a relação que se procurava:
∞ = i x i x i
γ
γ
γ
* , , (21)Podem consultar-se em apêndice (ver Tabela A.2) outras relações entre os coeficientes de actividade de diferentes escalas e convenções.
2.2.
Equilíbrio químico de aminoácidos em solução
aquosa
Como se referiu no Capítulo 1, os aminoácidos e peptídeos apresentam na sua estrutura os grupos carboxilo e amina, grupos estes que em água se ionizam e dão origem a uma espécie dipolar com carga global zero, AA±, chamada de
zeuterião, e a espécies catiónicas e aniónicas, AA– e AA+ respectivamente.
Dependendo do pH a que se encontra a solução, as espécies AA– e AA+ podem
existir em maior ou menor quantidade, sendo que no ponto isoeléctrico predomina a espécie zeuteriónica, AA±. Estas moléculas apresentam um
momento dipolar elevado o que dá origem a interacções importantes quer entre os seus iões quer com solventes polares como a água.
Considerando então a ionização de um aminoácido genérico (AA) em água, podem escrever-se as seguintes equações de equilíbrio:
– 17 – ± ⇔ AA AA D K KD (R0) ± + + ⇔ H +AA AA 1 ξ K1 (R1) − + ± ⇔ H +AA AA 2 ξ K2 (R2) − +
+
⇔
H
OH
O
H
2 3 ξ KW (R3)Na reacção R0 forma-se o zeuterião que, ao aceitar um protão na reacção R1,
forma o catião AA+. O zeuterião pode também ceder um protão, reacção R
2,
dando origem ao anião AA– [17]. A reacção R
3 representa a autoprotólise da água.
As constantes de equilíbrio KD, K1, K2 e KW permitem escrever:
AA AA D a a K = ± (22) + ± + = AA AA H a a a K1 (23) ± − + = AA AA H a a a K2 (24) − + = H OH W a a K (25)
Para aminoácidos alifáticos o valor de KD é da ordem de 105 a 106 [18], o que
significa que quase todo o aminoácido AA presente em solução está na forma de
AA±, AA+ e AA–. Adoptando-se a convenção simétrica e a escala molal, vem que: i
m i
i
m
a
=
γ
*,×
, pelo que as constantes das equações (22)-(25) se podem exprimir como o produto de um termo de coeficientes de actividade por outro de molalidades. Por exemplo, para a equação (23) obtém-se:m AA AA H AA AA H AA AA H K K m m m a a a K * 1, 1, * * 1 = = × ≡ *× + ± + + ± + + ± + γ
γ
γ
γ
(26)– 18 –
2.3.
Modelos de coeficientes de actividade
Apresenta-se nesta secção uma breve descrição dos modelos de coeficientes de actividade de UNIFAC e Debye-Hückel, dado terem sido escolhidos neste trabalho para o estudo de aminoácidos. Termina-se com uma revisão de alguns modelos existentes na literatura para este efeito.
2.3.1.
Modelo UNIFAC
O modelo UNIFAC (UNIversal Functional Activity Coefficient) é um modelo semi-empírico, preditivo, desenvolvido por Fredeslund et al [19], baseado em termodinâmica molecular e bastante usado para descrever o equilíbrio líquido-vapor em sistemas de não-electrólitos. No entanto, várias extensões deste método foram sendo aplicadas em outros tipos de soluções, nomeadamente poliméricas e electrolíticas. O objectivo de métodos preditivos como o UNIFAC é utilizar informação obtida a partir de sistemas conhecidos (i.e., parâmetros) para prever o comportamento de sistemas distintos para os quais não exista informação disponível [20].
