Electron transfer and docking between cytochrome
cd
1
nitrite reductase
and different redox partners
—
A comparative study
Humberto A. Pedroso
a,1, Célia M. Silveira
a,b,1, Rui M. Almeida
a, Ana Almeida
a, Stéphane Besson
a,c,
Isabel Moura
a, José J.G. Moura
a, M. Gabriela Almeida
a,d,⁎
aUCIBIO, REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal bInstituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Av. da República, 2780-157 Oeiras, Portugal
cEscola de Psicologia e Ciências da Vida, Departamento de Ciências da Vida, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande, 1749-024 Lisboa, Portugal dCentro de Investigação Interdisciplinar Egas Moniz (CiiEM), Instituto Superior de Ciências da Saúde Egas Moniz, 2829-511 Caparica, Portugal
a b s t r a c t
a r t i c l e
i n f o
Article history:
Received 19 January 2016
Received in revised form 30 March 2016 Accepted 27 April 2016
Available online 28 April 2016
Cytochromecd1nitrite reductases (cd1NiRs) catalyze the reduction of nitrite to nitric oxide in denitrifying
bacte-ria, such asMarinobacter hydrocarbonoclasticus. Previous work demonstrated that the enzymatic activity depends on a structural pre-activation triggered by the entry of electrons through the electron transfer (ET) domain, which houses a hemeccenter. The catalytic activity ofM. hydrocarbonoclasticus cd1NiR (Mhcd1NiR) was tested
by mediated electrochemistry, using small ET proteins and chemical redox mediators. The rate of enzymatic re-action depends on the nature of the redox partner, with cytochrome (cyt)c552providing the highest value. In
sit-uations where cytc552is replaced by either a biological (cytcfrom horse heart) or a chemical mediator the
catalytic response was only observed at very low scan rates, suggesting that the intermolecular ET rate is much slower.
Molecular docking simulations with the 3D model structure ofMhcd1NiR and cytc552or cytcshowed that
hydro-phobic interactions favor the formation of complexes where the hemecdomain of the enzyme is the principal docking site. However, only in the case of cytc552the preferential areas of contact and Fe–Fe distances between
hemecgroups of the redox partners allow establishing competent ET pathways. The coupling of the enzyme with chemical redox mediators was also found not to be energetically favorable. These results indicate that although low activity functional complexes can be formed betweenMhcd1NiR and different types of redox mediators,
ef-ficient ET is only observed with the putative physiological electron donor cytc552.
© 2016 Elsevier B.V. All rights reserved.
Keywords:
Cytochromecd1nitrite reductase Cytochromec552
Intermolecular electron transfer Mediated electrochemistry Molecular coupling Electronic pathways
1. Introduction
Theflow of electrons to and from enzymes that catalyze redox reac-tions is typically provided by soluble electron transfer (ET) proteins, such as monohemic cytochromes (cyt) and cupredoxins. For the inter-molecular ET reaction to occur, short-lived protein–protein complexes
are formed. The inter-protein interactions are generally based on elec-trostatic and hydrophobic matching patches that lead to and stabilize the complexes. They also determine the molecular recognition, kinetic efficiency and specificity among reaction partners[1–3]. Diffuse com-plexes and multiple orientations are allowed, as long as the distance be-tween the redox centers enables fast ET[1,3–5]. Nonetheless, other protein couples may progress to afinal lower-energy arrangement after initial pre-orientation interactions[3,5,6].
Some proteins have the ability to react with alternative redox part-ners, and even non-physiological ones, of quite different structures.
For example, superoxide reductase has rubredoxin and desulforedoxin as ET partners, while plastocyanin and cytb6are both involved in the
electron transport between the cytochromebfcomplex and photosys-tem I during photosynthesis[7,8]. Likewise, several electron shuttle pro-teins, such as the small iron–sulfur ferredoxin, may be involved in a large number of ET processes, interacting in slightly differing ways with each reaction partner[4,9]. Such promiscuity has been associated with the small size of the interfacial regions, the relatively nonspecific interactions and the transient nature of ET complexes[3,6,7].
In some cases the molecular interactions in ET complexes set off structural rearrangements that lead to alterations in the protein proper-ties. For instance, the association of ferredoxin–NADP+reductase with
ferredoxin was shown to induce residue reorientation in the latter that may be partially responsible for a shift in its redox potential[4]. Likewise, mitochondrial cytcinteraction with cytochromecoxidase re-sults in a conformational change that affects its redox properties[10]. Another example is the pairing of cytochrome P450 reductase with cy-tochrome P450. The former undergoes large structural modifications in the course of the ET reactions, that are most likely involved in the cata-lytic cycle[11].
⁎ Corresponding author.
E-mail address:[email protected](M.G. Almeida). 1 These authors contributed equally to the work.
http://dx.doi.org/10.1016/j.bbabio.2016.04.279
0005-2728/© 2016 Elsevier B.V. All rights reserved.
Contents lists available atScienceDirect
Biochimica et Biophysica Acta
In this work we studied the ET to cytochromecd1nitrite reductase
(cd1NiR) fromMarinobacter hydrocarbonoclasticus(Mhcd1NiR) from
physiological and non-physiological electron donors. The enzyme can be found in the periplasm of several types of denitrifying bacteria and is involved in respiratory nitrite reduction, converting nitrite to nitric oxide in a one electron reaction[12–15]. Three-dimensional crystal structures have been determined for cd1NiR from Pseudomonas
aeruginosaandParacoccus pantotrophus(Pacd1NiR andPpcd1NiR,
re-spectively)[16,17]. The enzyme is a homodimer containing two heme cofactors in each of its 60 kDa subunits. The monomers are organized in two domains, the largest of which is the catalytic one–the site of
ni-trite reduction–harboring a non-covalently boundd1heme. The
small-er ET domain contains acheme cofactor and is the site of electron entry on the enzyme, transferred thereafter to the active site. Despite the high sequence identity between the enzymes from the two organisms (70%) [13]notable differences have been reported at the level of heme axial li-gation in the oxidized states; however, both have similar catalytically competent reduced forms. Heme ligand switching is required for en-zyme activation and is accompanied by conformational rearrangements that take part of the catalytic cycle and may be triggered by contact with electron donor partners[18,19].
In vivo cd1NiR accepts electrons from small periplasmicc-type
cyto-chromes (e.g. cytc551, cytc552) or copper proteins (e.g. pseudoazurin,
azurin)[20]. Some bacterial species, (e.g.P. pantotrophus) have both types of ET proteins (cytc550and pseudoazurin) andPpcd1NiR was
shown to react with either one. The interactions betweencd1NiR and
its redox partners were classified as pseudo-specific since it can accept electrons from proteins of quite different structures and redox centers [21,22]. Earlierin vitrokinetic studies have also demonstrated that cd1NiR can accept electrons from ET proteins of different organisms,
in-cluding eukaryotic cytochromes (horse heart cytc) but, with few exceptions, the reactivity is slower[13,22–26]. In addition, the ki-netic properties ofcd1NiR have been studied using small redox
ac-tive molecules as electron donors (e.g. ascorbate, tetramethyl-phenylenediamine, viologens and quinones)[23,24,27,28]. Later work, mostly using electrochemical methods, has shown that more than the thermodynamic properties, the charge of the electron shuttles and their structural features may be important for enzyme/ET partner recognition[18,29]. For example,Mhcd1NiR could discriminate
between its putative physiological electron donor, cytc552(a 20 kDa
dimericc-type cytochrome) and other soluble electron carrier cyto-chromes also purified from the bacterium's periplasm[18]. Neverthe-less, that same cytc552could be used as reaction partner forPacd1NiR,
supporting the earlier considerations about the enzyme's pseudo-specific interactions with electron donor proteins[29].
