Modulation of HIV-1-host cell pathways : from virus entry to latency
241
0
0
Texto
(2) II.
(3) Ana Catarina Taborda Godinho dos Santos was a recipient of a PhD fellowship (SFRH/BD/81265/2011) from Fundação para a Ciência e Tecnologia (FCT), Lisboa, Portugal.. III.
(4) The results described in this thesis were included in manuscripts already published or in preparation, in peer-reviewed journals, and in posters in international and national scientific meetings:. Godinho-Santos A, Hance AJ, Gonçalves J and Mammano F. (2016) CIB1 and CIB2 are HIV-1 helper factors involved in viral entry. Sci. Rep. 6, 30927; doi: 10.1038/srep30927. Godinho-Santos A, Hance AJ, Gonçalves J and Mammano F. (2016) EZH2: helper or restriction factor? Manuscript in preparation.. Godinho-Santos A, Hance AJ, Gonçalves J and Mammano F. (2016) Characterization of the role of CIB1 and CIB2 as HIV‑1 helper factors. Frontiers of Retrovirology Conference 2016. Erlangen, Germany 12-14 September 2016 Retrovirology 2016, 13(Suppl 1): P5. Godinho-Santos A, Hance AJ, Gonçalves J and Mammano F. (2016) CIB1 and CIB2 are HIV-1 helper factors and their modulation influences envelope-mediated viral entry. 6th European Virology Conference and Expo. Madrid, Spain March 10-12, 2016 Virol-mycol 2016, 5:1 (Suppl) http://dx.doi.org/10.4172/2161-0517.C1.009. Godinho-Santos A, Hance AJ, Gonçalves J and Mammano F. (2015) CIB1 and CIB2 are HIV-1 helper factors and their modulation influences envelope-mediated viral entry. ePoster – 5th World Congress on Virology, Atlanta, USA. December 07-09, 2015 J Antivir Antiretrovir, 7:4. http://dx.doi.org/10.4172/1948-5964.C1.025_027. Brito P, Rato S, Santos AC, Resende L, Ramos A, Moita L, Ferreira J, Gonçalves J. (2011). DNA-PK, a cofactor in HIV-1 driven LTR-transcription. Cellular Microbiology and Pathogenesis S9:8 - MicroBiotec'11: Braga, Portugal. 1 - 3 December 2011. https://microbiotec11.wordpress.com/. IV.
(5) V.
(6) VI.
(7) “Under pressure, you don’t rise to the occasion, you sink to the level of your training. That’s why we train so hard.”. An anonymous US Navy SEAL. VII.
(8) VIII.
(9) Acknowledges Agradecimentos. Em primeiro lugar, ao Professor Doutor João Gonçalves, pela oportunidade de realizar o meu projeto de doutoramento no seu laboratório, pela confiança e pela orientação dada nos oito anos em que fiz parte da sua equipa de investigação. I deeply thank François Clavel, Fabrizio Mammano, Allan Hance and Sylvie Rato, without you this thesis would not be possible. To François, thank you for seeing me as an independent and wit person and for allowing me to stay in your Unit for so long time. To Fabrizio, thank you for all the support, the commitment and the trust you had in me all these months. To Allan, thank you for believing in me and in my abilities, and for making this believe "contagious". Your wisdom, your thoughts and your personality will never be forgotten and time to time I hope I can seek for more advices. To Sylvie, thank you for not only be part of my second family but also for helping me with this project. All the laughs and tears and drinks and foods we shared with the girls are some of my favorite moments. I would not be able to finish this thesis without your advices, corrections and suggestions, both in the bench and in the “paper”. Aos meus colegas de laboratório, em especial à Andreia e ao Luís, por me terem ajudado nas coisas mais básicas e existenciais possíveis nos primeiros anos. Ao André e às Catarinas, por me aturarem (muito) e por todo o apoio técnico, musical e emocional que não foi esquecido passado estes anos todos. À Soraia, à Mariana e ao Pedro P. pela ajuda diária. À Paula, pelas dicas e ensinamentos sem os quais não teria conseguido. IX.
