Setembro, 2015
Diogo Ruas Fernandes dos Reis
Licenciatura em Engenharia Química e BioquímicaDesenvolvimento, implementação e validação de
métodos cromatográficos
Dissertação para obtenção do Grau de Mestre em Engenharia Química e Bioquímica
Orientadora: Engª Paula Pereira, Responsável Laboratório de Controlo de Qualidade, Sapec Agro S.A
Júri:
Presidente: Prof. Mário Fernando José Eusébio, Professor Auxiliar
Vogais: Eng. José Fernando Trindade Neves, Director Industrial, Sapec Agro S.A.
Engª Paula Pereira, Responsável Laboratório de Controlo de Qualidade, Sapec Agro S.A
Desenvolvimento, implementação e validação de métodos cromatográficos
Copyright © Diogo Ruas Fernandes dos Reis, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa.
v
Agradecimentos
Começo por agradecer à Sapec Agro pela oportunidade de estagiar no laboratório de controlo de qualidade.
À engenheira Paula Pereira por toda a simpatia e constante disponibilidade.
Agradecer também à Isabela Silva por todo o conhecimento transmitido durante os seis meses de estágio.
A todos os analistas e restantes funcionários que tive o prazer de conhecer, que se mostraram sempre disponíveis para qualquer dúvida que pudesse ter.
À professora Isabel Coelhoso.
Uma referência a todos os colegas de curso que tive o prazer de conhecer durante este período.
vii
Resumo
O presente trabalho, decorreu no Laboratório de controlo de qualidade da Sapec Agro e teve como objetivo a validação de três métodos analíticos. Para avaliar a validade de um método é necessário recorrer à análise dos parâmetros de validação
O primeiro trabalho consistiu num método para a quantificação de impurezas em A por cromatografia líquida de alta eficiência (HPLC). Os resultados obtidos para os parâmetros avaliados (seletividade, gama de trabalho, precisão e exatidão), estão dentro dos critérios estabelecidos. Concluiu-se que o método de ensaio é válido, porém.o seu uso em rotina está dependente de estudos de reprodutibilidade.
O segundo trabalho trata-se da validação de um método para a quantificação de B em C por analisador de energia térmica (TEA). Os parâmetros de validação avaliados foram: a gama de trabalho, precisão e exatidão. Os resultados obtidos estão dentro dos critérios estabelecidos, conclui-se que o método é válido.
O terceiro trabalho consistiu na avaliação de desempenho de dois HPLC de marcas diferentes (Waters e Agilent) com o objetivo de validar um método acreditado no cromatógrafo Waters. Concluiu-se que os cromatógrafos têm desempenhos semelhantes.
ix
Abstract
The present work, took place at Sapec Agro’s Quality Control Laboratory and aimed to
validate three analytical methods scope of the present work is to validate three analytical methods. To assess the validity of a method is necessary to analyze the validation parameters
The first assay relates to a method to quantify impurities in A by high performance liquid chromatography (HPLC). The results of the evaluated parameters (selectivity, range of work, precision and accuracy), are in agree with the acceptance criteria. It was concluded that the test method is valid, its use in routine although is dependent reproducibility studies.
The second assay it’s the validation method for the quantitation of B in C by thermal energy analyzer (TEA). The evaluated validation parameters were: the working range, precision and accuracy. The results obtained are within the established criteria, it is concluded that the method is valid.
The third assay aimed to evaluate the performance of two different brands of HPLC (Waters and Agilent) in order to validate a method believed the chromatograph Waters. It was concluded that chromatographers have similar performances
xi
Índice
AGRADECIMENTOS ...V RESUMO ... VII ABSTRACT ... IX ÍNDICE ... XI
1. INTRODUÇÃO ... 1
1.1. GRUPO SAPEC ... 1
1.2FITOQUÍMICA ... 2
1.3. VALIDAÇÃO DE MÉTODOS ANALÍTICOS ... 4
1.3.1 Seletividade/Especificidade[3] ... 4
1.3.2. Gama de trabalho e linearidade ... 4
1.3.3. Limites de Sensibilidade: limites de deteção e de quantificação[5] ... 8
1.3.4. Precisão ... 9
1.3.5. Exatidão[5] ... 9
1.4. CROMATOGRAFIA LÍQUIDA DE ALTA EFICIÊNCIA (HPLC) ... 10
1.4.1. Sistema de bombagem ...11
1.4.2 Injetor ...11
1.4.3 Coluna ...12
1.4.4. Detetor ...13
1.5ANALISADOR DE ENERGIA TÉRMICA (TEA)–DETEÇÃO DE NITROSAMINAS[7] ... 14
1.6. DEFINIÇÃO DE OBJETIVOS ... 16
2.MATERIAIS UTILIZADOS ... 17
2.1.REAGENTES ... 17
2.2. MATERIAL ... 18
3. VALIDAÇÃO DE UM MÉTODO PARA A QUANTIFICAÇÃO DE IMPUREZAS EM A 19 3.1.PROCEDIMENTO EXPERIMENTAL ... 19
3.2. RESULTADOS E DISCUSSÃO ... 22
3.2.1. Estudo da seletividade ...22
3.2.2. Gama de trabalho ...23
3.3. PRECISÃO ... 25
3.4. EXATIDÃO ... 27
4. VALIDAÇÃO DE UM MÉTODO PARA A QUANTIFICAÇÃO DE B EM C ... 29
4.1.PROCEDIMENTO ... 29
xii
4.1.2. Preparação de amostras ...30
4.1.3. Preparação do meio reacional ...30
4.2. RESULTADOS E DISCUSSÃO ... 31
4.2.1. Gama de trabalho ...31
4.3. PRECISÃO ... 32
4.4. EXATIDÃO ... 33
5. ESTUDO DE VALIDAÇÃO COM VISTA À PASSAGEM DE MÉTODOS ACREDITADOS PARA UM CROMATÓGRAFO WATERS ... 35
5.1.PROCEDIMENTO ... 35
5.1.1. Preparação de soluções padrão de calibração/controlo ...36
5.1.2. Preparação de amostras ...37
5.1.3. Condições Operatórias...38
5.2. RESULTADOS E DISCUSSÃO ... 39
5.2.1. Gama de trabalho ...39
5.2.2. Limites de Cálculo ...40
5.2.1. Exatidão ...41
5.2.2. Precisão ...42
6. CONCLUSÕES ... 43
7. BIBLIOGRAFIA ... 45
8. ANEXOS ... 47
1
1.
Introdução
1.1.
Grupo Sapec
O presente trabalho esteve englobado num estágio de seis meses efetuado no laboratório de controlo de qualidade da Sapec Agro.