Finally, several unsuccessful attempts, including our own work, were made in order to use electrode surfaces as redox partners for cd1NiR[18,29,30]. Recently we have shown that it is possible to wire
the enzyme onto alkanethiol based self-assembled monolayer modified electrodes.cd1NiR is immobilized in a redox active state, as shown by
the spectroscopic data, however, no electrochemical signals were ob-served either in the absence or presence of nitrite, probably due to an unfavorable orientation of the protein, structural alterations or the in-ability of the electrode system to induce the important conformational changes required for activation/catalysis[31].
Herein we have used a combination of electrochemical methods and molecular docking studies, and a variety of redox mediators, to provide a new insight into the ET reactions that lead to the activation of the cd1NiR fromM. hydrocarbonoclasticus.
2. Experimental
2.1. Reagents and proteins
Mhcd1NiR (12.5μM in 50 mM Tris–HCl buffer, pH 8 and 150 mM NaCl,
unless stated otherwise) and cytc552(50μM in 50 mM Tris–HCl buffer,
pH 8 and 150 mM NaCl) were purified fromM. hydrocarbonoclasticus cells as previously described[32,33]. Horse heart cyt c, phenazine methosulfate (PMS) and phenosafranin were purchased from Sigma-Aldrich. Other reagents, also acquired from Sigma-Aldrich, were analytical grade and used without further purification. Solutions were prepared with deionized water (18 MΩ·cm) from a Millipore MilliQ water purifi ca-tion system.
2.2. Electrode preparation
The working pyrolytic graphite electrodes were polished with alu-mina slurry (0.3 and 1μm, from Buehler), rinsed with water and placed in an ultrasounds bath for 5 min. Finally the electrodes were rinsed with 30% ethanol solution and dried with compressed air.
The experiments were performed using a membrane electrode, prepared as described previously elsewhere[34]. For assays with small electron transfer proteins, mixtures of enzyme and ET protein werefirst prepared in a 1:4 M ratio. Next, a small volume of this mixture (6μL) was entrapped between the electrode surface and a square piece (ca. 1 cm2) of dialysis membrane (MW cut-off
3500 Da, Spectra/Por). A rubber o-ring was thenfitted around the
electrode body tofix the proteinfilm and the membrane to the elec-trode. In the experiments with chemical redox mediators, only the enzyme was imprisoned on the electrode surface in this manner, the mediators were added to the supporting electrolyte in 4:1 stoi-chiometric proportion with the enzyme.
2.3. Electrochemical measurements
The electrochemical assays were performed in a conventional three electrode cell composed of a Ag/AgCl reference electrode (197 mVvsNHE), a platinum counter electrode (both from Radiom-eter) and a homemade pyrolytic graphite working electrode (∅= 4 mm). The experiments were performed using an Autolab 12 potentiostat (Eco-Chemie) controlled by the NOVA software (version 1.6).
The electrochemical cells were cleaned in a 25% sulfuric acid solu-tion in order to remove residual nitrite prior to the experiments. The supporting electrolyte was 100 mM KCl in 50 mM MES buffer pH 6.3 (for determination of ET constants) or pH 6.5 (kinetic assays). The
electrolyte solution (20 mL) was thoroughly purged with argon be-fore the experiments to deplete dissolved oxygen; an argon atmo-sphere was also maintained inside the electrochemical cell during the assays by continuouslyflushing argon onto the surface of the solution.
In kinetic experiments, small volumes of nitrite standard solutions were consecutively added to the electrochemical cell. Assays were per-formed using a scan rate of 20 mV/s, unless stated otherwise. All poten-tials were quotedversusthe Ag/AgCl reference electrode.
2.4. In silico docking
Molecular docking studies were performed using a homology model ofMhcd1NiR. The structure template,Pacd1NiR (PDB ID: 1HZV), and the
amino acid sequence ofMhcd1NiR were used as inputs on I-TASSER[35,
36]. The template was chosen because of its identity percentage (72.0%), knowing that amino acid sequences with identities larger than 50% gen-erate three-dimensional structures with RMSD no bigger than 1 Å[37]. Furthermore, the template structure was crystallized at acidic pH and exhibits a domain swing, bringing closer the ET heme (c), and the cata-lytic heme (d1), which is thought to facilitate active center regeneration
[38]. Electrostatic surface potential was calculated using Adaptive Poisson–Boltzmann Solver (APBS) software[39].
The model structure was then used to generate docking poses with the structure of the physiological ET partner, the M. hydrocarbonoclasticuscytc552dimer (PDB ID: 1CNO) and one other
heme-bearing molecule, horse heart cytc(PDB ID: 1HRC). The BiGGER algorithm[40,41]generates models by searching the six-dimensional space of the probe (in the present case, the small ET cytochrome) in re-lation to the target (Mhcd1NiR). Solutions are then ranked according to
a series of parameters, including electrostatic energy minimization, changes in accessible solvent area, and geometric parameters.
Top solutions, meeting the criteria that the Fe–Fe distances between
target and probe molecules should be no more than 20 Å apart, were further analyzed in terms of interaction interfaces using PDBePISA, to assess the amino acid residues that were predominantly present in the interface of the better docking poses. ET pathways were calculated
Table 1
Electrochemical characterization of the redox mediator systems in the presence of
Mhcd1NiR. Electrolyte: 100 mM KCl, 50 mM MES buffer, pH 6.3. Scan rate range: 5–200 mV/s. Comparison of redox potentials with reference values is given in parenthesis.
Redox mediator ΔEp (mV) Ic/Ia E0′(mV)vsNHE
Cytc552 110 1.1 246 ± 4 (250[45])
Cytc 90 1.1 283 ± 9 (279[29])
PMS 56 1.2 82 ± 4 (80)
Phenosafranin 61 1.0 −244 ± 11 (−255)
Fig. 3.Electrochemically mediated activity ofMhcd1NiR. Step 1 involves the reversible reduction of the mediator molecule at the electrode surface, step 2 consists in the homogeneous intermolecular ET reaction between the mediator andMhcd1NiR yielding the reduced (red) catalytically active enzyme and regenerating the oxidized (ox) form of the mediator molecule, step 3 corresponds to the irreversible catalytic reduction of nitrite byMhcd1NiR which returns to the oxidized state,finally step 4 indicates the diffusion of product and substrate through the membrane electrode.
using the HARLEM PATHWAYS software[42]and evaluated in terms of the electron coupling parameter of the Marcus equation.