(10) acabar esta tese (mais uma), assim como a bolsa de investigação que me ofereceu alguma serenidade nestes últimos meses. Aos meus colegas da Faculdade de Farmácia, principalmente aqueles que não se esqueceram de mim enquanto estive fora. Seja pelas horas partilhadas no P3, na sala de cultura ou no café, ou pela ajuda diária durante o meu percurso na faculdade. Em particular, ao Pedro Borrego por todas as conversas sérias, mas positivas, que tivemos nos últimos meses (mas as horas no P3 estão bem vívidas na memória), e ao Pedro Rodrigues por todas as gargalhadas que vou sentir falta. Agradeço ainda aos meus amigos, pelas gargalhadas e lágrimas partilhadas nos bons e maus momentos que definiram a pessoa que sou hoje. Em especial, à Carlota e à Diana, por estarem sempre presentes quando precisei. Vocês são as irmãs que nunca tive e que, ao mesmo tempo, terei para sempre. Sem nunca esquecer os “vossos” sobrinhos e o cunhado Ludwik que farão sempre parte da minha família. À Sara, pela amizade constante mesmo a milhares de quilómetros de distância, e por me fazer acreditar que coisas boas acontecem a boas pessoas. Ao Ricardo e à Ângela por me ouvirem nas minhas queixas sobre o futuro e o mestrado e as mil e uma coisas que fiz no último ano. À Stefanie, ao José e à Catarina pelas gargalhadas e pela ajuda imprescindível durante o Executive Master em Gestão, sem nunca esquecer que sou (e continuarei a ser nos próximos meses) “refugiada”. À Mila e ao João pelos momentos em Londres e na Quinta do Hespanhol. Ao Vasco, ao Renato, à Cheila, à Sofia Romano e à Rita por me receberem de volta de braços abertos, pelas gargalhadas, “parvoíces” e afins (vocês sabem do que falo). Á Joana, à Sofia Narciso e à Sofia Cerqueira por me fazerem companhia no laboratório nos últimos meses. À Ana pelas histórias trazidas de New Castle. À Cátia, que sempre esteve presente, nem que fosse para me ouvir reclamar e fazer-me rir. À Madalena, por me fazer acreditar que eu tenho tempo para descobrir o meu rumo e por me dar também um rumo de morada, algo que nunca será esquecido nem diminuído.. X.
(11) To my second family, the one I got from the 2 years and half I spent in Paris. Thank you to Azaria, for all the laughs, tears and moments shared during these years, by velib, by train, car or walking (nothing else is needed to be said, right?). To Ju, thank you for everything, I will not forget our moments from staying at your place to all the hours shared in the lab and outside (and my French phone!!). To Débora, for all the positive energy, for the help in the lab and for letting me stay at you place. To Béné, thank you for the cupcakes and for the workshops we did at your place, we had a lot of fun. To Julie, for always being available to help me with the smallest and boring details in the lab. To Anthony, for the conversations in French/Portuguese about everything. To Sentob, for all the world conversations and laughs. To everyone in U941 Inserm, thank you for making me feel welcome to the lab without any constrains as if I belonged there from the start. E obrigada à Oriane pelas horas a aprender francês na companhia de um copo de vinho, e também ao Nico por me receberem tão bem em vossa casa. Por último, mas não menos importantes, à minha família, por me apoiar incondicionalmente em todos os aspetos da minha vida. Em particular, ao meu Pai e à minha Mãe, por me ensinarem o que é carinho e felicidade, por todos os sermões que me chamaram à realidade, por todos os sonhos que incentivaram, por toda a educação e oportunidades que me proporcionaram, por todo o orgulho que sempre demonstraram. À Hortense, à Joana e ao Francisco pelos jantares de domingo (e almoços de segunda) acompanhados de muitas gargalhadas. Ao meu marido Ivo, por todo o apoio e amor que me dedicou, por todos os sacrifícios que nunca esquecerei, por todos os momentos inesquecíveis partilhados. Por todas as vezes que me disseste ‘Sim’ quando quiseste dizer ‘Não’: às minhas partidas, às idas ao laboratório a horas inoportunas, às minhas viagens para Paris, aos meus desvaneios de stress durante este último ano.. Agradeço ainda à Fundação para a Ciência e Tecnologia por me ter concedido a bolsa de doutoramento sem a qual esta tese não seria possível.. XI.
(12) XII.
(13) Abstract. AIDS represents a major global problem on human health due to increased number of HIV-1 resistant variants to antiretroviral drugs in infected individuals. HIV infection occurs following a series of virus-host interactions that not only lead to viral replication but also result in the generation of latently infected CD4+ T cells. Thus, targeting host cell factors involved in HIV-1 replication might be an alternative strategy to the currently available antiviral agents. Here we present strategies that allow the study of the function of specific cellular genes in HIV replication cycle through gene silencing. Using shRNA or siRNA constructs, we knockdown the expression of DNA-PKcs, EZH2, CIB2 and CIB1 in different cell lines and primary cells to verify the impact of the downmodulation of these candidates on HIV-1 replication. We observed a reduction of viral proteins expression and virus production, indicating for the first time that CIB1 is a helper factor for HIV-1, in addition to CIB2, EZH2 and DNA-PKcs. We also demonstrate that reduced expression of these proteins affected HIV-2 replication, suggesting them as HIV2 helper factors for the first time. Following this evaluation, we assessed the steps of the viral cycle that could be affected by these helper factors. EZH2 down-modulation did not impair HIV-1 replication in primary CD4+ T lymphocytes and facilitated viral reactivation in latently infected cell lines, suggesting that EZH2 is not indeed a helper factor for HIV-1. Both CIB1 and CIB2 act on early steps of the viral life cycle, facilitating HIV-1 entry in natural target cells in part through modulation of relevant cell surface molecules. Overall, this research work provides new insights for the complex host-HIV interactions instigating further studies that could raise new possibilities for antiviral strategies. Keywords: HIV; helper factors; RNAi; latency; entry. XIII.