O Grupo Sapec, fundado em 1926, começõu por explorar as minas de pirite, mais tarde desenvolveu as suas actividades na produção e comercialização de produtos para a agricultura, tais como: produção e comercialização de adubos, de agroquímicos, de sementes e rações para animais. Atualmente, o Grupo Sapec gere os seguintes negócios: produção para a agricultura, distribuição de produtos químicos e tratamento de resíduos industriais, logística, distribuição e trading de agro-alimentares e a produção de energia renovável. [1]
2
1.2 Fitoquímica
Fitoquímica, ou química das plantas, é um ramo da química orgânica de extrema importância na identificação de compostos químicos sintetizados pelas plantas, muitos deles com a função de as proteger das mais diversas ameaças ambientais.
A indústria química desenvolveu produtos, conhecidos por pesticidas, que fossem constiuídos por compostos fitoquímicos na sua matriz, com a finalidade de os aplicar em plantações agrícolas e obter um maior rendimento das mesmas.
No laboratório de controlo e qualidade da Sapec Agro são efetuadas análises ao teor de matéria ativa dos produtos formulados na fábrica, que carecem de extrema importância, pois são produtos que são maioritariamente aplicados em solos agrícolas e caso não se encontrem dentro dos limites aceitáveis, poderão implicar problemas a nível ambiental.
3 Tabela 1 - Matérias ativas referidas no trabalho
Matéria ativa
(A)
Herbicida pós emergente Previne o crescimento de ervas
daninhas,por inibição da enzima acetalatosintase responsável pela
divisão celular nas plantas.
(C)
Herbicida pré emergente Não seletivo
Controla o aparecimento de uma vasta gama de ervas e plantas. Herbicida mais vendido a nível
mundial.
(G) Herbicida pré e/ou pós emergente
Seletivo
(I)
Fungicida Controla a ação do fungo Phytophthora infestans, responsável
pelo míldio-da-batateira
(J)
Inseticida
4
1.3.
Validação de métodos analíticos
A validação de um método analítco consiste em garantir que os resultados obtidos após o mesmo ser aplicado em análises de rotina são confiáveis. Todo o processo de validação de um método analítico passa por diferentes etapas.
A primeira fase é necessário definir qual será o âmbito do método que se pretende validar[3]:
Quais os compostos a analisar?
Que equipamento utilizar? Quais as condições operatórias? Quais os reagentes a utilizar?
Após a etapa de desenvolvimetnto, é necessário avaliar um conjunto de parâmetros que permitam avaliar o desempenho do método.
1.3.1 Seletividade/Especificidade[3]
Os termos seletividade e especificidade estão geralmente interligados. A especificidade de um método analítico, consiste na capacidade do mesmo emitir uma resposta para um único analito; enquanto que seletividade refere-se à capacidade de um método identificar com exatidão um analito, mesmo na presença de componentes secundários como por exemplo, precursores sintéticos ou enantiómeros.
1.3.2. Gama de trabalho e linearidade
Antes de iniciar um processo de validação, é necessário definir uma gama de trabalho. A gama de trabalho consiste no intervalo de concentrações, para o qual o método terá capacidade de garantir com eficácia, a quantificação de um analito.
A linearidade de um método cromatográfico, pode ser descrita como a existência de proporcionalidade entre o sinal experimental e a concentração de analito numa amostra, numa gama de trabalho estabelecida [4]. Avalia-se a linearidade de um método através da quantificação de um conjunto de padrões de calibração, a fim de se obter uma reta de calibração.
5
1.3.2.1. Coeficiente de correlação
O coeficiente de correlação (R) é um parâmetro que demonstra o ajuste da reta de calibração aos resultados experimentais obtidos.
Equação 1
Cada reta de calibração é formada por N conjuntos de pares ordenados (x,y): xi representa a concentração dos padrões utilizados, enquanto yi a intensidade do sinal experimental
Este parâmetro, porém, não é conclusivo quanto à linearidade do método, sendo apenas um indicador do grau de ajustamento dos resultados. Num processo de validação, as retas de calibração devem ter valores de coeficientes de correlação superiores a 0,995.
1.3.2.2. Análise de resíduos
A análise dos resíduos, representa o desvio que os valores experimentais apresentam em relação aos esperados.
Equação 2
6
1.3.2.3. Teste de Mandel
O teste de Mandel avalia a linearidade tendo em consideração o ajuste linear como o ajuste polinomial dos dados experimentais.
Equação 3
Equação 4
S(y/x) e S(y/x)2º representam o desvio padrão referente ao ajuste linear e polinomial, respetivamente. As constantes a, b e c referem-se às constantes de grau zero, um e dois.
Equação 5
Equação 6
7
1.3.2.4. Teste de Rikilt
Outra via usada na avaliação da linearidade de um método é o teste de Rikilt.
Para tal determinou-se o fator de resposta, FR (y/x), para cada par ordenado da reta de calibração que corresponde ao quociente entre o sinal instrumental e a sua concentração correspondente.
Equação 7
Em que i corresponde a cada ponto da reta de calibração. Posteriormente calculou-se o fator de resposta médio.
Equação 8
Onde n representa o número de padrões de calibração. Para uma linearidade perfeita, cada um dos fatores de resposta individuais teriam de ser iguais ao fator de resposta médio, ou seja, a razão percentual ser igual a 100%; para admitir linearidade à reta definiu-se que os valores não devem apresentar um desvio superior a 10%, isto é, terão de estar compreendidos entre 90% e 110%.
1.3.2.5. Homogeneidade de variâncias[5]
O ajuste da gama de trabalho pode ser avaliado através do teste de homogeneidade de variâncias.
Este teste consiste na determinação das variâncias associadas ao primeiro (i=1) e último padrão (i=N) da gama de trabalho.
8 Sendo i o número do padrão; j o número de repetições máximo para cada padrão; a média do sinal experimental de cada solução padrão; ni o número de réplicas por padrão.
Para verificar se existem diferenças significativas entre as variâncias nos limites da gama de trabalho, procedeu-se ao cálculo do valor teste PG.
, quando
, quando
Compara-se o valor de PG com o valor tabelado da distribuição de Fisher/Snedecor. Se PG≤F(n1-1 ,nN-1, 99%), as diferenças de variâncias não são significativas e a gama de trabalho bem ajustada.
Se PG>F(n1-1 ,nN-1, 99%), as diferenças de variâncias são significativas, nesta situação a solução passa por reduzir a gama de trabalho.
1.3.3. Limites de Sensibilidade: limites de deteção e de
quantificação[5]
O limite de deteção é a menor concentração de um analito que pode ser detetada numa amostra, mas não necessariamente quantificada.
O limite de quantificação, refere-se à menor concentração de um analito capaz de ser quantificada com um grau de precisão e exatidão aceitável.
Equação 10
Equação 11
9
1.3.4. Precisão
Esta análise tem como finalidade a observação da variabilidade de uma medição, num curto espaço de tempo quando sujeita às mesmas condições operatórias.
O limite de repetibilidade (r) é um parâmetro que permite avaliar a repetibilidade de um método, é o valor abaixo do qual se deve situar a diferença absoluta entre dois resultados do ensaio[5].