To assess docking poses of the smaller, non-proteic ET partners, the structures of phenosafranine and PMS were taken from PubChem and
energy-minimized prior to docking using UCSF Chimera[43]. The struc-tures were then docked using the Autodock Vina algorithm[44]. Top structures were analyzed in terms of the coupling constant term of the Marcus equation using HARLEM PATHWAYS[42].
3. Results and discussion
3.1. Mediated electrochemistry of M. hydrocarbonoclasticus cd1NiR
3.1.1. Physiological electron donor cytochrome c552
Firstly, the mediated electrochemical response ofMhcd1NiR was
measured in the presence of its putative physiological ET partner, the small redox protein cytc552. Both proteins were confined to the
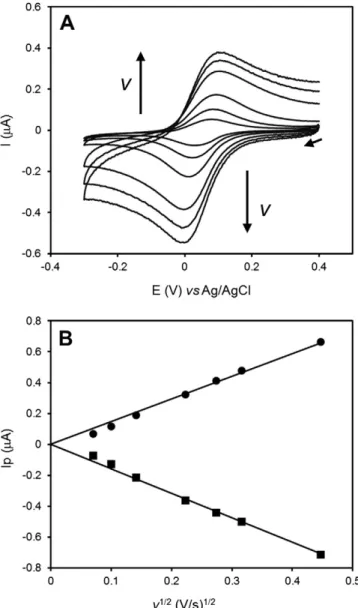
pyrolyt-ic graphite electrode surface through the use of a dialysis membrane. The bioelectrode was characterized in the absence of nitrite, in scan rates ranging from 5 to 200 mV/s (Fig. 1A).
The cyclic voltammograms (CVs) show a pair of peaks assigned to the heterogeneous oxidation/reduction of cytc552. No direct
electro-chemical response ofMhcd1NiR could be obtained in these conditions.
The ET reaction could be classified as quasi-reversible, with cathodic/an-odic peak separation values up to 110 mV and peak intensity ratios close to one (1.1). The midpoint reduction potential E0′of cytc
552, estimated
from the average of the cathodic and anodic peak positions, was 246 ± 4 mVvsNHE, similar to the value reported in solution[45]. The peak currents varied linearly with the square root of the scan rate, which is consistent with a diffusion controlled electrochemical process (Fig. 1B). This contrasts with previous works, by Santos et al. (cytc552)
and Lojou et al. (cytc551), where thin-layer voltammetry laws were
obeyed[29,46]and could be explained by the additional presence of the enzyme behind the dialysis membrane. In fact, it increases the thick-ness of the diffusion layer and may difficult cytc552access to the
electrode; thus the heterogeneous ET reaction is controlled by the trans-port of the cytc552molecules at all scan rates.
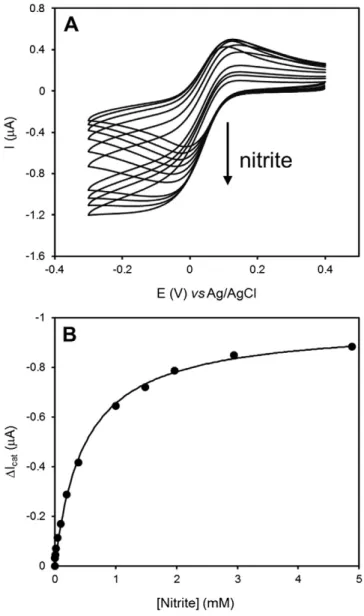
Next, the catalytic activity of the cytc552/Mhcd1NiR electrode was
measured in the presence of nitrite, at a scan rate of 20 mV/s (Fig. 2). Upon adding the substrate to the electrochemical cell, the cathodic cur-rents increased as a consequence of the enzymatic reduction of nitrite and the regeneration of the oxidized form of the mediator molecule, cytc552, according to an EC′electrochemical mechanism (Fig. 3).
Under substrate saturation conditions (above 1 mM of nitrite), typical sigmoidal shape current–potential curves were obtained (increased
ca-thodic peak currents and absence of anodic peaks), indicating that cyt c552is able to efficiently shuttle electrons betweenMhcd1NiR and the
electrode surface. The plot of catalytic current (cathodic peaks) as a function of nitrite concentration could befitted to a Michaelis–Menten
type kinetics curve (Fig. 2B) with KMappand Imaxapp values of 0.48 ±
0.03 mM and−0.97 ± 0.02μA, respectively. Due to the immobilization conditions, the measured parameters are apparent kinetic constants. The KMappvalue denotes the substrate concentration at half maximum
current (Imaxapp) and should be influenced by the rate of substrate diffusion
(Fig. 3, step 4) across the membrane coat and inside the cytc552and
Mhcd1NiR layer (external and internal diffusion, respectively)[47].
The KMappis similar to the one reported previously, with the enzyme
and electron donor co-entrapped in a polymeric matrix (photopolymerizable polyvinyl alcohol)[48]. However, it is ca. two or-ders of magnitude higher than the KMfor the pair cytc551/Pacd1NiR
also measured using a similar dialysis membrane electrode[29]. Taking
into consideration the comparison between these three different exper-imental setups we hypothesize thatMhcd1NiR may have a lower affinity
to nitrite. Unfortunately, due to the immobilization effects we cannot deconvolute the contribution of the enzyme kinetics to KMapp. Therefore,
we should also consider that different surface charges inPacd1NiR and
Mhcd1NiR (cf.Section 3.2.1) may confer alternative preferential enzyme
orientations towards the (mostly) negatively charged electrode[49] and thus slightly impair substrate diffusion inside the cytc552/Mhcd1NiR
layer. In fact, the surface of the model structure ofMhcd1NiR presents a
predominantly negative charge when compared to the mostly hydro-phobic nature ofPacd1NiR (cf.Section 3.2.1).
The intermolecular ET rate between cytc552andMhcd1NiR (Fig. 3,
step 2) was determined according to the method described previously [18,29], based on the theoretical model proposed by Nicholson and Shain[50]. The catalytic CVs were measured at different scan rates, using various concentrations of enzyme while keeping the mediator amount constant. The experimental conditions were set so that the en-zymatic reaction was irreversible, and pseudo-first order, by using a sat-urating concentration of nitrite (5 mM); at reducing potentials, this shifts the equilibrium of the overall reaction, presented inFig. 3, to-wards the reduction ofMhcd1NiR by accumulating reduced cytc552on
the surface of the electrode. Using this method, the second-order rate constant for the reaction between the enzyme and the electron donor protein was determined to be 1.32 × 105M−1s−1. The value is similar
to the one previously measured by Lopes et al. with bothMhcd1NiR and
cyt c552 in solution, using thiol modified gold electrodes (4.1 ±
0.1 × 105M−1s−1) and demonstrates the high ef
ficiency of the ET reac-tion between the two partner proteins[18]. The intermolecular rate
constant values are in good agreement with previous electrochemical determination for thePacd1NiR/azurin pair; however, they are one
order of magnitude lower when compared with the ET between Pacd1NiR and cytc551(cf. Table S1).