(14) XIV.
(15) Resumo. A. síndrome. da. imunodeficiência. adquirida. (SIDA). caracteriza-se. pelo. aparecimento de infeções oportunistas e determinados tipos de tumores causado pela destruição progressiva do sistema imunológico. O Vírus da Imunodeficiência Humana (VIH; do inglês Human Immunodeficiency Virus, HIV) é o agente etiológico da SIDA, existindo dois tipos: VIH-1 (Vírus da Imunodeficiência Humana tipo 1) e VIH-2 (Vírus da Imunodeficiência Humana tipo 2). Atualmente 36,7 milhões de pessoas vivem infetadas com o VIH-1, tendo sido descritos 2,1 milhões de novos casos em 2015. Por esta razão, existe um contínuo esforço em desenvolver estratégias eficientes que consigam neutralizar o vírus no seu hospedeiro. Como todos os vírus, o VIH-1 depende da maquinaria celular para conseguir replicar-se. A fase precoce do ciclo de replicação viral inicia-se com a ligação de um virião infeccioso ao receptor celular de superfície CD4 através da glicoproteína de superfície do vírus, gp120. Esta proteína sofre alterações conformacionais que permitem o reconhecimento dos co-receptores, em particular CCR5 e CXCR4, possibilitando a fusão do invólucro viral com a membrana celular e consequentemente a entrada viral. No citoplasma, o RNA genómico viral em cadeia simples é convertido num intermediário de DNA de cadeia dupla pela transcriptase reversa (RT). Este intermediário é então transportado para o núcleo na forma de complexo de pré-integração (pre-integration complex, PIC), e integrado no genoma da célula por ação da integrase (IN). Na fase tardia do ciclo, os genes do DNA proviral são transcritos e traduzidos pela maquinaria da célula hospedeira em proteínas virais. As proteínas estruturais são transportadas para a membrana plasmática onde se acumulam. A poliproteína Gag liga-se ao RNA genómico. XV.
(16) do vírus e então interage com a membrana plasmática, que é forçada a se projetar e eventualmente a se separar, formando-se uma partícula que é libertada para o exterior da célula infetada. A maturação do virião por ação da protease resulta na reorganização nuclear e a aquisição da infecciosidade viral. Existe um vasto repertório de proteínas celulares que podem assistir na replicação viral ao serem sequestradas pelo vírus. Assim, o uso destes fatores de ajuda do VIH-1 como alvos terapêuticos pode ser uma alternativa aos compostos antivirais disponíveis. Este facto impulsionou estudos cujo objetivo era a identificação de fatores celulares associados à replicação do VIH-1. Recentemente, vários autores exploraram um fenómeno biológico no qual pequenas moléculas de RNA de cadeia dupla (double stranded RNA, dsRNA) levam à degradação de RNAs mensageiros (do inglês, messenger RNAs, mRNAs). Este processo chamado de RNA de interferência (RNA interference, RNAi) tornou-se uma ferramenta útil, podendo ser usados pequenas moléculas de RNAs de interferência (small interfering RNAs, siRNAs) ou pequenos RNAs com formato em hairpin (short hairpin RNAs, shRNAs). Vários estudos conseguiram identificar numerosos fatores adjuvantes envolvidos na replicação viral através de siRNAs e shRNAs. No entanto, existe pouca sobreposição dos fatores detetados nos vários estudos e alguns fatores muito conhecidos não foram identificados. Mais, a caracterização da maioria destes fatores ainda é limitada. Tendo em conta todos estes aspectos, este estudo tem como objetivo comparar diferentes estratégias para identificar fatores celulares envolvidos no ciclo viral do HIV. Para proceder a esta comparação selecionámos um fator adjuvante do HIV-1 conhecido e identificado em diferentes estudos, DNA-PKcs (do inglês DNA-dependent protein kinase catalytic subunit), que já foi sugerido participar em duas etapas do ciclo viral – a integração e a transcrição. Para além do DNA-PKcs, foram também selecionadas as proteínas CIB2 e EZH2, anteriormente identificadas num estudo baseado em shRNAs como fatores adjuvantes do HIV-1. O presente estudo pretende ainda caracterizar a contribuição das proteínas. XVI.