Equação 12
Sendo t o valor da distribuição t -Student (1,96) para um nível de confiança de 95%; e S o desvio padrão.
Outro método de verificar a repetibilidade é através do cálculo do coeficiente de variação.
Equação 13
Estabeleceu-se como critério de aceitação que o valor do coefiente de variação deve ser inferior a 10%.
1.3.5. Exatidão[5]
A exatidão é definida como a concordância entre um resultado de um ensaio e o valor de referência considerado como verdadeiro. No presente trabalho este parâmetro foi analisado, através de ensaios de recuperação, que consiste na adição de um padrão de calibração com uma concentração conhecida (reforço) a uma amostra.
Equação 14
10 recuperação aproximadamente de 100%. Considerou-se, como critério de aceitação, recuperações que apresentassem desvios entre 90%-110% em relação ao valor expectável.
1.4.
Cromatografia líquida de alta eficiência
(HPLC)
A cromatografia foi definida pela primeira vez no início do século XX devido ao trabalho do botânico russo, Mikhail S. Tswett. O seu trabalho visionário consistia na separação de compostos, extraídos das plantas, com o auxílio de um solvente (fase móvel) e uma coluna com um determinado enchimento (fase estacionária)[6]. Desde então têm sido desenvolvidas várias técnicas cromatográficas, que permitam analisar e quantificar com sucesso um maior número de compostos.
A cromatografia líquida de alta eficiência é uma das mais importantes técnicas analíticas usadas atualmente, permitindo realizar análises qualitativas e quantitativas de compostos orgânicos em misturas multicomponente.Consiste num sistema completamente automatizado constituído, por uma coluna de cromatografia (fase estacionária), um sistema de bombagem, um injetor e um detetor.[3]
11
1.4.1.
Sistema de bombagem
O sistema de bombagem de um HPLC deve ser capaz de fazer chegar a fase móvel a todo o equipamento. Pode gerar pressões acima dos 6000 psi, com caudais que poderão variar dos 0.1 mL/min até aos 10 mL/min [7].
A eluição da fase móvel num HPLC pode ser feita de uma de duas maneiras: eluição isocrática, quando a composição da fase móvel se mantém constante ao longo do processo de separação; ou eluição por gradiente caso exista variação da composição da fase móvel.
1.4.2 Injetor
A fase da injeção representa o momento em que a amostra que se deseja analisar é introduzida na fase móvel do sistema. Os cromatógrafos usados em HPLC, em geral, utilizam
injetores em “loop”. Este sistema é de uma elevada eficiência, pois garante uma boa
reprodutibilidade, isto é, o volume de injeção praticamente não sofre alterações entre injeções. [7].
Figura 2 – Esquema de um injetor em “loop”[8]
Quando se encontra na posição “load”, a seringa lava e enche o “loop” com a amostra a pressão atmosférica; quando a válvula é rodada para a posição “inject” o conteúdo do “loop” é
12
1.4.3 Coluna
A coluna cromatográfica, ou fase estacionária, é o local onde efetivamente ocorre a separação de uma amostra.
As colunas utilizadas em HPLC, em geral, são constituídas por aço inoxidável e um enchimento composto por partículas de sílica. Apresentam comprimentos que poderão variar entre os 10 e 30cm e diâmetros internos de 2 a 5mm. Recorrentemente, a coluna principal encontra-se protegida por uma pré-coluna, cuja função é adsorver impurezas que possam existir em amostras e/ou solventes.[7]
Devido às diferentes afinidades eletrostáticas entre as moléculas da amostra e as que constituem o enchimento da coluna, irá ocorrer uma separação entre os diferentes componentes da amostra no interior da coluna.
13
1.4.4.
Detetor
Após a fase móvel percorrer todo o comprimento da coluna, irá passar por um detetor. O cromatograma gerado a partir dos sinais elétricos provenientes do detetor, permitirá quantificar um analito presente numa amostra, por integração da área do pico correspondente.
Existem vários tipos de detetores usados em cromatografia, porém o mais utilizado em cromatografia líquida é o detetor de díodos (DAD).
Uma lâmpada de tungsténio emite uma radiação visível que ao entrar numa lâmpada de deutério, passa a ter radiação ultravioleta (além da visível). Esta radiação policromática entra numa célula (flow cell), resultando na separação da radiação em vários comprimentos de onda, cujas intensidades são quantificadas por um conjunto de fotodíodos. Uma das vantagens no uso de um DAD está no facto de se poder analisar uma amostra a vários comprimentos de onda, simultâneamante.[7]
14
1.5 Analisador de Energia Térmica (TEA)
–
Deteção de Nitrosaminas[7]
O analisador de energia térmica, é um equipamento que permite identificar e quantificar compostos que contenham azoto, após estes serem submetidos a uma reação de pirólise a temperatura controlada, com vista à produção de monóxido de azoto (NO). O gás produzido é então conduzido para um compartimento arrefecido onde irá reagir com ozono, levando à formação de dióxido de azoto em estado excitado (NO2*). Este composto intermediário sofre um decaimento até ao seu estado fundamental por quimioluminescência, emitindo uma radiação num comprimento de onda característico que após ser filtrada, passa por um detetor. A intensidade do sinal experimental é proporcional à concentração de composto nitroso existente na amostra.
15 Figura 6 - Reações que ocorrem no interior do TEA[8]
Ao invés de existir uma reação de pirólise de modo a libertar o grupo N=O da nitrosamina, esta reação poderá ser desempenhada através de uma reação química. A libertação de monóxido de azoto ocorre através de uma reação de refluxo num meio reacional constituído por ácido bromídrico (HBr) em ácido acético. A amostra é injetada manualmente no meio reacional levando à formação de monóxido de azoto, de uma amina secundária e uma molécula de bromo. O monóxido de carbono é conduzido por uma corrente gasosa, para o interior do analisador de energia térmica. As reações que ocorrem tanto no meio reacional como no equipamento estão representadas em seguida.
16
1.6.
Definição de objetivos
No primeiro trabalho pretende-se validar um método no cromatógrafo Waters, cuja finalidade é a de identificar e quantificar as impurezas, sintetizadas durante a formulação do produto técnico A. A avaliação do desempenho do método terá em consideração a análise de parâmetros de validação como: seletividade, linearidade, estudos de precisão e ensaios de recuperações.
Em seguida, realizar-se-à a validação de um método com vista à quantificação de B, uma impureza sintetizada durante a formulação do produto técnico C, utilizando um analisador de energia térmica. Os parâmetros avaliados são idênticos aos do primeiro trabalho.
