w w w . r b h h . o r g
Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
Case
Report
Compound
heterozygous
state
of

-thalassemia
with
IVS1-5
(G
→
C)
mutation
and
Indian
deletion-inversion
G
␥
(A
␥␦
)
0
-thalassemia
in
eastern
India
Snehadhini
Dehury
1,
Prasanta
Purohit
1,
Satyabrata
Meher,
Kishalaya
Das,
Siris
Patel
∗VeerSurendraSaiMedicalCollege,Burla,India
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received21October2014 Accepted11December2014 Availableonline12May2015
Introduction
Ithasrecentlybeenestimatedthateachyear,morethanseven millionbabies worldwideare born witheitheracongenital abnormalityor ageneticdisease.1 Hemoglobinopathiesare
thecommonestautosomalhereditarydisordersandpresenta majorpublichealthprobleminIndia.Theoverallprevalence ofthe-thalassemiatraitis2.78%butthisvariesfrom1.48 to3.64%indifferentstatesofIndia2comparedtothecarrier
frequencyinBrazil(1%).3Recentlywereportedthatthecarrier
frequencyofthe-thalassemiagenewiththeIVS1-5(G→C)
mutation in western districts of Odisha, India is 3.75%.4,5
In India, the IVS1-5 (G→C) mutation isthe mostcommon
-thalassemiamutation.However,theIVS1-5(G→C)mutation
alongwithothermutations,includingIVS1-1(G→T),Cd41/42
(-TCTT),Cd 8/9and a619 basepair deletion, accounts for >90%ofmutationscausing-thalassemia.InBrazil,theCd39 (C→T)mutationisthemostprevalentcauseof-thalassemia
∗ Correspondingauthorat:Qr.3R/27,DoctorsColony,768017Burla,Dist.Sambalpur,Odisha,India.
E-mailaddress:[email protected](S.Patel).
1 Bothauthorssharefirstauthorship.
followedbytheIVSI-6(T→C),andIVSI-110(G→A)mutations.6
Amapshowingzonaldistributionof-thalassemiamutations inIndiaisdepictedinFigure1.
TheVeerSurendraSaiMedicalCollegeandHospital,Burla, Odisha isatertiary carereferral hospitalcateringfor west-ernOdishaaswellaseasterndistrictsofChhattisgarhstate. Under acomprehensivesicklecellcareprogram,wescreen outpatientandhospitalizedcasesintheInstituteforSickle CellDiseaseandotherhemoglobinopathiesattheSickleCell ClinicandMolecularBiologyLaboratory.
Compound heterozygotes for -thalassemia and struc-turalhemoglobin(Hb)variantsusuallypresentwithasevere form of the disease. Here, we report a case, a com-pound heterozygotefor the-thalassemiamutationIVS1-5 (G→C)andIndiandeletion-inversionG␥(A␥␦)0-thalassemia
(HbVarID-1038) from thedistrictofBargarhinthestateof Odisha,India.ThemolecularstructureoftheIndian deletion-inversion[G␥(A␥␦)0-thal]iscausedbyamajorrearrangement
http://dx.doi.org/10.1016/j.bjhh.2014.12.002
North India
West India
Central India
East India
South India
IVSI-5(G>C) IVSI-1(G>T) Codon 8/9(+G) Codon 30(G>C) Codon 5(–CT) –88(C>T) –90(C>T) IVSI-1(G>A) –28(A>G)
619-bp deletion Codon 41/42(–TCTT) Codon 15(G>C) Cap site +1(A>C) Codon 16(–C) Codon 15(–T) Poly A site(T>C) IVSII-837(T>G) Others/unknown
Figure1–MapofIndiadepictingthezonaldistributionof-thalassemiamutations.
includinganinversionofthesequencebetweenthe3′endof
theA␥geneandtheIVS-IIregionofthe-globingene,followed bytwodeletions(total8.5kb)oftheflankingDNAsequence. ThisisthefirstcasereportofsuchacasefromeasternIndia.