(17) CIB2 e EZH2 no ciclo replicativo, assim como caracterizar a potencial contribuição de um homólogo de CIB2, a proteína CIB1. CIB1 e CIB2 pertencem a uma família de proteínas capazes de se ligarem ao cálcio e a integrinas, como o seu nome indica (Calcium- and integrin-binding family member 1 or 2, CIB1 or CIB2). Estas proteínas são expressas em vários tecidos humanos e foram identificadas em vários compartimentos celulares. Uma vez que conseguem interagir com diversos fatores celulares, desde integrinas a fatores de transcrição, as proteínas CIB foram implicadas em processos tão distintos como proliferação celular, reparação de DNA em dupla cadeia, ativação de canais iónicos, organização do citoesqueleto, e ativação de integrinas. EZH2 (do inglês Enhancer of zeste homolog 2) constitui a subunidade catalítica do complexo repressivo Polycomb (do inglês Polycomb repressive complex 2, PCR2) que é responsável por silenciamento génico através da trimetilação da histona 3 no resíduo de lisina 27. Uma vez que este complexo PCR2 influencia um grande conjunto de genes, vários processos são afetados pela EZH2 como ciclo celular, diferenciação e pluripotência. EZH2 tem sido associado a transformações neoplásticas de vários tipos celulares, o que é consistente com a sua elevada expressão em tecidos proliferativos. Apesar de reconhecido como um repressor associado a PCR2, EZH2 já foi demonstrado como ativador de genes quando sobre expresso.. Neste trabalho apresentamos diferentes estratégias que permitem estudar a função de genes celulares específicos no ciclo de vida do HIV através do seu silenciamento. Com o uso de shRNAs e siRNAs, a expressão dos vários candidatos foi silenciada em diferentes linhas celulares e em células primárias humanas antes de serem expostas à infeção viral. A biblioteca de shRNAs utilizada, clonada em plasmídeos lentivirais, permitiu a produção de lentivírus que expressam os shRNAs (shRNA lentivirus) e que após transdução levam à integração do shRNA no genoma da célula, tornando a sua expressão constitutiva. Para comparar estratégias com shRNA, foi XVII.
(18) utilizada a linha celular Jurkat, uma linha celular imortalizada de linfócitos T de modo a mimetizar o hospedeiro natural do HIV‑1. Para a construção de clones shRNA, procedeuse à transdução de células Jurkat utilizando vectores lentivirais que expressam shRNAs individuais para os genes em estudo: DNA-PKcs, EZH2, CIB2 e CIB1. Os clones individuais foram obtidos pelo método de ClonaCell-TCS e selecionados em meio suplementado com 2 μg/mL de puromicina. Comparámos esta estratégia com a transdução transiente de células Jurkat de modo a obtermos uma população mista. Tanto os clones como as populações celulares obtidas foram expostas a partículas HIV-1 para verificar o impacto da redução de expressão destes candidatos na replicação viral. Podemos observar uma redução na expressão das proteínas virais tal como na produção viral, indicando pela primeira vez que CIB1 é de facto um fator adjuvante do HIV-1 em células Jurkat, para além de CIB2 e EZH2. As proteínas CIB2 e EZH2, assim como CIB1 e DNA-PKcs, também demonstraram ter um papel importante no ciclo replicativo do HIV2, como se pode observar pela redução da quantidade viral detetada no sobrenadante dos clones infetados. Demonstramos ainda que tanto siRNAs como shRNAs são eficientes a silenciar os transcritos de DNA-PKcs, constituindo-se como ferramentas válidas para o estudo de fatores adjuvantes em diferentes células. Utilizando populações transduzidas com shRNAs, fomos caracterizar a contribuição das proteínas EZH2, CIB2 e CIB1 no ciclo replicativo do HIV-1. Relativamente ao EZH2, os nossos ensaios em linhas celulares latentes e em células T primárias sugerem uma função restritiva deste fator e não um papel adjuvante como observado na linha celular Jurkat. Quando EZH2 tem uma expressão reduzida em linhas celulares latentes, observa-se uma maior reativação do vírus integrado no genoma celular destas células em comparação com populações não transduzidas. Uma vez que o silenciamento de EZH2 facilita a reativação do HIV-1, a função desta proteína no ciclo viral pode passar pela alteração da cromatina de modo a permitir a integração viral ou a subsequente transcrição do genoma viral. Estas observações são consistentes com os. XVIII.