17
2.Materiais utilizados
2.1. Reagentes
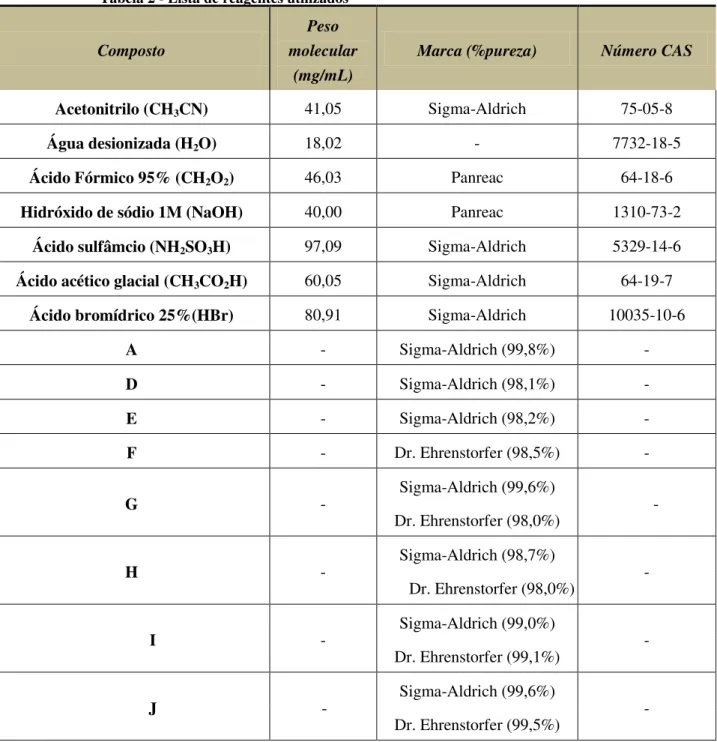
Tabela 2 - Lista de reagentes utilizados
Composto
Peso molecular
(mg/mL)
Marca (%pureza) Número CAS
Acetonitrilo (CH3CN) 41,05 Sigma-Aldrich 75-05-8
Água desionizada (H2O) 18,02 - 7732-18-5
Ácido Fórmico 95% (CH2O2) 46,03 Panreac 64-18-6
Hidróxido de sódio 1M (NaOH) 40,00 Panreac 1310-73-2
Ácido sulfâmcio (NH2SO3H) 97,09 Sigma-Aldrich 5329-14-6 Ácido acético glacial (CH3CO2H) 60,05 Sigma-Aldrich 64-19-7
Ácido bromídrico 25%(HBr) 80,91 Sigma-Aldrich 10035-10-6
A - Sigma-Aldrich (99,8%) -
D - Sigma-Aldrich (98,1%) -
E - Sigma-Aldrich (98,2%) -
F - Dr. Ehrenstorfer (98,5%) -
G - Sigma-Aldrich (99,6%) -
Dr. Ehrenstorfer (98,0%)
H - Sigma-Aldrich (98,7%) -
Dr. Ehrenstorfer (98,0%)
I - Sigma-Aldrich (99,0%) -
Dr. Ehrenstorfer (99,1%)
J - Sigma-Aldrich (99,6%) -
18
2.2.
Material
Proceder-se-à a uma enumeração do material de laboratório utilizado nos diversos estudos: balões volumétricos (5mL, 10mL, 20mL, 25mL, 50mL, 100mL, 250mL, 500mL); micropipeta; pipeta graduada (5mL); vials, copos de precipitação, condensador, balão de fluxo redondo, esferas de ebulição, seringa (10μL); coluna de cromatografia Zorbax SB-C3 250x4,6nm 5μm, Zorbax 4.6x100mm, Agilent rapid resolution 3,5mm
19
3.
Validação de um método para a
quantificação de impurezas em A
3.1. Procedimento experimental
Dividiu-se o procedimento de validação deste método em três fases distintas.
Numa primeira fase, analisou-se seletividade do método; em seguida avaliou-se o ajuste da gama de trabalho e estudou-se a precisão em condições de repetibilidade; por fim avaliou-se a exatidão do método, recorrendo a ensaios de recuperação.
Prepararam-se soluções padrão primárias de 25 mL para cada analito, dissolveram-se 19,8 mg, 20,3 mg e 20,7 mg de A, impureza 1 e impureza 2, respetivamente, em acetonitrilo. As três soluções foram introduzidas num banho de ultra-sons, de modo a garantir a dissolução completa dos analitos.
Prepararam-se soluções padrão intermédias para cada um dos analitos, pipetando 0,5mL da solução primária para um balão de 25mL, aferindo com acetonitrilo.
Procedeu-se à preparação de uma solução padrão conjunta, pipetando 0,5mL de cada uma das soluções intermédias para um único vial.
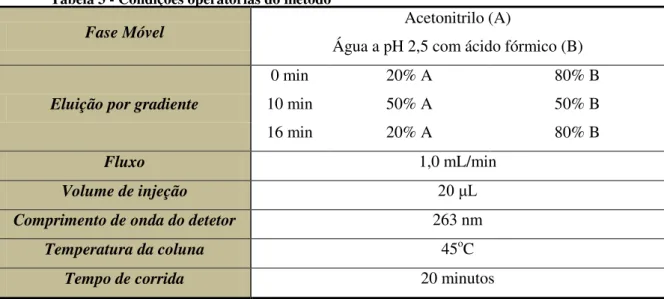
As condições operatórias do cromatógrafo estão descritas na Tabela 2.
Tabela 3 - Condições operatórias do método
Fase Móvel Acetonitrilo (A)
Água a pH 2,5 com ácido fórmico (B)
Eluição por gradiente
0 min 20% A 80% B
10 min 50% A 50% B
16 min 20% A 80% B
Fluxo 1,0 mL/min
Volume de injeção 20 μL
Comprimento de onda do detetor 263 nm
Temperatura da coluna 45oC
20 Na segunda fase do estudo, foram preparadas soluções padrão primárias, pesaram-se 54,0mg e 78,8 mg de impureza 1 e impureza 2, respetivamente, em balões de 10 mL usando acetonitrilo como solvente.
A solução padrão conjunta foi preparada pipetando 1,43mL e 2,48mL das soluções primárias de impureza 1 e impureza 2, respetivamente, para um balão de 20mL aferindo o volume com uma mistura de acetonitrilo e água a pH 2,5 com ácido fórmico (20:80).
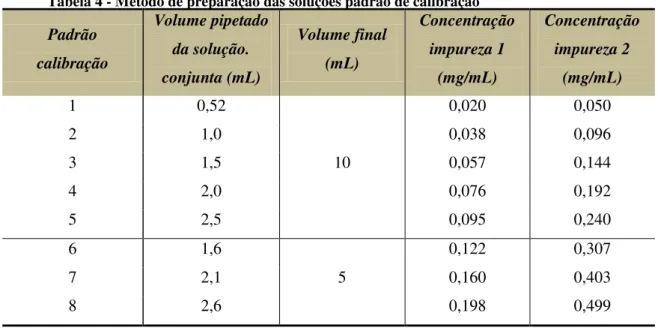
Procedeu-se à preparação das soluções padrão de calibração, como demonstrado na Tabela 3.