Case
report
An18-year-oldfemalepatient(height147cmandweight40kg) belongingtothe‘Chasa’castewasadmittedtothe Depart-mentofMedicineofVeerSurendraSaiMedicalCollegeand Hospital,Burla,Odisha,Indiawithsevereanemia,but with nohistoryofanyothermedicalcomplaints.HerHblevelwas 2g/dLatthetimeofadmissiontothehospital.Aftera trans-fusionofthreebloodunits,theHblevelreached7.8g/dL.The erythrocytesedimentationratewas12mm/h.Other parame-tersofbloodindicesafterthebloodtransfusionwerewhite bloodcellcount(WBC):10.9×109cells/L;redbloodcells(RBC):
3.35×106/L;hematocrit(HCT):23.9%;meancorpuscular vol-ume (MCV): 71.7fL; mean cell hemoglobin (MCH): 20.6pg; meancellhemoglobinconcentration(MCHC):29.0g/dL;and
platelets (PLT): 149×109/L. Peripheral blood smear
exam-ination showed microcytic, hypochromic anemia, marked anisopoikilocytosis, many microcytes, few macrocytes and fragmented RBCs. Stainingby the Kleihauer–Betke method demonstrated pancellular distribution of fetal hemoglobin (HbF).Ultrasonographyexaminationrevealedhepatomegaly (16.5cm)andsplenomegaly(14.5cm).AnX-rayofchest(P-A view)andX-rayofbothhipjoints(A-Pview)showedno abnor-malities.
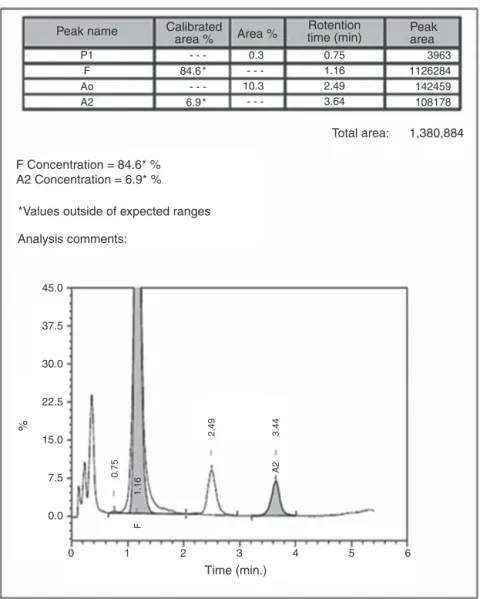
Automated high-performance liquid chromatography usingthe-ThalassemiaShortProgramonBio-RadVariant-II systemshowedvariousfractionsofHbwitharaisedlevelof HbF(84.6%)and anHb A2 concentrationof6.9%(Figure2).
Parents’ studiesrevealed that hermother hada highHb F level (13%)whereasherfatherhadhigh HbA2 level (5.2%).
All hematological parameters of the case and herparents are showninTable1. Screeningforthecommonmolecular determinants of raisedHb F, the Indian deletion-inversion G␥(A␥␦)0-thalassemia and hereditary persistence of fetal
0 0.0 7.5 15.0 22.5 30.0 37.5
%
45.0
*Values outside of expected ranges F Concentration = 84.6* % A2 Concentration = 6.9* %
Peak name Peak
area Calibrated
area %
Rotention time (min) Area %
Analysis comments:
Total area: 1,380,884
3963 1126284 142459 108178 0.75
1.16 2.49 3.64 P1
F Ao A2
0.3 -10.3
-84.6 * -6.9 *
1 2 3
2.49
0.75
1.16
F
A2
3.44
Time (min.)
4 5 6
Figure2–Highperformanceliquidchromatograminthecaseofcompoundheterozygousstatefor-thalassemiawith IVS1-5(G→C)mutationandIndiandeletion-inversionG␥(A␥␦)0thalassemia(chromatogramoneyearafterhospitalization
atfollow-up).