A ideia principal dos métodos de contribuição de grupos é que apesar de existirem milhares de compostos químicos com interesse para a indústria, o número de grupos funcionais que os compõem é muito inferior. Utiliza-se então o princípio de que uma propriedade física de um componente é a soma das contribuições de cada um dos grupos funcionais da molécula para essa mesma propriedade. No entanto, qualquer que seja o método de contribuição de grupos, produzirá resultados aproximados, uma vez que a contribuição de um determinado grupo numa molécula não é exactamente a mesma que a contribuição desse mesmo grupo noutra. Outro pressuposto é que a contribuição de um grupo dentro de uma molécula é independente da contribuição de outro grupo na mesma molécula [20]. Quanto menor o número de grupos em que a molécula é dividida, maior a exactidão do método. No limite, pode considerar-se toda a molécula como um grupo, perdendo-se no entanto as vantagens desta abordagem. Para ter utilidade prática, deve ser efectuada uma divisão sensata da molécula: o número de grupos deve permanecer pequeno, mas não tão pequeno
– 19 – que se percam os efeitos da estrutura molecular para as propriedades da mistura.
Usando as interacções entre cada um dos grupos funcionais presentes nas moléculas da mistura é então possível calcular o coeficiente de actividade de cada um dos componentes.
O método original de UNIFAC, proposto por Fredeslund et al. [19], propõe o cálculo do coeficiente de actividade da espécie i na convenção simétrica e escala de fracção molar, como sendo a combinação de dois termos, um termo combinatorial e um termo residual:
R C UNIFAC ln ln ln
γ
i =γ
i +γ
i (27)O primeiro é igual ao termo combinatorial do método UNIQUAC e representa o desvio à idealidade devido às diferenças de tamanho e forma das moléculas em solução. Neste trabalho é usada a correcção de Kikic et al. [21], introduzida para soluções diluídas:
Θ
Φ
−
+
Θ
Φ
−
Φ
−
+
Φ
=
i i i i i i c i i c i C iq
x
x
1
5
ln
1
ln
ln
γ
(28) em que,∑
= Φ j j j i i c i x r x r 3 2 3 2 (29)∑
= Φ j j j i i i x r x r (30)∑
= Θ j j j i i i x q x q (31)Nas equações anteriores o índice j representa o número total de moléculas presentes na solução. Os parâmetros ri e qi são, respectivamente, volume de van
der Waals e área superficial das moléculas e são calculados pelos parâmetros de área e volume de cada um dos grupos k que constituem a molécula, Rk e Qk:
– 20 –
∑
= k k i k i R rν
(32)∑
= k k i k i Q qν
(33)onde o índice k representa o número de diferentes grupos funcionais na molécula
i e
ν
ki é o número de grupos k presentes na molécula i. Na Tabela A.3 emapêndice são apresentados todos os parâmetros Rk e Qk utilizados nas equações
(32) e (33).
O termo residual,
γ
iR, é conhecido como o termo energético e tem em conta as interacções energéticas de curto alcance entre grupos. É calculado por:(
)
∑
Γ − Γ = k i k k i k i ln ln lnγ
Rν
(34)em que Γké o coeficiente de actividade do grupo k na solução e Γkié o coeficiente de actividade do grupo k numa solução contendo apenas moléculas do tipo i.
O coeficiente de actividade Γk é calculado por:
Θ Θ − Θ − = Γ
∑
∑
∑
m n nm n km m m mk m k k Qτ
τ
τ
ln 1 ln (35)onde os índices m e n correspondem a todos os grupos presentes em solução. A partir da equação (36) obtém-se a fracção de área do grupo m, Θm:
∑
= Θ n n n m m m X Q X Q (36)∑ ∑
∑
=
j n j i n j j i m mx
x
X
ν
ν
(37)– 21 – − = T amn nm exp
τ
(38)onde T representa a temperatura absoluta da mistura e amné o parâmetro de interacção entre os grupos m e n, sendo uma constante ajustável neste trabalho. Importa ainda referir que as equações (35)-(38) são também usadas no cálculo de
i k
Γ
ln .
2.3.2.