Tabela 4 - Método de preparação das soluções padrão de calibração
Padrão calibração Volume pipetado da solução. conjunta (mL) Volume final (mL) Concentração impureza 1 (mg/mL) Concentração impureza 2 (mg/mL)
1 0,52
10
0,020 0,050
2 1,0 0,038 0,096
3 1,5 0,057 0,144
4 2,0 0,076 0,192
5 2,5 0,095 0,240
6 1,6
5
0,122 0,307
7 2,1 0,160 0,403
21 Após estudada a linearidade e a precisão do método, procedeu-se à análise da exatidão do mesmo, para tal foram feitos ensaios de recuperação.
Pesou-se 1029,2 mg de uma amostra real de A para um balão de 50mL, designou-se esta solução de branco. De seguida prepararam-se os ensaios de recuperação, como descrito na tabela 4 da seguinte forma:
Tabela 5 - Método de preparação dos ensaios de recuperação
Ensaio de recuperação Volume adicionado da sol. branco (mL)
Volume adicionado
da sol. conjunta (mL) Volume final (mL) Nível 1
5
0,52
10
Nível 2 1,0
Nível 3 2,5
Na preparação destas soluções usou-se como solvente uma mistura de acetonitrilo e água a pH 2,5 com ácido fórmico (20:80).
22
3.2.
Resultados e discussão
3.2.1. Estudo da seletividade
Após a conclusão da sequência de trabalho para o primeiro plano de trabalho, procedeu-se à análise dos cromatogramas obtidos, de modo a aferir a seletividade do método.
Figura 8 - Representação dos cromatogramas correspondentes às soluções intermédias da impureza 1 (a), impureza 2 (b), A (c) e solução conjunta (d)
Os picos obtidos para cada analito encontram-se bem resolvidos, sendo possível identificá-los com exatidão. Observa-se que os tempos de retenção para cada analito encontram-se bem definidos:
Impureza 1: 3,5 minutos Impureza 2: 3,9 minutos A: 12,3 minutos
23
3.2.2. Gama de trabalho
3.2.2.1. Parâmetros de Calibração
A gama de trabalho que se pretende validar é de 0,020 a 0,198 mg/mL para a impureza 1 e de 0,050 a 0,499 mg/mL para a impureza 2. Os parâmetros de calibração obtidos encontram-se representados na Tabela 5.
Tabela 6 - Parâmetros de calibração
Parâmetro Impureza 1 Impureza 2 Impureza 2 (corrigida)
Nº padrões 8 8 6
Declive 13051,4 55375,7 63257,2
Ordenada na origem -2,8 1671,6 488,9
Coeficiente de correlação (R) 0,9999 0,9907 0,9992
Limite de Quantificação (mg/mL) 0,0080 0,1617 0,0424
O coeficiente de correlação para a reta de calibração da impureza 1 encontra-se dentro dos critérios de aceitação estabelecidos (>0,995); para a impureza 2 este critério não é atingido.
Analisando o limite de quantificação para as duas retas, constata-se que para a impureza 2, este valor encontra-se acima do limite inferior da gama de trabalho (0,050mg/mL), que se pretende validar. O limite de quantificação tem de ser sempre inferior ao padrão de menor concentração, logo para no caso impureza 2 este critério não é verificado.
Optou-se então por reduzir a gama de trabalho da impureza 2 para apenas seis padrões de calibração; desta forma obteve-se um limite de quantificação dentro do critério definido, assim como um melhor ajuste da reta aos dados experimentais.
24 Gráfico 1 - Representação das retas de calibração para cada impureza
Todos os testes estatísticos aplicados aos resultados da impureza 2, têm por base os dados referentes à gama de trabalho corrigida.
3.2.2.2. Linearidade
Os testes aplicados com vista ao estudo da linearidade do método encontram-se sumarizados na Tabela 6.
Tabela 7 - Testes aplicados na avaliação da linearidade
Teste Impureza 1 Impureza 2
Análise de resíduos -2,0% a +1,6% -7,6% a +3,3%
Teste de Mandel (VT) 0,35 33,22
F (1, N-3, 99%) 16,26 34,12
Teste de Rikilt 96,5% a 101,2% 94,1% a 103,1%
Os resíduos não apresentam desvios significativos, isto é, estão dentro do limite de aceitação estabelecido para este parâmetro (±10%).
Aplicou-se o teste de Mandel aos dados experimentais, constata-se que os valores de VT são inferiores a F (1, N-3, 99%).
25
3.2.2.3.
Homogeneidade de variâncias
Avaliou-se a homogeneidade de variâncias, injetando cinco vezes os padrões de calibração dos extremos da reta de calibração de cada impureza. Como já referido, a homogeneidade de variâncias conclui se a gama de trabalho está bem ajustada caso o valor de PG seja inferior ao valor do teste F para um nível de confiança de 99%. Consultando uma tabela de Fisher-Snedecor, para as condições impostas, o valor do teste é aproximadamente 15,98.
Os valores do teste PG foram de 8,22 e 15,10 para as impureza 1 e impureza 2, respetivamente. Os resultados obtidos confirmam o bom ajuste linear já evidenciado pelos testes determinados anteriormente.
3.3.
Precisão
Avaliou-se a precisão do método recorrendo ao cálculo do coeficiente de variação de repetibilidade. Fizeram-se cinco injeções (réplicas) consecutivas dos padrões de calibração correspondentes ao extremo inferior, extremo superior e a meio da gama de trabalho.
Tabela 8 - Resultados do estudo de precisão
Impureza 1 Impureza 2
Nº injeção Área P1 (mAU) Área P4 (mAU) Área P8 (mAU) Área P1 (mAU) Área P4 (mAU) Área P6 (mAU)
1 255,39 989,11 2576,73 3372,28 12865,62 19551,17
2 254,70 990,51 2574,42 3380,31 12894,67 19581,09
3 253,20 990,59 2576,25 3375,17 12892,01 19574,10
4 252,60 990,45 2573,22 3385,14 12898,29 19614,63
5 253,49 990,30 2568,56 3388,25 12898,75 19607,87
Limite de
repetibilidade (r) 3,26 1,75 9,36 19,04 39,56 73,98 |Xi-Xi-1|máximo 1,50 1,40 4,66 9,97 29,05 40,53
CV (%) 0,45 0,062 0,13 0,20 0,11 0,13
26 consecutivos apresentem uma diferença absoluta (|Xi-Xi-1|) inferior ao limite de repetibilidade. Comparando os valores de |Xi-Xi-1|máximo com o limite de repetibilidade, verifica-se
27
3.4.
Exatidão
O estudo da exatidão do método foi avaliado através de ensaios de recuperação. Os parâmetros de calibração usados neste plano de trabalho encontram-se sumarizados na Tabela 8.