(19) resultados obtidos em células primárias que mostram que EZH2 não é um fator adjuvante do HIV-1 em linfócitos T, que constituem o alvo principal do vírus. Os nossos resultados confirmam que tanto CIB1 como CIB2 são fatores adjuvantes do HIV-1 não só em linhas celulares como em células T primárias. Conseguimos demonstrar que CIB1 e CIB2 são importantes na entrada do HIV-1, uma vez que o seu silenciamento reduz o número de eventos de fusão vírus-célula assim como a eficiência de transmissão célula-a-célula. Para além disso, a expressão à superfície dos co-receptores CXCR4 e CCR5 e da integrina α4β7 é reduzida em células onde CIB1 e CIB2 estão silenciados. Estes dados são coerentes com a função celular destas proteínas relacionada com integrinas. É plausível admitir que CIB1 e CIB2 tenham um papel na ativação ou ligação de integrinas que possam ativar outros intervenientes na entrada do vírus como a integrina LFA-1.. Os resultados apresentados nesta tese mostram as potencialidades da tecnologia do RNAi para a compreensão da interação entre fatores celulares e o HIV, e proporciona novos conhecimentos nos fatores previamente identificados CIB2 e EZH2, assim como CIB1. Deste modo, este trabalho incentiva novos estudos que poderão levar a novas estratégias antivirais mais eficientes contra a infeção por HIV.. Palavras-chave: HIV; fatores adjuvantes; RNAi; entrada; latência. XIX.
(20) XX.
(21) Abbreviations and Symbols. 6HB. Six-helix bundle. Abs. Absorbance. AEBP2. Zinc-finger Adipocyte enhancer-binding protein 2. AFF1. AF4/FMR2 family protein 1. Ago2. Argonaute protein 2. AIDS. Acquired Immunodeficiency Syndrome. Akt. Protein kinase B. ALIX. ALG2‑interacting protein X. AME. Amphotericin B methyl ester. AP-1. Activator protein-1. APOBEC3. Apolipoprotein B-editing catalytic polypeptide 3 proteins. Arg. Arginine. ART. Antiretroviral therapy. ASK1. Apoptosis signal-regulating kinase 1. ATCC. American Type Culture Collection. BAF. Barrier-to-Autointegration Factor. BlaM. Beta-lactamase. BSA. Bovine serum albumin. BST-2. Bone Marrow Stromal Cell Antigen 2 or Tetherin. CA. Capsid. 2+. Ca. Calcium ion. CaBPs. Calcium-binding proteins. CaM. Calmodulin. CBP. Nuclear cap-binding protein subunit. CCF2. Cephalosporin core linking 7-hydroxycoumarin to fluorescein. CCL. Chemokine (C-C motif) ligand. CCR. Chemokine receptor. CD11a. Integrin subunit alpha L. CD184. Cluster of differentiation 184 or Chemokine (C-X-C motif) receptor 4. CD4. Cluster of differentiation 4. CD40L. Cluster of differentiation 40 ligand. CDK9. Cyclin-dependent protein kinase-9. XXI.
(22) cDNA. Complementary DNA. CFSE. Carboxyfluorescein succinimidyl ester. CIB. Calcium- and integrin-binding protein. CIB1. Calcium- and integrin-binding protein 1. CIB2. Calcium- and integrin-binding protein 2. COMM D1. Copper metabolism (Murr1) domain-containing protein 1. CpG. Region rich in CG content with phosphodiester bonds. cPPT. Central PPT. CPRG. Chlorophenol red-β-D-galactopyranoside. CRM1. Chromosome maintenance-1. CT. Cytoplasmic tail. CTD. Carboxy-terminal domain. CTR9. Paf1/RNA Polymerase II Complex Component. CTS. Central terminal sequence. CXCR4. Chemokine (C-X-C motif) receptor 4. CypA. Cyclophilin A protein. DC. Dendritic Cell. DC-SIGN. DC-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin. DDX1. DEAD/H-Box Helicase 1. DDX3. DEAD/H-Box Helicase 3. DGCR8. DiGeorge Syndrome Critical Region Gene 8. DICER1. Double-stranded RNA-specific endoribonuclease type III. DMEM. Dulbecco’s minimal essential medium. DNA. Deoxyribonucleic acid. DNA-PK. DNA-dependent protein kinase. DNA-PKcs. DNA-dependent protein kinase catalytic subunit. dsDNA. Double-stranded DNA. dsRNA. Double-stranded RNA. EED. Embryonic ectoderm development. eIF4G. Eukaryotic translation initiation factor 4 G. eIF5A. Eukaryotic translation initiation factor 5 A. ELISA. Enzyme-Linked Imunosorbent Assay. Env. Envelope protein. ER. Endoplasmic reticulum. ERK1/2. Extracellular signal-regulated protein kinases 1 and 2. ESCRT. Endosomal sorting complex required for transport. EZH2. Enhancer of Zeste Homologue 2. FAK. Focal Adhesion Kinase. FBS. Fetal bovine serum. FDA. Food and Drug Administration. XXII.