3.1.2. Alternative redox mediators
Next, the catalytic activity ofMhcd1NiR was tested with alternative
electron donor partners, namely horse heart cytc, a versatile ET protein involved in several cellular processes, and small redox mediator mole-cules (PMS and phenosafranin). Cytcwas co-entrapped with the en-zyme on the electrode surface, such as in the assays with cytc552.
Conversely, due to their small size, the chemical redox mediators could not be immobilized in this manner, so they were added to the supporting electrolyte solution, being able to freely diffuse through the dialysis membrane retainingMhcd1NiR on the surface of the
pyro-lytic graphite electrode.
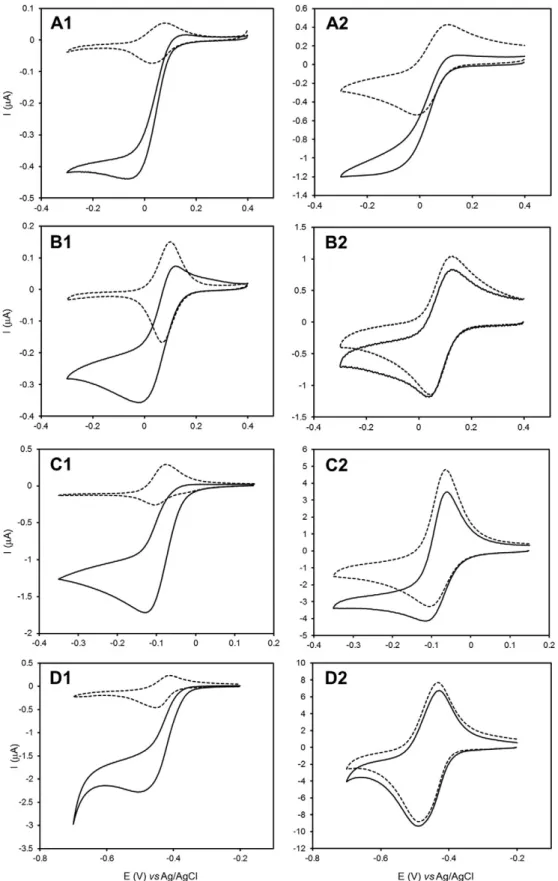
The electrochemical response of the different electron carriers was first evaluated in non-catalytic conditions, in the presence ofMhcd1NiR
only. The CVs of the mediators showed a pair of symmetric peaks, corre-sponding to reversible one electron redox reactions at the electrode sur-face (Fig. S1, left panels). In control experiments performed in the absence ofMhcd1NiR and dialysis membrane the electrochemical
sig-nals of the redox mediators were similar (results not shown). The sys-tems were classified as quasi-reversible and the midpoint redox potentials (E0′) were determined for each mediator at the experimental conditions used (Table 1). For comparison purposes the response of cyt c552electrodes is also included (Fig. S1A1).
The CVs of mediator/Mhcd1NiR systems, measured without and with
5 mM of nitrite, are represented inFig. 4for 5 and 200 mV/s (cf. Fig. S1 for additional scan rates). At the lower scan rates tested (below 20 mV/s), and under substrate saturation conditions, typical sigmoidal shape current–potential curves are obtained with all mediator species, indicative of the catalytic response ofMhcd1NiR to nitrite (Fig. 4, left
panels). However, no catalytic currents could be detected upon increas-ing the scan rate; in fact, above 20 mV/s, the CVs measured with nitrite are superimposable to those measured in the absence of the enzyme's substrate (Fig. 4, right panels). In opposition, the cytc552/Mhcd1NiR CVs
in the presence of nitrite display high catalytic currents and sigmoidal shaped curves regardless of the scan rate used (Fig. 4A1–2). These results could indicate that the electron donor/Mhcd1NiR complexes may not be
able to efficiently exchange electrons with the electrochemical interface despite establishing intermolecular ET. However, this is an unlikely possibility since the CVs of the redox mediators are almost identical in the presence and absence of nitrite at the higher scan rates, suggesting the heterogeneous ET was not affected. Instead we propose that the kinet-ics of the intermolecular ET between the electron donor andMhcd1NiR
(Fig. 3, step 2) is highly dependent on the type of redox mediator. While at the slower scan rates the enzymatic reaction proceeds normally, when the time scale of the experiment is shortened (increased scan rate) the quasi-reversible electrochemical reaction (Fig. 3, step 1) becomes too fast to allow the subsequent chemical reaction with nitrite to occur (Fig. 3, step 3). Therefore, the process of intermolecular ET toMhcd1NiR is not
rapid enough with any of the alternative redox mediators tested and prevents high turnover rates. Consequently, the theoretical model used for rate constant calculations was not verified (data not shown); so our attempt to determine the rates of intermolecular ET was unsuccessful. This study contrasts with previous reports on the electrochemical behavior ofPacd1NiR which did not display catalytic activity with
chemi-cal electron donors norc-type cytochromes of elevated isoelectric point (although good catalytic responses could be obtained with less positively charged proteins at optimum pH, ~6.5)[29]. Here we show that,first of all cytc552is the best electron donor forMhcd1NiR; however, other biological
and non-proteic electron carriers can serve as reducing agents and trigger catalytic activity if the assay is performed in relatively long time windows (scan rates between 5 and 20 mV/s). Very importantly, this does not depend on the redox potential, charge and structural properties of the mediator molecule. We postulate that, despite providing the driving force for the reaction, the alternative redox mediators do not mimic specific interactions between the physiological electron donor and the enzyme and thus are poor electron sources. In conclusion, the biological activity ofMhcd1NiR is governed by molecular complementarity with
the redox partner, which is best satisfied by cytc552.
In order to understand the reasons behind the different reaction rates, the interaction of the ET molecules withMhcd1NiR was studied
by molecular docking.
3.2. In silico docking
3.2.1. Electron transfer proteins
Since its 3D structure is not yet available, a homology model of Mhcd1NiR was constructed based on the P. aeruginosa enzyme
(Pacd1NiR). The RMSD between the model and the template structure
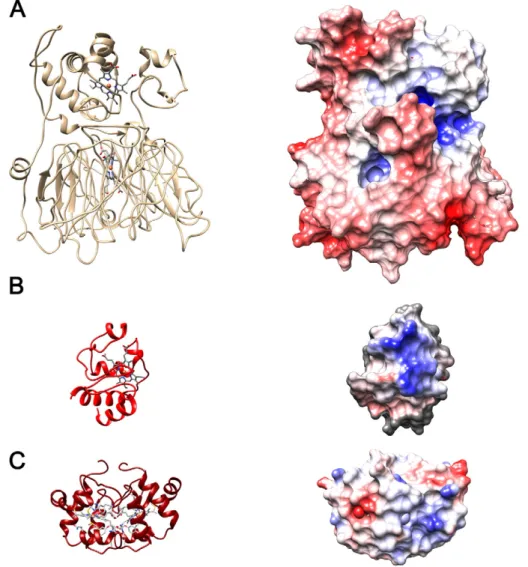
(1HZV) is 0.226 Å. Surface charge distributions for the model structure and the protein ET partners (cytc552and cytc) are shown inFig. 5.