Tabela 9 - Parâmetros de calibração (estudo de exatidão) Parâmetro Impureza 1 Impureza 2 Gama de trabalho
(mg/mL) 0,019 – 0,200 0,048 – 0,310
Declive 12596,71 61973,31
Ordenada na
origem 28,00 627,64
Coeficiente de
correlação 0,9992 0,9992
Após a calibração, é possível determinar os resultados que efetivamente permitem avaliar a exatidão do método. Os resultados obtidos estão representados na Tabela 9.
Tabela 10 - Resultados dos ensaios de recuperação
Impureza 1 Impureza 2
Concentração da
amostra real (mg/mL) 0.017 0,26
P1 (Nível I) P2 (Nível II) P5 (Nível III) P1 (Nível I) P2 (Nível II) P5 (Nível III) Concentração do
reforço (mg/mL) 0.019 0.038 0.095 0,048 0,096 0,24 Concentração amostra
reforçada (mg/mL) 0.038 0.056 0.115 0,299 0,344 0,467
Recuperação (%) 108,5% 102,2% 103,1% 90,1% - -
Decisão OK OK OK OK Diluir a
amostra
28 Para a impureza 1, determinou-se que a concentração presente na amostra é de 0,017 mg/mL. Idealmente, de modo a obter uma recuperação de 100%, a concentração da amostra reforçada teria de ser a soma entre a concentração da amostra real e a concentração do reforço. Constata-se que os três níveis de fortificação estão dentro do intervalo de aceitação (90%-100%).
29
4.
Validação de um método para a
quantificação de B em C
4.1. Procedimento
Este procedimento experimental foi repetido por três vezes. Na secção de resultados analisar-se-ão os resultados experimentais correspondentes às três sequências de trabalho.
4.1.1. Preparação de soluções
Começou-se por preparar as soluções padrão primárias, pesando cerca de 7mg do padrão F, para dois balões de 100mL aferindo com água desionizada.
Prepararam-se duas soluções intermédias (controlo e calibração), pipetando 5 mL da solução primária para um balão de 100mL aferindo com uma solução de hidróxido de sódio saturada em ácido sulfâmico (100mg/mL) (solvente).
Tabela 11 - Preparação de soluções padrão de calibração/controlo
Padrão Volume pipetado da solução. intermédia (mL) Volume final (mL) Concentração de NNG (mg/L)
Pad. Cal. 1 0,20
10
0,063
Pad. Cal. 2 0,25 0,079
Pad. Cal. 3 0,30 0,095
Pad. Cal. 4 0,40 0,126
Pad. Cal. 5 0,50 0,158
Pad. Cont. 1 0,20 0,063
Pad. Cont. 3 0,30 0,095
30
4.1.2. Preparação de amostras
Pesou-se cerca de 2000mg de uma amostra de C para um balão de 10mL, aferindo com solvente. Após passar pelo banho de ultra-sons, filtra-se a solução e pipeta-se 2,5mL para um balão de 5mL, aferindo com solvente.
O ensaio de recuperação preparou-se análogamente à solução anterior, com a diferença de se ter fortificado com 0,15mL da solução intermédia de calibração, equivalente ao padrão de calibração 3.
4.1.3. Preparação do meio reacional
Figura 9 - Meio reacional na deteção de nitrosaminas
Num balão de 250mL de fundo redondo com três pescoços (reator), colocou-se 100 mL de ácido acético, algumas esferas de ebulição e um agitador magnético. No pescoço central encontra-se o condensador principal, nos laterais coloca-encontra-se o tubo de carga (adição de HBr) e no outro um septo (injeção de padrões/amostras).
Usou-se um sistema de refrigeração, de forma a manter a temperatura no condensador a cerca de 6°C. Foi ligada à saída dondensador um tubo de lavagem de gases cheio com 100 mL de água, sendo este tubo de lavagem ligado ao analisador de energia térmica (TEA).
31 Após o equipamento estabilizar, pode-se dar início à injeção das amostras.
4.2.
Resultados e discussão
4.2.1. Gama de trabalho
A gama de trabalho que se pretende validar neste estudo, permite quantificar B para concentrações entre 0,063 a 0,16 ppm (μg/mL). Após injetados todos os padrões e amostras no meio reacional, obteve-se um cromatograma semelhante ao da Figura 8.
Figura 10 - Cromatograma de absorção parauma sequência de trabalho
Após injetados todos as soluções padrão, obtiveram-se as respetivas retas tas de calibração. Os parâmetros que permitem aferir o bom ajuste da reta aos resultados experimentais, encontram-se sumarizados na Tabela 11.
Tabela 12 – Parâmetros de calibração e limite de quantificação para cada sequência de trabalho.
Parâmetro 1ª Seq. 2ª Seq. 3ª Seq.
Declive 83517,9 70065,5 73147
Ordenada na origem (uV) 585,7 2209,1 1558,8
Coeficiente de correlação (R) 0,9984 0,9981 0,9967
32 Os coeficientes de correlação (R) obtidos, indicam que a reta se encontra bem ajustada, visto serem valores acima de 0,995; quanto ao limite de quantificação está igualmente dentro dos requisitos (inferior ao padrão de menor concentração – 0,063 ppm), não sendo necessário efetuar qualquer ajuste aos limites da gama de trabalho.
4.2.1.1. Linearidade
Os resultados dos testes usados na avaliação da linearidade, encontram-se sumarizados na Tabela 12.
Tabela 13 - Resultados dos testes para a avaliação da linearidade
Teste 1ª Seq. 2ª Seq. 3ª Seq.
Análise de resíduos -2,1% a 1,4% -1,8% - 2,5% -2,6% – 3,2%
Teste de Mandel (VT) -1,99
F (1, N-3, 99%) 98,50
Constata-se que os resíduos para as três sequências de trabalho não ultrapassam o critério imposto (±10%).
Pelo teste de Mandel, confirma-se a boa linearidade do método uma vez que o valor do teste (VT) é consideravelmente inferior a F (1, N-3, 99%).
4.3.
Precisão
A precisão do método foi estudada em condições de precisão intermédia, analisando as áreas obtidas dos padrões de controlo, para três sequências de trabalho realizadas em dias diferentes.
Tabela 14 - Resultados do estudo da precisão
Sequência de trabalho P1 (uV) P3 (uV) P5 (uV)
1ª 5931,7 8654,3 13432,5
2ª 6982,3 8924,6 13633,9
3ª 6122,4 9321,3 13947,1
33 Analisando os coeficientes de variação dos padrões de controlo, verifica-se que se encontram abaixo do critério de aceitação estabelecido (<10%), o que é tido como uma boa precisão.
4.4.
Exatidão
A exatidão do método estudou-se através de ensaios de recuperação. Os resultados obtidos apresentam-se na Tabela 14.