Modelo de Debye-Hückel
Os aminoácidos são electrólitos fracos em solução aquosa. De modo a incluir as interacções electrostáticas (de longo alcance) que se estabelecem entre as espécies carregadas em solução (AA±, AA+, AA–, H+ e OH–) no desvio à idealidade,
vai recorrer-se ao modelo de Debye-Hückel apresentado a seguir.
Lei limite de Debye-Hückel
É sabido que iões com cargas opostas se atraem mutuamente. Como resultado, é mais provável encontrar aniões perto de catiões e vice-versa. A energia eléctrica do ião central e, consequentemente, o seu potencial químico é reduzido em resultado das interacções com a sua atmosfera iónica de carga oposta [22]. Esta diminuição de energia corresponde à diferença entre a energia livre de Gibbs e o seu valor em solução ideal, podendo representar-se por
±
γ
ln
RT . Mesmo em soluções muito diluídas, as forças atractivas e repulsivas são significativas, pelo que a sua contribuição para os desvios à idealidade deve ser contabilizada.
Peter Debye e Erich Hückel desenvolveram, em 1923, um modelo simples para soluções de electrólitos e obtiveram expressões para os coeficientes de actividade de espécies iónicas, representados por
γ
+eγ
−. Neste modelo os iões são tratados simplesmente como esferas rígidas carregadas e com diâmetro fixo.O modelo inicialmente proposto por eles permite calcular coeficientes de actividade de iões em soluções muito diluídas, tomando a designação de lei limite de Debye-Hückel [22]:
2 1
– 22 –
em que z+ e z− são as cargas eléctricas do catião e do anião, respectivamente, a constante A vale 0,509 para soluções aquosas a 25ºC e I representa a força iónica da solução dada por:
∑
= 2 2 1 i iz m I (40)Neste caso, a força iónica tende para zero a diluição infinita. A equação de Debye-Hückel mostrou ser aplicável para forças iónicas muito baixas, inferiores a 1 mmol/kg.
Extensão da lei de Debye-Hückel
Para soluções onde a força iónica é mais elevada, o coeficiente de actividade da espécie i pode ser estimado por uma extensão da lei de Debye-Hückel (usada neste trabalho): I Ba I Azi i + − = ∗ 1 log 2 DH ,
γ
(41)em que zi é a carga da espécie i e a é a distância de maior aproximação entre as
espécies i e j, representada na Figura 2.3.
– 23 –
2.3.3.
Revisão de modelos de coeficientes de actividade
para aminoácidos
Há várias décadas que são conhecidos estudos experimentais de medição de coeficientes de actividade de aminoácidos [23-27], dados que foram utilizados neste trabalho (ver secção 3.5). No entanto, quanto a modelos termodinâmicos capazes de prever o seu comportamento em solução, só nos últimos vinte anos foram surgindo e sendo publicados alguns resultados. Segundo Macedo [28], os modelos publicados podem distinguir-se em duas grandes categorias:
1. Modelos que apenas consideram as interacções de curto alcance entre as
partículas, considerando o aminoácido em água apenas na sua forma zeuteriónica, como os modelos publicados por Gupta e Heidemman [17], Kuramochi et al. [29] e Xu et al. [30].
No trabalho publicado por Gupta e Heidemann [17] é usado o método UNIFAC para alguns aminoácidos e antibióticos. Neste modelo é considerado o equilíbrio químico e usadas as respectivas constantes de ionização. Foram ainda introduzidos dois novos grupos, correspondentes às moléculas da glicina e prolina.
No trabalho de Kuramochi et al. [29] foi usado o modelo UNIFAC e foram introduzidos novos grupos. O zeuterião é considerado uma molécula neutra e é proposta uma alteração ao método UNIFAC de Larsen et al [31] de modo a ser estendido a soluções de biomoléculas. Para que o método representasse correctamente os coeficientes de actividade foram definidos novos grupos. Nesse trabalho, os aminoácidos foram divididos em quatro grupos: α-CH, NH2, COOH e
sc-CH2, e ainda foi introduzido o novo grupo CONH para os peptídeos.