Mhcd1NiR is mostly negatively-charged (red surface patches) with a
hydrophobic patch (white) surrounding the heme moiety of the ET do-main, and one positively-charged and one negatively-charged patch in its vicinity. Comparatively, the electrostatic surface ofMhcd1NiR is
more negatively charged than that ofP. aeruginosa. Cytc552is mostly
neutral around the heme center, with a moderately positive patch near-by; horse heart cytcalso shares some of these features. It was previously proposed that, inP. aeruginosaandP. pantotrophus,cd1NiR and its
elec-tron donors share complementary traits in terms of surface charge
distributions around their ET centers[20,21,51]. It is thought that the oppositely-charged patches allow the formation of encounter ET com-plexes, pre-orienting the interacting molecules towards a geometry that allows efficient electron exchange[52].
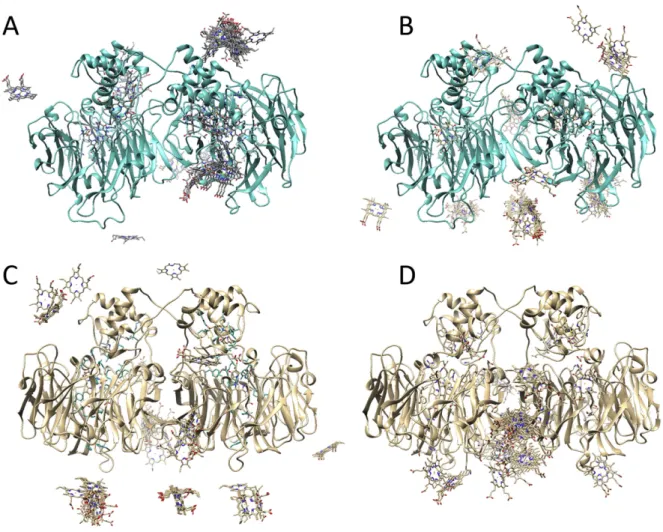
Fig. 6exhibits the top 100 hydrophobic (panels A and C) and top 100 electrostatic (panels B and D) energy minimization solutions in thein silicodocking betweenMhcd1NiR and cytc552(top) or cytc(bottom).
For simplicity, only the heme moieties of the interacting small cyt mol-ecules are shown, to highlight their relative orientation towards the tar-get. These data suggest a clear preference for orientations that lead to a feasible ET pathway governed by hydrophobic interactions, with ener-gies in the vicinity of−8.0 kcal/mol. Electrostatic energy minimization produces complexes that are theoretically more stable, but inefficient in terms of ET (distance between redox centers mostly above 20 Å). For the interaction betweenMhcd1NiR and cytc, one can readily observe
that relevant solutions that could transfer electrons between typec hemes are scarce, with a clear preference for orientations that are far from the ET center on the enzyme (Fig. 6C) or located at the monomer-monomer contact interface (Fig. 6D), which are inefficient for the proposed role played by the cyt molecule. This may be related
to surface charge distributions in the enzyme and ET proteins.Mhcd1NiR
possesses both a positively- and a negatively-charged patch in the vicin-ity of its ET center, and no charge near the heme surface. Cytcis mostly positively-charged, which may lead the molecule away from the ET cen-ter and onto more favorable conformations that help minimize electro-static repulsions. Cytc552is slightly less charged, which may assist in the
formation of stable, apolar interactions that bring the ET centers into closer contact. The importance of the surface characteristics was also observed in the pairing ofPacd1NiR and donor proteins, where polar
and hydrophobic interactions were suggested to be essential in the for-mation of the pseudo-specific ET complexes[29,51,53]. For example, changing the charge around the hydrophobic patch in the small cyt c551significantly affected its ability to react withPacd1NiR[54]. In this
context, we also performed docking simulations with cytc551with
bothPacd1NiR andMhcd1NiR using the BiGGER algorithm (Fig. S2). As
expected, the results indicate that cytochromec551binds preferentially
nearPacd1NiR's hemec, thereby validating the docking procedure used
in our work. Conversely, the same cytc551binds far fromMhcd1NiR's
hemec, suggesting that it should be a poor ET partner for this enzyme. This is not surprising since cytc551has a ring of positively charged
res-idues (mainly lysines) near the hydrophobic patch surrounding the heme pocket; this is thought to play a role in the functional coupling withPacd1NiR[51,54]but, as seen for cytc, it may hinder the approach
to theMhcd1NiR ET center.
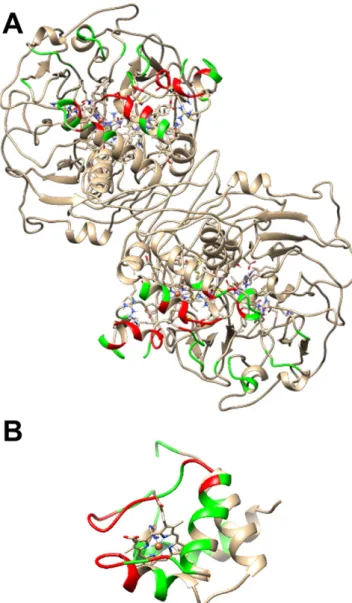
A manual thresholdfilter was applied for the top hydrophobic solu-tions—the Fe–Fe distance between the hemecof cytc552, and hemecof
Mhcd1NiR should be below the 20 Å range. This generated a subset
com-prising only 19 solutions, which were analyzed in further detail using the PDBePISA algorithm[55]to determine the residues involved in the interaction interface (Fig. 7), and the HARLEM suite[42]to determine possible ET pathways and respective coupling energies. From the range of possible solutions, one can acknowledge that the coupling values of the Marcus equation range between the 10−3and 10−5orders
of magnitude, and mainly involve non-bonded jumps of one electron between hemecof the small cyt molecule towards one of the following Mhcd1NiR residues: Glu26, Ala27 or Lys30, progressing through
cova-lent bonds to Ala25, Ala28 or Pro29, respectively, and from there to-wards hemecofMhcd1NiR via another non-bonded jump. Residues
that are involved in the interaction surface in more than 50% of the pro-posed solutions include 24, 25, 28, 32, 112–114 and 130 (colored red),
which include a mix of uncharged residues (Ala, Pro, Ser, Tyr, Gly) and one Aspartate residue. Amino acids in their vicinity, which are present
in some interaction interfaces (less than 50%) are colored green (Fig. 7A). For cytc552, residues include 1, 36, 38, 41, 42, 45, 48, 49, 51,
65, 68–70, and 73 (red) and those in their vicinity (green). The majority of these residues are either apolar (Ala and Val, comprising 50%) or pos-itively charged (Lys and Gln, aprox. 35%), which will allow the forma-tion of a productive ET complex between Mhcd1NiR and cyt c552
(Fig. 7B). The electronic coupling decay values are in line with previous results described in the literature for fast ET in multi-heme cytochromes [56]and between cytfand copper-containing plastocyanin[57]. Re-garding the complexes formed betweenMhcd1NiR and cytc, the relative
orientations place the heme groups at a large distance, above 20 Å, which is considered the upper limit for efficient ET, as has been ob-served in a number of other physiologic ET complexes (e.g. between plastocyanin and cytochromeffromNostocsp. PCC 7119[58]). This cor-roborates experimental electrochemistry results, i.e., slow ET and low catalytic activity with this partner protein. One should mention that de-spite the shift of about 0.7 pH units between the electrochemical and docking assays, in this pH range only histidine side chains are prone to change their charge. At the interaction interface ofMhcd1NiR, just
His142 may play a part (Fig. S3). Nonetheless, we have evaluated the surface charge distribution using APBS software at pH 6.3 and pH 7.0 (Fig. S3A and B, respectively). Only small changes were observed, which should be irrelevant in terms of ranking electrostatic energy min-imization candidates. Furthermore, amino acid residues involved in the interactions betweenMhcd1NiR and cytc552are mostly hydrophobic in
nature and therefore, less sensitive to pH changes.