Tabela 15 - Resultados do estudo de exatidão
Seq. de trabalho
Concentração da amostra
(mg/L)
Concentração do reforço
(mg/L)
Concentração da amostra reforçada
(mg/L)
Recuperação (%)
1ª 0,1742
0,095
0,263 93,5
2ª 0,1551 0,246 96,0
3ª 0,1374 0,241 109,5
35
5.
Estudo de validação com vista à passagem
de métodos acreditados para um
cromatógrafo
Waters
5.1. Procedimento
Foram analisadas quatro matérias ativas neste trabalho: G, H, I e J. O seguinte procedimento foi aplicado a dois pares de matérias ativas, G/H e I/J. As mesmas amostras foram analisadas em ambos os cromatógrafos (Waters e Agilent).
A seguinte descrição refere-se ao par G/H, procedendo-se análogamente para os restantes pares.
36
5.1.1.
Preparação
de
soluções
padrão
de
calibração/controlo
Iniciou-se por preparar as soluções padrão primárias de G e H, uma de calibração e outra de controlo (4 soluções), pesando cerca de 20mg de padrão para um balão de 10mL aferindo com acetonitrilo.
37 A partir das soluções conjuntas, preparou-se as soluções padrão de calibração e de controlo, como descrito na Tabela 15
Tabela 16 - Preparação das soluções padrão de calibração/controlo
Padrão Volume pipetado da solução. intermédia (mL) Volume final (mL) Concentração estimada (mg/mL)
Pad. Cal. 1 1,5
10
0,0060
Pad. Cal. 2 1,8 0,0072
Pad. Cal. 3 2,2 0,0088
Pad. Cal. 4 2,5 0,0100
Pad. Cal. 5 2,8 0,0112
Pad. Cal. 6 3,2 0,0128
Pad. Cal. 7 3,5 0,0140
Pad. Cont. 1 1,5 0,0060
Pad. Cont. 4 2,5 0,0100
Pad. Cont. 7 3,5 0,0140
5.1.2. Preparação de amostras
As soluções de amostras reais usadas neste estudo foram preparadas da mesma forma como são preparadas pelos analistas do laboratório de controlo de qualidade, isto é, a partir do método acreditado pela norma ISO17025. O método de preparação é explicado em seguida.
.Para a amostra de H pesou-se 200,3mg de amostra num balão de 50mL e aferiu-se com acetonitrilo; após passar pelo banho ultra-sons, filtra-se a solução e pipeta-se 0,5mL para um balão de 50mL, aferindo com acetonitrilo:água+TFA(0,1%) (50:50).
Para a amostra de G pesou-se 280,6mg de amostra num balão de 100mL e aferiu-se com acetonitrilo; após passar pelo banho ultra-sons, filtra-se a solução e pipeta-se 0,5mL para um balão de 50mL, aferindo com acetonitrilo:água+TFA(0,1%) (50:50).
Para a amostra de I pesou-se 161,8mg de amostra num balão de 50mL e aferiu-se com acetonitrilo; após passar pelo banho ultra-sons, filtra-se a solução e pipetapipeta-se-se 1mL para um balão de 10mL, aferindo com acetonitrilo:água+TFA(0,1%) (50:50).
38 Para a solução de recuperação, foi usado um branco de matéria ativa, que consiste numa amostra do produto em análise sem matéria ativa. A solução da amostra de branco foi fortificada com o equivalente a um padrão de calibração 4 (0,0100 mg/mL).
5.1.3. Condições Operatórias
As condições operatórias dos cromatógrafos, estão representadas na tabela 16.
Tabela 17 - Condições operatórias implementadas
Fase Móvel Acetonitrilo (A)
Acetonitrilo:Água ultra pura com 0,1% TFA (v/v) (50:50) (B)
Eluição por gradiente
T (min) 0 5,0 11,1 15,0 %A 50 100 100 50 %B 50 0 0 50
Fluxo 0,5 mL/min
Volume de injeção 20 μL
Comprimento de onda do detetor
G: 225nm H: 285nm I: 242nm J: 230nm
Temperatura da coluna 30oC
Tempo de corrida 15 minutos
39
5.2.
Resultados e discussão
5.2.1. Gama de trabalho
Iniciou-se por estudar a gama de trabalho e dos seus parâmetros.
Tabela 18 - Tabela comparativa dos parâmetros/testes que avaliam a gama de trabalho Matéria Ativa
(Gama de trabalho) Parâmetro HPLC Agilent HPLC Waters Critérios de aceitação
G
(0,00613 - 0,01430 mg/mL)
R 0,9999 0,9998
R>0,995
CV<10%
Resíduos(±10%)
VT< F(5,4,99%) = 21,20
CV (%) 0,43% 0,68%
Resíduos (%) -0,8% – +1,0% -0,8 – +1,0 Teste Mandel
(VT) 0,07 2,63
H (0,00607 –
0,01415 mg/mL)
R 0,9998 0,9997
CV (%) 0,64% 0,78%
Resíduos (%) -0,6% a +0,9% -0,8% a +1,0% Teste Mandel
(VT) 1,70 2,27
I
(0,00603 – 0,01407 mg/mL)
R 0,9986 0,9985
CV (%) 1,67% 1,75%
Resíduos (%) -0,8% a +1,0% -0,8% a +1,0% Teste Mandel
(VT) 0,46 0,16
J (0,00616 – 0,01436 mg/mL)
R 1,0000 0,9998
CV (%) 0,30% 0,59%
Resíduos (%) -0,4% a +0,4% -0,7% a +0,7% Teste Mandel
40 Todos os parâmetros e testes estatísticos usados na avaliação da linearidade das retas de calibração das matérias ativas, em ambos os cromatógrafos, estão dentro dos critérios de aceitação.
5.2.2. Limites de Cálculo
Determinou-se o limite de quantificação para cada matéria ativa, com base nos resultados obtidos pelos cromatógrafos Agilent e Waters.
Tabela 19 - Tabela comparativa para o limite de quantificação Matéria Ativa
(Gama de trabalho)
HPLC Agilent
HPLC
Waters Critério de aceitação
G
(0,00613 - 0,01430 mg/mL) 0,00040 0,00069
LQ < Padrão de menor concentração H (0,00607 – 0,01415
mg/mL) 0,00066 0,00079 I
(0,00603 – 0,01407 mg/mL) 0,00168 0,00176 J (0,00616 – 0,01436 mg/mL) 0,00030 0,00061
41
5.2.1.
Exatidão
A exatidão do método foi avaliada através de ensaios de recuperação, os resultados apresentam-se na Tabela 19
Tabela 20 - Tabela comparativa entre os ensaios de recuperação
Matéria Ativa HPLC Agilent
HPLC
Waters Critério de aceitação
G 100,6% 101,1%
90% - 110%
H 101,0% 101,5%
I 107,1% 103,7%
J 100,1% 99,1%
42
5.2.2.
Precisão
A precisão foi analisada fazendo 6 injeções consecutivas nos dois cromatógrafos em simultâneo. Os resultados estão representados na Tabela 20.