(23) G9a. Histone H3-K9 Methyltransferase 3. Gag. Group-specific-antigen protein. GAPDH. Glyceraldehyde-3-phosphate dehydrogenase. GATA4. GATA-Binding Protein 4. GFP. Green fluorescent protein. GM2A. GM2 ganglioside activator. GM-3. Ganglioside. gp120. Surface glycoprotein gp120 of HIV-1. gp125. Surface glycoprotein gp125 of HIV-2. gp32. Transmembrane glycoprotein gp32 of HIV-2. gp41. Transmembrane glycoprotein gp41 of HIV-1. GPCRs. G-protein-coupled receptors. GPI. glycosylphosphatidylinositol. GRKs. GPCR kinases. Gβγ. G (guanosine nucleotide-binding proteins) beta-gamma complex. H3K27. Lysine 27 of histone 3. H3K27me3. Trimethylated lysine 27 of histone 3. HAART. Highly Active Antiretroviral Therapy. HATs. Histone acetylases. HDAC1. Histone deacetylase 1. HEK. Human Embryonic Kidney. HeLa. Cervical cancer cells from Henrietta Lacks. HeLa-P4. HeLa-CD4-LTR-β-gal. HeLaP4C5. HeLa-CD4-CCR5-LTR-β-gal. HIV. Human Immunodeficiency Virus. HIV-1. Human Immunodeficiency Virus type 1. HIV-2. Human Immunodeficiency Virus type 2. HMG. High mobility group proteins. HMTs. Histone methyltransferases. HR-C. Carboxy-terminal helical region. HR-N. Amino-terminal helical region. HRP. Horseradish peroxidase. HRP-2. Hepatoma-derived growth factor related protein 2. Hsp70. Heat shock protein 70. Hsp90. Heat shock protein 90. ICAM 1. Inter Cellular Adhesion Molecule 1. IFITMs. Interferon Induced Transmembrane Proteins. IFN. Interferon. IFN-α. Interferon-alpha. iGag. Intracellular Gag. XXIII.
(24) IL-2. Interleukin-2. IN. Integrase. Ini1. Integrase Interactor 1 Protein. InsP3. Inositol 1,4,5-triphosphate. InsP3R. Inositol 1,4,5-triphosphate receptor. IRES. Internal ribosome entry site. ISG15. IFN-stimulated gene 15. JNK. c-Jun N-terminal kinase. KIP. DNA-PKcs interacting protein. KIP2. DNA-PKcs interacting protein 2. KSHV. Kaposi’s Sarcoma-Associated Herpesvirus. Ku70/Ku80. Ku heterodimer. Lck. Lymphocyte Cell-Specific Protein-Tyrosine Kinase. LEDGF/p75. Lens epithelium-derived growth factor and coactivator protein p75. LFA-1. Leukocyte Antigen Function-1. LSF. Late SV40 factor. LTR. Long-terminal repeats. Lys. Lysine. MA. Matrix. mAb. Monoclonal antibody. MAPK. Mitogen-activated protein kinases. MFI. Mean fluorescence intensity. Mg2+. Magnesium ion. miRNAs. MicroRNAs. MOI. Multiplicity of infection. mRNA. Messenger RNA. MTT. 3-(4,5-dimethylthiazolyl-2-yl)-2,5-diphenyltetrazolium bromide. NC. Nucleocapsid. Nef. Negative regulator factor. NFAT. Nuclear factor of activated T cells. NF-κB. Nuclear factor kappa B. NHEJ. Non-homologous end joining. Ni. Non-infected. NLS. Nuclear localization signal. NMDAR. N-methylo-D-aspartate receptor. NNRTIs. Non-Nucleoside Reverse Transcriptase Inhibitors. NRTIs. Nucleoside Reverse Transcriptase Inhibitors. NT. Non-transduced. Nuc. Nucleosome. NUP153. Nucleoporin 153kDa. XXIV.