No trabalho publicado por Xu et al. [30] foi usado o modelo de Wilson para soluções aquosas de polímeros para calcular os coeficientes de actividade e as solubilidades de aminoácidos e peptídeos em água, usando dois parâmetros ajustáveis por aminoácido. Nos parâmetros de energia foi introduzida a influência da temperatura, podendo obter-se, segundo os autores, uma melhor representação da solubilidade a temperaturas elevadas [30].
– 24 –
2. Modelos que, para além das interacções de curto alcance, têm em conta as interacções de longo alcance devidas às forças electrostáticas entre iões. Para isso usam a equação de Debye-Hückel. Nesta abordagem inserem-se os trabalhos publicados por Peres e Macedo [32], Pinho et al. [33] e o modelo desenvolvida no âmbito desta dissertação.
No artigo de Pinho et al. [33] foi combinado o modelo UNIFAC modificado por Kikic et al. [21] com a equação de Debye-Hückel. Novos grupos iónicos foram definidos, tendo em conta as cargas das espécies em solução, e os dados experimentais da literatura foram usados para ajustar os parâmetros de interacção entre esses novos grupos e os já existentes, obtendo-se resultados satisfatórios como se verá no Capítulo 4.
O modelo de Peres e Macedo (1994) [32] utiliza os modelos de Debye-Hückel e de UNIQUAC para descrever o equilíbrio sólido-líquido de aminoácidos/peptídeos em soluções aquosas. Tal como nos modelos anteriormente indicados, os dados experimentais de coeficientes de actividade foram usados para optimizar novos parâmetros, obtendo-se também resultados satisfatórios.
– 25 –
3.
MODELO PROPOSTO PARA COEFICIENTES DE
ACTIVIDADE DE AMINOÁCIDOS
Neste capítulo apresenta-se o desenvolvimento do modelo proposto neste trabalho para calcular coeficientes de actividade de aminoácidos e peptídeos em solução aquosa. É um modelo que integra interacções de curto e de longo alcance, traduzidas pelas equações de UNIFAC e Debye-Hückel, respectivamente. Faz-se também uma descrição do programa e do algoritmo utilizados na optimização dos parâmetros do modelo. Apresenta-se ainda a base de dados compilada.
3.1.
Cálculo do equilíbrio químico
O equilíbrio químico de aminoácidos em solução aquosa encontra-se descrito na secção 2.2. O cálculo deste equilíbrio é imprescindível para se conhecer a concentração de cada espécie em solução, pelo que será abordado em primeiro lugar.
De modo a simplificar as expressões a desenvolver, adopta-se a notação seguinte para as espécies envolvidas: AA± ≡ 1, AA+ ≡ 2, AA– ≡ 3, H+ ≡ 4 e OH– ≡ 5.