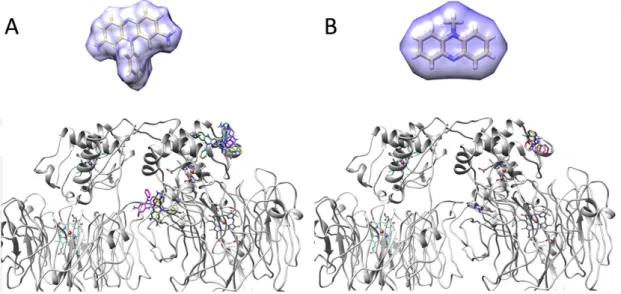
3.2.2. Chemical redox mediators
The small ligands phenosafranin (Fig. 8A) and PMS (Fig. 8B) were analyzed in terms of their binding poses relative toMhcd1NiR using
Autodock Vina[44]. They show a small cluster of solutions in the vicinity ofMhcd1NiR hemec, but also interaction sites scattered across the
sur-face of the enzyme, with similar binding energies among them. The cou-pling decay values, calculated by PATHWAYS, are in the order of 10−5or
lower, for the ET transfer between the redox mediator andMhcd1NiR
hemec. Again, these results are in agreement with those obtained using electrochemical techniques, wherein one observed slow ET rates for all the chemical redox mediators being studied. The binding posi-tions and respective energies along with the coupling decays demon-strate that the alternative redox mediators do not interact in an efficient manner withMhcd1NiR.
4. Conclusion
Efficient docking between redox partners is crucial in biological ET processes. As generally observed with many redox enzymes,Mhcd1NiR
was shown to be activated by structurally different physiological and non-physiological donors, including small redox active molecules, which can be explained by pseudo-specific interactions. However, our electrochemical and molecular docking studies show thatMhcd1NiR is
clearly less efficient with any molecule other than cytc552. In fact, the
partnering of cytc552withMhcd1NiR triggers both high nitrite reducing
activities and intermolecular ET rates. All remaining biological and chemical electron donors tested are poor ET partners as the catalytic ac-tivity can only be detected electrochemically at very low scan rates (slow reaction times). Supporting these data, the docking studies show thatMhcd1NiR accommodates cyt c552 preferentially in the
hemecregion, i.e., the enzyme's ET domain, thereby enabling the for-mation of productive ET complexes. Hydrophobic-type interactions in-volving a majority of uncharged and apolar residues favor the electronically functional couples. In contrast, the spatial positions with the alternative electron donors are not compatible with rapid ET.
Therefore, the complementarity betweenMhcd1NiR and cytc552
sur-faces governs the docking and determines the efficiency of the ET, dem-onstrating an unusually high level of selectivity. The prompt enzyme activation in the presence of only cytc552emphasizes the physiological
role of this small redox protein. Because catalytic activities were not comparably high, the optimal ET pathways toMhcd1NiR could not be
mimicked by other electron carriers. This may justify the inability of the enzyme to be activated by direct contact with electroactive surfaces as reported in previous works[29,31].
Transparency Document
TheTransparency documentassociated with this article can be found, in online version.
Acknowledgments
The authors acknowledge funding from UCIBIO@REQUIMTE Pest-C/ EQB/LA0006/2013. CM Silveira and RM Almeida thank thefinancial sup-port from Fundação para a Ciência e Tecnologia (Postdoctoral fellow-ships SFRH/BPD/79566/2011 and SFRH/BPD/80293/2011).
Appendix A. Supplementary data
Supplementary data to this article can be found online athttp://dx. doi.org/10.1016/j.bbabio.2016.04.279.
References
[1] I. Díaz-Moreno, R. Hulsker, P. Skubak, J.M. Foerster, D. Cavazzini, M.G. Finiguerra, A. Díaz-Quintana, B. Moreno-Beltrán, G.-L. Rossi, G.M. Ullmann, N.S. Pannu, M.A. De la Rosa, M. Ubbink, The dynamic complex of cytochromec6and cytochromefstudied with paramagnetic NMR spectroscopy, Biochim. Biophys. Acta Bioenerg. 1837 (2014) 1305–1315,http://dx.doi.org/10.1016/j.bbabio.2014.03.009.
[2] P.B. Crowley, M.A. Carrondo, The architecture of the binding site in redox protein complexes: implications for fast dissociation, Proteins 55 (2004) 603–612,http:// dx.doi.org/10.1002/prot.20043.
[3] J. Schilder, M. Ubbink, Formation of transient protein complexes, Curr. Opin. Struct. Biol. 23 (2013) 911–918,http://dx.doi.org/10.1016/j.sbi.2013.07.009.
[4] R.E. Blankenship, It takes two to tango, Nat. Struct. Biol. 8 (2001) 94–95,http://dx. doi.org/10.1038/84191.
[5] S. Scanu, J.M. Foerster, G.M. Ullmann, M. Ubbink, Role of hydrophobic interactions in the encounter complex formation of the plastocyanin and cytochromefcomplex re-vealed by paramagnetic NMR spectroscopy, J. Am. Chem. Soc. 135 (2013) 7681–7692,http://dx.doi.org/10.1021/ja4015452.
[6] M.D. Vlasie, R. Fernández-Busnadiego, M. Prudêncio, M. Ubbink, Conformation of pseudoazurin in the 152 kDa electron transfer complex with nitrite reductase deter-mined by paramagnetic NMR, J. Mol. Biol. 375 (2008) 1405–1415,http://dx.doi.org/ 10.1016/j.jmb.2007.11.056.
[7] P.B. Crowley, M. Ubbink, Close encounters of the transient kind: protein interactions in the photosynthetic redox chain investigated by NMR spectroscopy, Acc. Chem. Res. 36 (2003) 723–730,http://dx.doi.org/10.1021/ar0200955.
[8] R.M. Almeida, P. Turano, I. Moura, J.J. Moura, S.R. Pauleta, Superoxide reductase: dif-ferent interaction modes with its two redox partners, Chembiochem 14 (2013) 1858–1866,http://dx.doi.org/10.1002/cbic.201300196.