Tabela 21 - Tabela comparativa para o estudo de precisão
Matéria Ativa Parâmetro HPLC Agilent HPLC Waters Critérios de aceitação G
Conc. Média (mg/mL) 0,009098 0,009093
Amplitude relativa < 5%
CV < 10% Amplitude relativa (%) 0,053%
CV (%) 0,217% 0,410%
H
Média (mg/mL) 0,01060 0,01067 Amplitude relativa (%) 0,67%
CV (%) 0,029% 0,099%
I
Média (mg/mL) 0,01279 0,01235 Amplitude relativa (%) 3,49%
CV (%) 0,100 0,142
J
Média (mg/mL) 0,005899 0,006030 Amplitude relativa (%) 2,19
CV (%) 0,153 0,403
Determinou-se a concentração média para as seis injeções. Analisando o coeficiente de variação obtido, verifica-se que não existem grandes variações entre cada injeção para ambos os cromatógrafos.
A amplitude relativa entre as médias das concentrações não excede o critério de aceitação imposto.
Equação 15
43
6.
Conclusões
Na validação do método para análise de impurezas do A em HPLC:
O método apresenta uma boa seletividade, uma vez que todos os analitos se encontram bem identificados nos cromatogramas, sendo um indício que as condições instrumentais aplicadas são ótimas para a análise destes compostos.
A gama de trabalho validada para ambas as impurezas apresenta um bom ajuste ao dados experimentais, o que traduz ao método uma boa linearidade.
Tendo em conta os valores do limite de repetibilidade (r) e do coeficiente de variação (CV), conclui-se que o método é preciso. Quanto à exatidão, os ensaios de recuperação confirmam a veracidade dos resultados.Para os parâmetros analisados, este método é considerado válido.
Na validação do método para análise de B em C,todos o parâmetros avaliados passaram nos critérios de aceitação. A gama de trabalho encontra-se bem ajustada; a precisão que foi estudada em condições de precisão intermédia apresenta baixos níveis de variabilidade.Tendo em conta os parâmetros avaliados para este método, considera-se o método válido.
A principal conclusão a reter do estudo de validação com vista à passagem de métodos acreditados para o Waters, consiste no facto de os dois equipamentos apresentarem desempenhos muito semelhantes em todos os parâmetros avaliados e para todas as matérias ativas estudadas.
As concentrações de matéria ativa presentes em amostras reais, apresentam diferenças mínimas entre os dois equipamentos.
45
7.
Bibliografia
[1] http://www.sapecagro.pt/internet/empresa/ (consultado em 26/11/2015)
[2]http://diarioagrario.blogspot.pt/2013/03/sapec-agro-apresentou-evolucao-de.html (consultado em 26/11/2015)
[3] Waksmundzka-Hajnos, Monika e Joseph Sherma. High performance liquid chromatography
in phytochemical analysis. CRC Press, 2011.
[4] Singh, Ranjit. “HPLC method development and validation- an overview.” Journal of
Pharmaceutical Education and Research (2013): Vol.1, 26-33.
[5] RELACRE. Guia RELACRE 13 - Validação de métodos internos de ensaio em análise
química. RELACRE
[6]Waters Corporation. Beginers Guide to liquid chromatography.
[7]Skoog, Douglas A., et al. Fundamentals of analytical chemistry. Brooks/Cole, 2004.
[8]Rodrigues, Alexandre M.; Formação em métodos cromatográficos, 2013
[9]http://www.teamcag.com/support/theory/chroma/hplc_bas_at/detectors/dadPrinciple.html
(consultado em 13 Julho de 2014)
[10] Landon, Mark, et al.;The Importance of the Analysis of Nitrosamines by a Thermal Energy
Analyser (TEA) and Their Link to Cancer
[11] Corradini, J.; Handbook of HPLC, CRC Press, 2010
[12] Haley, Elizabeth Ann (1997) The application of the thermal energy analyser to the analysis
47
8.
Anexos
8.1. Estudo de validação de um método para a
quantificação de impurezas em A
8.1.1. Estudo da gama de trabalho
Tabela 22 – Resultados para a impureza 1 e 2 Concentração
(mg/mL)
Média Área (mAU)
0,020 250,4
0,038 490,7
0,057 753,9
0,076 990,5
0,095 1236,5
0,122 1576,7
0,160 2100,2
0,198 2570,0
8.1.2. Linearidade
Tabela 23 – Análise de resíduos para a impureza 1
Concentração (mg/mL) Área de Pico (mAU)
Área de Pico
estimada (mAU) Resíduos
0,020 250,38200 255,41228 -2,0
0,038 490,68400 493,77415 -0,6
0,057 753,87050 742,06777 1,6
0,076 990,47500 990,36139 0,0
0,095 1236,51300 1238,65500 -0,2
0,122 1576,73750 1586,26607 -0,6
0,160 2100,18050 2082,85331 0,8
0,198 2569,98800 2579,44 -0,4
Concentração (mg/mL)
Média Área (mAU)
0,050 3369,8
0,096 6523,1
0,144 9907,3
0,192 12840,7
0,240 15702,9
0,307 19657,1
0,403 24210,7
48 Tabela 24 – Análise de resíduos para a impureza 2
Concentração (mg/mL) Área de Pico (mAU) Área de Pico
estimada (mAU) Resíduos
0,050 3369,81350 3645,15691 -7,6
0,096 6523,10300 6558,62886 -0,5
0,144 9907,32800 9593,49548 3,3
0,192 12840,67100 12628,36209 1,7
0,241 15702,93050 15663,22870 0,3
![Figura 1 – Esquema representativo de um HPLC [4]](https://thumb-eu.123doks.com/thumbv2/123dok_br/16695850.743817/20.892.288.703.668.979/figura-esquema-representativo-de-um-hplc.webp)
![Figura 2 – Esquema de um injetor em “loop”[8]](https://thumb-eu.123doks.com/thumbv2/123dok_br/16695850.743817/21.892.328.661.611.822/figura-esquema-de-um-injetor-em-loop.webp)
![Figura 3 – Exemplo de colunas de cromatografia usadas em HPLC (esquerda); imagem microsópica do enchimento.[5]](https://thumb-eu.123doks.com/thumbv2/123dok_br/16695850.743817/22.892.200.763.515.693/figura-exemplo-colunas-cromatografia-usadas-esquerda-microsópica-enchimento.webp)
![Figura 4 - Esquema representativo de um detetor de díodos[7]](https://thumb-eu.123doks.com/thumbv2/123dok_br/16695850.743817/23.892.201.753.522.757/figura-esquema-representativo-de-um-detetor-de-díodos.webp)

![Figura 7 - Reações que ocorrem no meio reacional [8]](https://thumb-eu.123doks.com/thumbv2/123dok_br/16695850.743817/25.892.339.649.136.397/figura-reações-que-ocorrem-no-meio-reacional.webp)