O número de moles de cada espécie pode calcular-se pelas relações estequiométricas correspondentes aos equilíbrios traduzidos pelas equações químicas R1 a R3 (ver secção 2.2):
∑
= + = ≡ ± 3 1 , 1 0 , 1 1 j j j AA n n nυ
ξ
(42)∑
= + = ≡ + 3 1 , 2 0 , 2 2 j j j AA n n nυ
ξ
(43)∑
= + = ≡ − 3 1 , 3 0 , 3 3 j j j AA n n nυ
ξ
(44)∑
= + = ≡ + 3 1 , 4 0 , 4 4 j j j H n n nυ
ξ
(45)– 26 –
∑
= + = ≡ − 3 1 , 5 0 , 5 5 j j j OH n n nυ
ξ
(46)onde
υ
i,j representa o coeficiente estequiométrico da espécie i na reacção j,ξ
j é oavanço da reacção j e ni,0 o número inicial de moles do componente i. Substituindo os coeficientes estequiométricos obtém-se:
2 1 0 , 1 1 =n +
ξ
−ξ
n (47) 1 0 , 2 2 =n −ξ
n (48) 2 0 , 3 3 =n +ξ
n (49) 3 2 1 0 , 4 4 =n +ξ
+ξ
+ξ
n (50) 3 0 , 5 5 =n +ξ
n (51)No início, apenas temos as espécies AA±, H+ e OH–, pelo que o número total de
moles, n0, é dado por:
∑
= + + = = NC i i n n n n n 1 0 , 5 0 , 4 0 , 1 0 , 0 (52)Dividindo as equações (47) a (51) pela massa de solvente, estas aparecem escritas em termos das molalidades de cada componente
(
mi =ni nsMs)
e doavanço intensivo de cada reacção j
(
φ
j =ξ
j nsMs)
:2 1 0 , 1 1=m +
φ
−φ
m (53) 1 2 = −φ
m (54) 2 3 =φ
m (55) 3 2 1 0 , 4 4 =m +φ
+φ
+φ
m (56) 3 0 , 5 5=
m
+
φ
m
(57)– 27 – Como as reacções em que o zeuterião intervém para originar o AA+ e o AA- são
pouco extensas, pode introduzir-se a aproximação:
0 , 1 2 1 0 , 1 1 m m m = +
φ
−φ
≅ (58)Substituindo as relações (54)-(58) nos equilíbrios (23)-(25) obtém-se:
(
)
1 3 2 1 0 , 1 , 1 1 , 1φ
φ
φ
φ
γ − + + ⋅ = ≡ m K K K m (59)(
)
0 , 1 3 2 1 2 , 2 2 , 2 m K K K m φ φ φ φ γ + + ⋅ = ≡ (60)(
1 2 3)
3 , ,φ
φ
φ
φ
γ⋅
+
+
=
≡
W W m WK
K
K
(61)onde se atendeu à notação definida na equação (26). A solução do sistema de equações não linear obtém-se facilmente por manipulação algébrica:
m m m m W
K
m
K
K
m
K
m
, 1 0 , 1 2 , 1 , 2 3 0 , 1 , 2 0 , 1 2 1+
⋅
⋅
+
⋅
=
φ
(62) m mK
K
m
, 1 1 , 2 2 0 , 1 2⋅
⋅
−
=
φ
φ
(63) m m W K K m , 1 1 , 0 , 1 3 ⋅ ⋅ − =φ
φ
(64)Assim, conhecendo-se a molalidade inicial do aminoácido, m1,0, as constantes de equilíbrio termodinâmicas
(
K1,K2,KW)
e as constantes expressas em termos decoeficientes de actividade
(
K1,γ,K2,γ,KW,γ)
calculam-seφ
1,φ
2 eφ
3 pelas equações (62)-(64), que substituídos nas equações (54)-(58) permitem determinar a concentração do sistema. Este cálculo é iterativo: inicia-se com coeficientes de actividade unitários e obtém-se uma primeira estimativa para mi; com estesvalores calculam-se os
γ
i’s que serão utilizados na obtenção de novas molalidades; o procedimento repete-se até se atingir convergência.– 28 –
3.2.
Descrição do modelo
O modelo proposto nesta dissertação compreende as interacções de curto e de longo alcance, traduzidas pelas equações de UNIFAC e Debye-Hückel, respectivamente. Desta forma, a função de Gibbs em excesso do sistema, na convenção assimétrica pode escrever-se como:
E E E G G G*, = UNIFAC*, + DH*, (65)
Em todas as publicações existentes na literatura, a contribuição das cargas do zeuterião, AA±, não foi tida em conta por ser uma molécula globalmente
neutra. No entanto, neste trabalho, o efeito das cargas dos grupos NH3+ e COO–
do AA± é considerado muito importante para os desvios à idealidade, pois grande
parte do aminoácido existe na forma AA±. Este facto levou-nosa introduzir esta
contribuição. Como se observa na Figura 3.1, os grupos iónicos interagem electrostaticamente com os restantes grupos em solução, quer sejam espécies positivas, negativas ou zeuteriónicas, afectando dessa forma o coeficiente de actividade do AA±.