Common Indian -thalassemia mutations were confirmed by multiplex amplification refractory mutation system (ARMS)-PCR using a previously described protocol.4
More-over, alpha globin gene deletions (␣−3.7 and ␣−4.2) were
investigated by gap-PCR.8 The DNA study showed father
asheterozygousforthe IVS1-5(G→C)mutationandmother
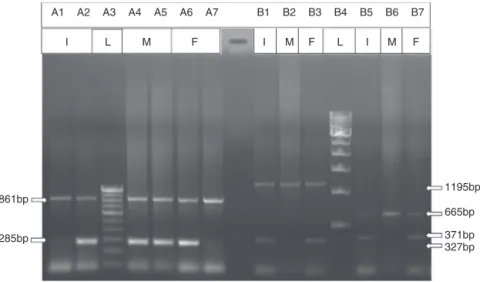
as a G␥(A␥␦)0-thalassemia carrier. The gel picture for
Indiandeletion-inversionG␥(A␥␦)0-thalassemiaisshownin
Figure3.Alpha-thalassemiawasnotobservedinanyofthem.
Writteninformedconsentwasobtainedfromthepatient alongwithherparentsandthestudywasapprovedby Institu-tionalEthicsCommitteeofVeerSurendraSaiMedicalCollege andHospital,Burla,Odisha,India.
Discussion
+-Thalassemiaisthesecondmostcommon hemoglobinopa-thy in our clinic’s population. We have reported earlier the clinical and molecular characteristics of G␥(A␥␦)0
-thalassemia in heterozygous as well as in compound
heterozygousstateswithHbS.InHbS/G␥(A␥␦)0-thalassemia
cases, the patients had repeated painful crises along with histories of blood transfusions.9 Indian deletion-inversion
G␥(A␥␦)0-thalassemia in the heterozygous form has been
reported in western India in the state of Maharashtra.10
Recently, Pandey et al. described seven cases with Indian deletion-inversion G␥(A␥␦)0-thalassemia, of which three
cases were co-inheritedwith -thalassemia withthe IVS1-5(G→C)mutation.11Thesethreecaseshadlowvaluesofboth
HbA2(3.2%,2.8%,and2.7%)andHbF(21.3%,10.7%and20.3%)
comparedtothe6.9%and84.6%ofHbA2andHbF,
respec-tivelyinthecurrentcase.Theirthreecasesweretransfusion dependentandshowedmoderateanemia.Ourcaseisthefirst caseofcompoundheterozygotestateof-thalassemiawith IVS1-5(G→C)mutationandG␥(A␥␦)0-thalassemiawithmild
A1 A2
861bp
285bp
1195bp
665bp
371bp 327bp A3 A4 A5 A6 A7 B1 B2 B3 B4 B5 B6 B7
I
I L M F M F L I M F
Figure3–Agarosegelelectrophoresisforthedetectionof-thalassemiawithIVS1-5(G→C)mutation(ARMSPCR)and
Indiandeletion-inversionG␥(A␥␦)0thalassemia(GAP-PCR).*I:index;M:mother;F:father;L:ladder.Detectionof
-thalassemiawithIVS1-5(G→C)mutationbyARMSPCR(LineA1,LineA4andLineA6fornormal-globingene,andLine
A2,LineA5andLineA7forIVS1-5[G→C]mutation).DetectionofIndiandeletion-inversionG␥(A␥␦)0thalassemiaby GAP-PCR(LineB1,LineB2andLineB3forBreakpoint-A,andLineB4,LineB5andLineB6forBreakpoint-B).
Hb Flevel (84.6%). Our observation ofhomogeneous Fcell distributionandanelevatedHbFlevelinperipheralblood, isinagreementtothe reportofWayeet al.inan African-American with compound heterozygosity of a Black form of(A␥␦)0-thalassemia and the
−29 (A→G) +-thalassemia mutation.12Weonlyinvestigatedtwotypesof␣-thalassemia
(␣−3.7and␣−4.2),themostprevalentinIndia.Anotherformof
␣-thalassemiamaybeafactorformilderclinicalpresentation ofthepatient.
Table1–Hematologicalinvestigationsinthecaseof
compoundheterozygousstatefor-thalassemiawith
IVS1-5(G→C)mutationandIndiandeletion-Inversion
G␥(A␥␦)0thalassemiawithherparents.