(25) NUP93. Nucleoporin 93kDa. NVP. Nevirapin. NXF1. Nuclear RNA export factor 1. p55Gag. Gag precursor protein of 55 kDa. PABP. Poly(A)-binding protein. PAF1. Polymerase associated factor 1. PAK1. P21-Activated Kinase 1. Pax3. Paired Box 3. PBAF. Polybromo and Brahma-related gene 1-associated factor remodeling complex. PBMCs. Peripheral blood mononuclear cells. PBS. Primer binding site. PBS 1x. Phosphate-Buffered Saline solution. PcG. Polycomb-group proteins. PCR. Polymerase chain reaction. PCR2. Polycomb repressive complex 2. PFA. Paraformaldehyde. PGK. Phosphoglycerate kinase. PI3k. Phosphatidylinositol 3-kinase. PIC. Pre-integration complex. PKC. Protein kinase C. PKC-ϑ. Protein kinase C isoform theta. PML. Promyelocytic Leukemia Protein. Pol. Polymerase. PPT. Polypurine tracts. PR. Protease. pri-miRNAs. primary microRNAs. PtdIns(4,5)P2. Phospholipid phosphatidylinositol-4,5 bisphosphate. P-TEFb. Positive transcription elongation factor b. qRT-PCR. Quantitative real time PCR. Rab11 FIP1C. Rab11 family-interacting protein 1 isoform C. Rac3. Ras-Related Botulinum Toxin Substrate 3. RanGTP. RAs-related GTP-binding nuclear protein. RbAp48/46. Retinoblastoma protein (Rb)-associated protein 48 and 46. Rev. Regulator of Virion. RISC. RNA-induced silencing complex. RNA. Ribonucleic Acid. RNA pol. RNA polymerase. RNAi. RNA interference. RNAi. RNA interference. RNase H. RiboNuclease enzyme H. XXV.
(26) RNASEN. RNase III nuclear or Drosha. RNP. Ribonucleoprotein. RPMI. Roswell Park Memorial Institute medium. RPMI-10. RPMI supplemented with 10% FBS. RRE. Rev responsive element. RT. Reverse transcriptase. RTC. Reverse transcriptional complex. Sam68. Src-Associated substrate in Mitosis of 68 kDa. Scr. SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase. SCRAM. Scramble. SDF-1. Stromal cell-derived factor 1. SDS-PAGE. Sodium dodecyl sulfate polyacrylamide gel electrophoresis. SETDB1. SET Domain Bifurcated 1. shRNAs. Short hairpin RNAs. SIDA. Síndrome da imunodeficiência adquirida. Siglec1. Sialic acid binding Ig like lectin 1. siRNAs. Small interfering RNAs. SIV. simian immunodeficiency virus. Sp1. Specificity protein 1. ssDNA. Single-stranded DNA. ssRNA. Single-stranded RNA. SU. Surface. SUV39H1. Suppressor of Variegation 3-9 Homolog 1. SUZ12. Suppressor of zeste 12 homolog. SV40. Simian vacuolating virus 40. SWI/SNF. Switching/Sucrose non-fermenting genes. TAR. Trans-activating response element. TAS1R2. Taste 1 Receptor Member 2. Tat. Trans-activator of Transcription. tDNA. Target DNA. TGN. Trans-Golgi Network. Th1. Type 1 T helper. Th2. Type 2 T helper. TLR. Toll-like receptors. TM. Transmembrane. TNF. Tumour necrosis factor. TNF-α. Tumour necrosis factor alpha. TNPO3. Transportin 3. TRIM. Tripartite Motif protein. TRIM5α. Tripartite Motif protein 5 alpha. XXVI.
(27) tRNA(Lys3). Transfer RNA Lys3. TSG101. Tumor Susceptibility gene 101. USH1J. Usher syndrome type 1J. Vif. Viral Infectivity Factor. VIH. Vírus da Imunodeficiência Humana. VIH-1. Vírus da Imunodeficiência Humana tipo 1. VIH-2. Vírus da Imunodeficiência Humana tipo 2. Vpr. Viral protein R. VPS4. Vacuolar protein sorting 4. Vpu. Viral protein U. Vpx. Viral protein X. VSV-G. Vesicular stomatitis virus glycoprotein. YY1. Yin and yang 1. ZFNs. Zinc-Finger nucleases. α4β7. Integrin Alpha 4 and Beta 7. αIIb. Integrin subunit alpha II b. β-gal. Beta-galactosidase. XXVII.
(28) XXVIII.
(29) Table of Contents. ACKNOWLEDGES / AGRADECIMENTOS ........................................................ IX ABSTRACT ...................................................................................................... XIII RESUMO ...........................................................................................................XV ABBREVIATIONS AND SYMBOLS .................................................................XXI TABLE OF CONTENTS ................................................................................. XXIX Index of Figuress .................................................................................................... XXXI. CHAPTER I: GENERAL INTRODUCTION .......................................................... 1 1.. Human Immunodeficiency Virus .......................................................................... 1 1.1. HIV as the etiological agent of AIDS ................................................................. 1 1.2. HIV taxonomy ................................................................................................... 2 1.3. HIV Characterization ........................................................................................ 3 1.3.1. Structure and Genome of HIV-1 ................................................................................ 3 1.3.2. Structure and Genome of HIV-2 ................................................................................ 6 1.4. HIV replication cycle ......................................................................................... 6 1.4.1. Early phase of HIV-1 cycle .......................................................................................... 7 Viral attachment .................................................................................. 7 Viral entry ............................................................................................ 8 Uncoating .......................................................................................... 10 Reverse Transcription ....................................................................... 12 Nuclear Import................................................................................... 14 Integration ......................................................................................... 14 1.4.2. Late phase of HIV-1 cycle .......................................................................................... 16 Transcription ..................................................................................... 16 Translation ........................................................................................ 18 Assembly .......................................................................................... 20 Budding and maturation .................................................................... 21. 2.. Antiviral Approaches against HIV ...................................................................... 22 2.1. Targeting viral proteins ................................................................................... 22 2.2. Host proteins as targets .................................................................................. 23 2.2.1. Restriction factors ......................................................................................................... 24 XXIX.