Partindo da equação (65) deriva-se o coeficiente de actividade não simétrico, obtendo-se: DH , , UNIFAC , , ,
ln
ln
ln
γ
i∗m=
γ
i∗m+
γ
i∗m (66)O termo de Debye-Hückel para o caso particular do zeuterião, ln
γ
AA∗,DH±,m, seráentão calculado por:
DH , , DH , , DH , , ln ln 3 ln
γ
AA∗ ±m =γ
COO∗ −m +γ
NH∗ +m (67)Grupos UNIFAC usados neste trabalho. Novo grupo mCH3.
Na Tabela 3.1 encontram-se listados os grupos que compõem as espécies zeuteriónicas de aminoácidos e peptídeos. Como se pode observar na Tabela 3.1 foi introduzido um novo grupo neste trabalho, chamado mCH3 (modified CH3),
– 29 – O NH3+ O -O NH3+ O -O N H3 + O -O N H3 + O H O NH2 O -AA± AA± AA+ AA– AA± Interacções electrostáticas entre cargas opostas
Figura 3.1: Interacções electrostáticas entre as espécies com carga.
Na Figura 3.2 encontram-se representados coeficientes de actividade experimentais em função da molalidade para 5 aminoácidos que diferem entre si pela presença de um ou mais grupos CH3 na cadeia lateral alifática (ver Tabela
1.2). A alanina, com o grupo R =CH3 (ver Figura 1.1), apresenta um coeficiente de actividade superior à glicina (R =H) e inferior ao ácido aminobutírico (R =CH2CH3). Prosseguindo para o ácido aminovalérico (R=CH2CH2CH3), o
seu coeficiente de actividade é superior ao do ácido aminobutírico e inferior ao da valina (R=CHCH3CH3). Como a única diferença entre este pequeno grupo de aminoácidos reside na estrutura da sua cadeia lateral, o comportamento distinto dos seus coeficientes de actividade pode ser atribuído ao grupo CH3 e aos
subgrupos CH2 e CH.
Por outro lado, os parâmetros originais UNIFAC para a interacção energética (amn) entre o grupo CH3 e os demais da mistura já publicados não são capazes de
descrever correctamente o andamento das curvas apresentadas na Figura 3.2. Pelo contrário, a adição de grupos CH3 faz diminuir o valor do coeficiente de
actividade, contrariando os dados experimentais. Por este motivo, define-se neste trabalho o grupo mCH3 (modified CH3), específico para este tipo de sistemas.
– 30 –
Parâmetros de interacção energética a optimizar
Os parâmetros de interacção energética entre os grupos que aparecem na Tabela 3.1 dividem-se em dois tipos: os que já estão publicados e os que foram definidos/redefinidos neste trabalho. Os valores dos primeiros foram tirados de tabelas existentes na literatura (ver Tabela A.4 no Apêndice); os restantes foram optimizados (ou fixados ao longo do trabalho) utilizando a base de dados da secção 3.3. Os pares ajustados são: H2O/CNH3+, CNH3+/H2O, COO–/H2O, COO–
/CNH3+, CNH3+/mCH3, mCH3/COO–, mCH3/H2O, H2O/mCH3, COO–/OH,
OH/CNH3+, OH/COO–, NH3+/OH, OH/CNH2+, H2O/CNH2+, CNH2+/H2O,
CONHCH2/COO–, CONHCH2/CNH3+, CONHCH2/mCH3 e mCH3/CONHCH2.
Tabela 3.1: Grupos constituintes da espécie zeuteriónica de aminoácidos e peptídeos.