Parameters Index(follow-up) Oneyearafter hospitalization
Father Mother
Completebloodcount
WBC(103/L) 13 13.9 9.2
RBC(106/L) 4.81 3.87 3.67 Hemoglobin(g/dL) 8.0 8.3 7.8
HCT(%) 32.3 30.4 29.7
MCV(fL) 67.2 78.6 80.9
MCH(pg) 16.6 21.4 21.3
MCHC(g/dL) 24.8 27.3 26.3
Platelets(103/L) 125 312 188
FractionsofHbvariantsbyHPLC
HbA0(%) 10.3 83 76.2
HbA2(%) 6.9 5.2 2.8
HbF(%) 84.6 0.3 13
WBC:white blood cells; RBC: redblood cells; HCT: hematocrit; MCV: mean cell volume; MCH: mean cell hemoglobin; MCHC: meancellhemoglobinconcentration;HPLC:high-performance liq-uidchromatography.
The Indian subcontinent has a heterogeneous
popula-tionwithdifferenthemoglobinopathies.TheseHbdisorders shouldbeincludedintheprenataldiagnosisofpatientswith
severe or mild thalassemia. The characterization of these
hemoglobinopathies willfacilitateapreventionandcontrol programofhemoglobinopathiesincludingthalassemiainthis region.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest
Acknowledgments
Thisstudy wassupportedbyresearchfundingfrom Indian
CouncilofMedicalResearch(ICMR),NewDelhi,Departmentof ScienceandTechnology(DST),NewDelhi,andNationalHealth
Mission(NHM),Odisha.
r
e
f
e
r
e
n
c
e
s
1.ChristianskonAC,HowsonCP,ModellB.Themarchofdimes
globalreportonbirthdefects:thehiddentollofdyingand
disabledchildren.WhitePlains,NewYork:MarchofDimes;
2006.
2.MohantyD,ColahRB,GorakshakarAC,PatelRZ,MasterDC,
MahantaJ,etal.Prevalenceof-thalassemiaandother
haemoglobinopathiesinsixcitiesinIndia:amulticentre
study.JCommunityGenet.2013;4(1):33–42.
3.RamalhoAS,SilvaRB,TeixeiraRC,CompriMB.Hemoglobin
screening:responseofaBraziliancommunitytooptional
programs.CadSaudePublica.1999;15(3):591–5.
4.MeherS,PatelDK,PatelS,DehuryS,PurohitP,DasK.
EpidemiologyofbetathalassaemiatraitineasternIndia.
5. PurohitP,MashonRS,PatelS,DehuryS,PattanayakC,DasK,
etal.ClinicalandmolecularcharacterizationofHbHofuin
easternIndia.IntJLabHaematol.2014;36:71–6.
6. FonsecaSF,KerbauyJ,EscrivaoC,FigueiredoMS,CancadoR,
ArrudaVR,etal.Geneticanalysisofbeta-thalassemiamajor
andbeta-thalassemiaintermediainBrazil.Hemoglobin.
1998;22(3):197–207.
7. CraigJE,BarnetsonRA,PriorJ,RavenJL,TheinSL.Rapid
detectionofdeletionscausing␦thalassemiaandhereditary
persistenceoffetalhemoglobinbyenzymaticamplification.
Blood.1994;83(6):1673–82.
8. ChongSS,BoehmCD,HiggsDR,CuttingGR.Single-tube
multiplex-PCRscreenforcommondeletionaldeterminantsof
␣-thalassemia.Blood.2000;95(1):360–2.
9. PatelDK,PatelM,MashonRS,PatelS,DashPM,DasBS.
Clinical&molecularcharacterisationofS
G␥(A␥␦)0-thalassemiaineasternIndia.Hemoglobin.
2010;34(6):604–9.
10.NadkarniA,WadiaM,GorakshakarA,KiyamaR,ColahRB,
MohantyD.Molecularcharacterizationofdb-thalassemia
andhereditarypersistenceoffetalhemoglobinintheIndian
population.Hemoglobin.2008;32(5):425–33.
11.PandeyS,PandeyS,RanjanR,MishraR,SharmaM,SaxenaR.
PhenotypicheterogeneityofAsianIndianinversiondeletions
G␥(A␥␦)0breakpointAandbreakpointB.IndJClinBiochem.
2013;28(1):98–101.
12.WayeJS,EngB,ColemanMB,SteinbergMH,AlterBP.␦
thalassemiainanAfrican-American:identificationofthe
deletionendpointsandPCR-baseddiagnosis.Hemoglobin.