[9] X. Xu, S.K. Kim, P. Schurmann, M. Hirasawa, J.N. Tripathy, J. Smith, D.B. Knaff, M. Ubbink, Ferredoxin/ferredoxin–thioredoxin reductase complex: complete NMR mapping of the interaction site on ferredoxin by gallium substitution, FEBS Lett. 580 (2006) 6714–6720,http://dx.doi.org/10.1016/j.febslet.2006.11.027. [10] N. Khare, D.M. Lovelace, C.M. Eggleston, M. Swenson, T.S. Magnuson, Redox-linked
conformation change and electron transfer between monohemec-type cyto-chromes and oxides, Geochim. Cosmochim. Acta 70 (2006) 4332–4342,http://dx. doi.org/10.1016/j.gca.2006.06.1561.
[11] L. Aigrain, D. Pompon, S. Morera, G. Truan, Structure of the open conformation of a functional chimeric NADPH cytochrome P450 reductase, EMBO Rep. 10 (2009) 742–747,http://dx.doi.org/10.1038/embor.2009.82.
[12] W.G. Zumft, Cell biology and molecular basis of denitrification, Microbiol. Mol. Biol. Rev. 61 (1997) 533–616.
[13] F. Cutruzzolà, Bacterial nitric oxide synthesis, Biochim. Biophys. Acta Bioenerg. 1411 (1999) 231–249,http://dx.doi.org/10.1016/S0005-2728(99)00017–1.
[14] B.A. Averill, Dissimilatory nitrite and nitric oxide reductases, Chem. Rev. 96 (1996) 2951–2964.
[15] S. Wherland, O. Farver, I. Pecht, Intramolecular electron transfer in nitrite reduc-tases, ChemPhysChem 6 (2005) 805–812, http://dx.doi.org/10.1002/cphc. 200400353.
[16] P.A. Williams, V. Fulop, E.F. Garman, N.F. Saunders, S.J. Ferguson, J. Hajdu, Haem-ligand switching during catalysis in crystals of a nitrogen-cycle enzyme, Nature 389 (1997) 406–412,http://dx.doi.org/10.1038/38775.
[17] D. Nurizzo, F. Cutruzzola, M. Arese, D. Bourgeois, M. Brunori, C. Cambillau, M. Tegoni, Conformational changes occurring upon reduction and NO binding in nitrite reduc-tase fromPseudomonas aeruginosa, Biochemistry 37 (1998) 13987–13996,http://dx. doi.org/10.1021/bi981348y.
[18]H. Lopes, S. Besson, I. Moura, J.J. Moura, Kinetics of inter- and intramolecular elec-tron transfer ofPseudomonas nauticacytochromecd1nitrite reductase: regulation
of the NO-bound end product, J. Biol. Inorg. Chem. 6 (2001) 55–62.
[19] I. Moura, S.R. Pauleta, J.J. Moura, Enzymatic activity mastered by altering metal coor-dination spheres, J. Biol. Inorg. Chem. 13 (2008) 1185–1195,http://dx.doi.org/10. 1007/s00775-008-0414-3.
[20]S. Ferguson, V. Fulop, Cytochromecd1nitrite reductase structure raises interesting
mechanistic questions, in: A. Holzenburg, N. Scrutton (Eds.), Enzyme-Catalyzed Electron and Radical Transfer, vol. 35, Springer, US 2000, pp. 519–540.
[21] P.A. Williams, V. Fulop, Y.C. Leung, C. Chan, J.W. Moir, G. Howlett, S.J. Ferguson, S.E. Radford, J. Hajdu, Pseudospecific docking surfaces on electron transfer proteins as il-lustrated by pseudoazurin, cytochromec550and cytochromecd1nitrite reductase,
Nat. Struct. Biol. 2 (1995) 975–982.
[22] C.D. Richter, J.W. Allen, C.W. Higham, A. Koppenhofer, R.S. Zajicek, N.J. Watmough, S.J. Ferguson, Cytochromecd1, reductive activation and kinetic analysis of a multi-functional respiratory enzyme, J. Biol. Chem. 277 (2002) 3093–3100,http://dx.doi. org/10.1074/jbc.M108944200.
[23] R. Timkovich, R. Dhesi, K.J. Martinkus, M.K. Robinson, T.M. Rea, Isolation of
Paracoccus denitrificanscytochromecd1: comparative kinetics with other nitrite
re-ductases, Arch. Biochem. Biophys. 215 (1982) 47–58.
[24] C.L. Hulse, J.M. Tiedje, B.A. Averill, A spectrophotometric assay for dissimilatory ni-trite reductases, Anal. Biochem. 172 (1988) 420–426.
[25]M.K. Robinson, K. Martinkus, P.J. Kennelly, R. Timkovich, Implications of the inte-grated rate law for the reactions ofParacoccus denitrificansnitrite reductase, Bio-chemistry 18 (1979) 3921–3926.
[26] M.C. Silvestrini, S. Falcinelli, I. Ciabatti, F. Cutruzzolà, M. Brunori,Pseudomonas aeruginosanitrite reductase (or cytochrome oxidase): an overview, Biochimie 76 (1994) 641–654,http://dx.doi.org/10.1016/0300-9084(94)90141–4.
[27] S.J. George, J.W. Allen, S.J. Ferguson, R.N. Thorneley, Time-resolved infrared spectroscopy reveals a stable ferric heme-NO intermediate in the reaction of
Paracoccus pantotrophuscytochromecd1nitrite reductase with nitrite, J. Biol. Chem. 275 (2000) 33231–33237,http://dx.doi.org/10.1074/jbc.M005033200. [28] F. Cutruzzola, K. Brown, E.K. Wilson, A. Bellelli, M. Arese, M. Tegoni, C. Cambillau,
M. Brunori, The nitrite reductase fromPseudomonas aeruginosa: essential role of two active-site histidines in the catalytic and structural properties, Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 2232–2237,http://dx.doi.org/10.1073/pnas. 041365298.
[29] E. Lojou, F. Cutruzzolà, M. Tegoni, P. Bianco, Electrochemical study of the intermolec-ular electron transfer toPseudomonas aeruginosacytochromecd1nitrite reductase, Electrochim. Acta 48 (2003) 1055–1064, http://dx.doi.org/10.1016/S0013-4686(02)00843–5.
[30] A. Serra, Isolamento e Caracterização de Enzimas Multihémicas de Origem Microbiana e sua Aplicação no Desenvolvimento de Biossensores (PhD Thesis) Universidade Nova de Lisboa, 2012 (Available:http://hdl.handle.net/10362/7627). [31] C.M. Silveira, P.O. Quintas, I. Moura, J.J. Moura, P. Hildebrandt, M.G. Almeida, S. Todorovic, SERR spectroelectrochemical study of cytochromecd1nitrite reductase co-immobilized with physiological redox partner cytochromec552on biocompatible metal electrodes, PLoS One 10 (2015) e0129940,http://dx.doi.org/10.1371/journal. pone.0129940.