Aminoácido/peptídeo Grupos constituintes
glicina CH2NH3+; COO– alanina mCH3; CHNH3+; COO–
ác. aminobutírico mCH3; mCH2; CHNH3+; COO- valina 2 × mCH3; mCH; CHNH3+; COO- ác. aminovalerico mCH3; 2 × mCH2;CHNH3+; COO-
hidroxiprolina CH2; 2 × CH; OH; CH2NH2+; COO- prolina 2 × CH2; CH; CH2NH2+; COO-
serina mCH2; OH; CHNH3+; COO- treonina mCH3; mCH; OH; CHNH3+; COO- alanilalanina 2 × mCH3; CONHCH; CHNH3+; COO-
alanilglicina mCH3; CHNH3+;CONHCH2; COO- glicilalanina mCH3; CONHCH; CH2NH3+; COO-
glicilglicina CH2NH3+; CONHCH2; COO- triglicina 2 × CONHCH2; CH2NH3+; COO-
– 31 – 0 0.5 1 1.5 2 2.5 3 3.5 0.7 0.8 0.9 1 1.1 1.2 molalidade C o e f. d e A c ti v id a d e , γ * glicina alanina ác.aminobutírico ác.aminovalérico valina
Figura 3.2: Coeficientes de actividade experimentais em função da molalidade para 5
aminoácidos de cadeia lateral alifática.
Coeficientes de actividade real e aparente
O coeficiente de actividade real de um aminoácido decorre do desvio à idealidade das diversas espécies que aparecem com a sua ionização. No entanto, os dados experimentais publicados são apresentados para o aminoácido não dissociado AA. Este valor passa a ser designado por aparente
( )
γ
m*,ap , em contraposição ao coeficiente real( )
γ
m* .Torna-se necessário relacionar os dois coeficientes. Para isso parte-se da igualdade das fugacidades para a solução real e aparente:
◊
◊
=
×
×
×
×
xf
x
x,f
px
γ
apγ
ap a ( 68)Atendendo a que, quando x→1, xap →1 e
γ
x=
γ
x,ap→
1
, conclui-se que◊ ◊
=
f
ap– 32 –
de actividade a diluição infinita, aparecem os coeficientes em convenção assimétrica: * ap * ap , x x
x
x
γ
γ
=
(69)O quociente entre a fracção molar real e a aparente é dado por:
(
)
AA AA AA AA AA AA AA AA AA AA AA AA m m n n n n n n n n n n n n x x ± ± − + ± ± − + ± ± = = + + = + + = total total ap real (70)Combinando as duas equações anteriores, obtém-se:
* * ap , x AA AA x m m
γ
γ
= ± ⋅ (71)e como nestes sistemas o valor de
γ
x* coincide com * mγ
(ver Tabela A.2), a relação final pretendida é: * * ap , m AA AA m m mγ
γ
= ± ⋅ (72)3.3.
Programa de cálculo
No desenvolvimento desta dissertação foram usados o software de cálculo
MATLAB e a aplicação de cálculo e base de dados termodinâmicos THERMOLIB
[34]. A estrutura do programa feito em MATLAB pode observar-se na Figura 3.3. A base do programa assenta no gestor de optimização representado pelo bloco 1. Neste gestor, são definidos os aminoácidos que irão ser ajustados e as estimativas iniciais dos parâmetros a optimizar, amn_iniciais. No bloco 1 é usada a
função fminsearch, uma função do MATLAB que usa o algoritmo de Nelder-Mead para procurar mínimos relativos; a função objectivo é dada por:
![Figura 1.2: Curva de titulação da alanina por adição de uma base. Adaptado de [8].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15929709.1094869/17.892.131.754.656.909/figura-curva-titulação-alanina-por-adição-base-adaptado.webp)

![Figura 1.5: Esquema do processo de produção de um aminoácido, por fermentação [12].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15929709.1094869/20.892.160.760.656.1030/figura-esquema-do-processo-produção-aminoácido-por-fermentação.webp)