[33] G. Fauque, J.J.G. Moura, S. Besson, L. Saraiva, I. Moura, Preliminary characterization of the cytochrome system in the marine denitrifying bacteriumPseudomonas nautica
617, Oceanis 18 (1992) 211–216.
[34] J. Haladjian, I. Thierry-Chef, P. Bianco, Permselective-membrane pyrolytic graphite electrode for the study of microvolumes of [2Fe–2S] ferredoxin, Talanta 43 (1996) 1125–1130,http://dx.doi.org/10.1016/0039-9140(96)01874–7.
[35] A. Roy, A. Kucukural, Y. Zhang, I-TASSER: a unified platform for automated protein structure and function prediction, Nat. Protoc. 5 (2010) 725–738,http://dx.doi. org/10.1038/nprot.2010.5.
[36] Y. Zhang, I-TASSER server for protein 3D structure prediction, BMC Bioinformatics 9 (2008) 40,http://dx.doi.org/10.1186/1471-2105-9-40.
[37] F. Kiefer, K. Arnold, M. Kunzli, L. Bordoli, T. Schwede, The SWISS-MODEL repository and associated resources, Nucleic Acids Res. 37 (2009) D387–D392,http://dx.doi. org/10.1093/nar/gkn750.
[38] K. Brown, V. Roig-Zamboni, F. Cutruzzola’, M. Arese, W. Sun, M. Brunori, C. Cambillau, M. Tegoni, Domain swing upon his to Ala mutation in nitrite reductase ofPseudomonas aeruginosa, J. Mol. Biol. 312 (2001) 541–554,http://dx.doi.org/10. 1006/jmbi.2001.4986.
[39] N.A. Baker, D. Sept, S. Joseph, M.J. Holst, J.A. McCammon, Electrostatics of nanosys-tems: application to microtubules and the ribosome, Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 10037–10041,http://dx.doi.org/10.1073/pnas.181342398.
[40] L. Krippahl, J.J. Moura, P.N. Palma, Modeling protein complexes with BiGGER, Pro-teins 52 (2003) 19–23,http://dx.doi.org/10.1002/prot.10387.
[41] P.N. Palma, L. Krippahl, J.E. Wampler, J.J. Moura, BiGGER: a new (soft) docking algo-rithm for predicting protein interactions, Proteins 39 (2000) 372–384.
[42] I.V. Kurnikov, HARLEM 1.0, Department of Chemistry, University of Pittsburgh, Pitts-burgh, PA, 2000.
[43] E.F. Pettersen, T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, T.E. Ferrin, UCSF Chimera—a visualization system for exploratory research and analysis, J. Comput. Chem. 25 (2004) 1605–1612,http://dx.doi.org/10.1002/jcc.20084. [44] O. Trott, A.J. Olson, AutoDock Vina: improving the speed and accuracy of docking
with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem. 31 (2010) 455–461,http://dx.doi.org/10.1002/jcc.21334.
[45] L.M. Saraiva, G. Fauque, S. Besson, I. Moura, Physico-chemical and spectroscopic properties of the monohemic cytochromec552fromPseudomonas nautica617, Eur.
J. Biochem. 224 (1994) 1011–1017.
[46] M.M.C. Santos, P.M.P. Sousa, M.L.S. Goncalves, L. Krippahl, J.J.G. Moura, E. Lojou, P. Bianco, Electrochemical studies on small electron transfer proteins using membrane electrodes, J. Electroanal. Chem. 541 (2003) 153–162,http://dx.doi.org/10.1016/ s0022-0728(02)01427–4.
[47] F. Scheller, F. Schubert, Physicochemical, biochemical and technological fundamen-tals of biosensors, in: F. Scheller, F. Schubert (Eds.), Techniques and Instrumentation in Analytical Chemistry - Biosensors, vol. 11, Elsevier, Amsterdam, The Netherlands 1992, pp. 7–84.
[48] A.S. Serra, S.R. Jorge, C.M. Silveira, J.J. Moura, E. Jubete, E. Ochoteco, G. Cabanero, H. Grande, M.G. Almeida, Cooperative use of cytochromecd1nitrite reductase and its redox partner cytochromec552to improve the selectivity of nitrite biosensing, Anal. Chim. Acta 693 (2011) 41–46,http://dx.doi.org/10.1016/j.aca.2011.03.029. [49] J.J. Davis, H.A.O. Hill, A.M. Bond, The application of electrochemical scanning
probe microscopy to the interpretation of metalloprotein voltammetry, Coord. Chem. Rev. 200–202 (2000) 411–442,http://dx.doi.org/10.1016/ S0010-8545(00)00321–0.
[50] R.S. Nicholson, I. Shain, Theory of stationary electrode polarography. Single scan and cyclic methods applied to reversible, irreversible, and kinetic systems, Anal. Chem. 36 (1964) 706–723,http://dx.doi.org/10.1021/ac60210a007.
[51] M. van de Kamp, M.C. Silvestrini, M. Brunori, J. Van Beeumen, F.C. Hali, G.W. Canters, Involvement of the hydrophobic patch of azurin in the electron-transfer reactions with cytochromec551and nitrite reductase, Eur. J. Biochem. 194 (1990) 109–118,
http://dx.doi.org/10.1111/j.1432-1033.1990.tb19434.x.
[52] M. Ubbink, The courtship of proteins: understanding the encounter complex, FEBS Lett. 583 (2009) 1060–1066,http://dx.doi.org/10.1016/j.febslet.2009.02.046. [53] K. Brown, D. Nurizzo, S. Besson, W. Shepard, J. Moura, I. Moura, M. Tegoni, C.
Cambillau, MAD structure ofPseudomonas nauticadimeric cytochromec552mimicks thec4dihemic cytochrome domain association, J. Mol. Biol. 289 (1999) 1017–1028,
http://dx.doi.org/10.1006/jmbi.1999.2838.
[54] F. Cutruzzolà, M. Arese, G. Ranghino, G. van Pouderoyen, G. Canters, M. Brunori,
Pseudomonas aeruginosacytochromec551: probing the role of the hydrophobic patch in electron transfer, J. Inorg. Biochem. 88 (2002) 353–361,http://dx.doi.org/ 10.1016/S0162-0134(01)00390–7.
[55] E. Krissinel, Crystal contacts as nature's docking solutions, J. Comput. Chem. 31 (2010) 133–143,http://dx.doi.org/10.1002/jcc.21303.
[56] P.M. Matias, L.M. Saraiva, C.M. Soares, A.V. Coelho, J. LeGall, M.A. Carrondo, Nine-haem cytochromecfromDesulfovibrio desulfuricansATCC 27774: primary sequence determination, crystallographic refinement at 1.8 and modelling studies of its inter-action with the tetrahaem cytochromec3, J. Biol. Inorg. Chem. 4 (1999) 478–494,
http://dx.doi.org/10.1007/s007750050334.
[57] F. Musiani, A. Dikiy, A.Y. Semenov, S. Ciurli, Structure of the intermolecular complex between plastocyanin and cytochromeffrom spinach, J. Biol. Chem. 280 (2005) 18833–18841,http://dx.doi.org/10.1074/jbc.M412760200.