(30) 2.2.2.. 2.2.3.. TRIM proteins .................................................................................... 24 APOBEC Protein Family .................................................................... 25 Tetherin ............................................................................................. 27 Helper factors................................................................................................................. 28 Receptor CD4 .................................................................................... 28 Co-receptors CCR5 and CXCR4 ....................................................... 29 LEDGF/p75........................................................................................ 31 RNAi as a tool to identify possible host targets ................................................... 32 Calcium- and integrin-binding family member 2 (CIB2) ...................... 35 Enhancer of zeste homolog 2 (EZH2) ................................................ 39. CHAPTER II: AIMS ............................................................................................ 43 CHAPTER III: METHODS .................................................................................. 45 CHAPTER IV: VALIDATION OF HELPER FACTORS ON HIV REPLICATION USING DIFFERENT DOWN-MODULATION APPROACHES ........................... 57 Abstract ..................................................................................................................... 58 Introduction ............................................................................................................... 60 Results ...................................................................................................................... 63 Discussion ................................................................................................................. 76 Acknowledgments ..................................................................................................... 80. CHAPTER V: EZH2: HELPER OR RESTRICTION FACTOR? ......................... 81 Abstract ..................................................................................................................... 82 Introduction ............................................................................................................... 84 Results ...................................................................................................................... 87 Discussion ................................................................................................................. 95 Acknowledgments ................................................................................................... 100. CHAPTER VI: CIB1 AND CIB2 ARE HIV-1 HELPER FACTORS INVOLVED IN VIRAL ENTRY ................................................................................................. 101 Abstract ................................................................................................................... 102 Introduction ............................................................................................................. 104 Results .................................................................................................................... 107 Discussion ............................................................................................................... 122 Acknowledgments ................................................................................................... 126. CHAPTER VII: CONCLUDING REMARKS AND FUTURE PERSPECTIVES 129 CHAPTER VIII: REFERENCES ....................................................................... 135 CHAPTER IX: ANNEXES ................................................................................ 163. XXX.
(31) Index of Figures. Figure 1. HIV-1 virion. .................................................................................................. 3 Figure 2. HIV-1 genome............................................................................................... 4 Figure 3. HIV-2 genome............................................................................................... 6 Figure 4. HIV-1 replication cycle. ................................................................................. 7 Figure 5. The HIV entry process.. ................................................................................ 9 Figure 6. Models of HIV-1 uncoating. ......................................................................... 11 Figure 7. HIV reverse transcription process... ............................................................ 13 Figure 8. Mechanisms of retroviral integration. .......................................................... 16 Figure 9. The mechanism of transcription activation .................................................. 18 Figure 10. Post-transcription regulation on HIV-1....................................................... 20 Figure 11. RNA silencing by miRNAs and siRNAs. .................................................... 34 Figure 12. Possible mechanisms of function for CIBs proteins. .................................. 37 Figure 13. Possible mechanisms of function for EZH2. .............................................. 41 Figure 14. Representation of shRNAs. ....................................................................... 46 Figure 15. Knockdown of DNA-PKcs in Jurkat T-cells.. .............................................. 64 Figure 16. Optimization of siRNA transfection to downmodulate DNA-PKcs in HeLa-P4 cells.. .......................................................................................................................... 65 Figure 17. Impairment of HIV-1 in DNA-PKcs-depleted HeLa-P4 cells....................... 66 Figure 18. Construction of shRNA cell clones. ........................................................... 67 Figure 19. mRNA knockdown in individual shRNA clones.. ........................................ 68 Figure 20. shRNA cell clones are resistant to HIV-1 infection.. .................................. 68 Figure 21. shRNA cell clones are resistant to HIV-2 infection. ................................... 69 Figure 22. Establishment of shRNA-transduced cell populations. .............................. 70 Figure 23. mRNA knockdown in shRNA cell populations. .......................................... 70 Figure 24. Comparison of shRNA cell populations during HIV infection ..................... 71 Figure 25. shRNA cell populations are resistant to HIV-1 infection ............................. 72 Figure 26. CIB1 mRNA knockdown in shRNA individual cell clones ........................... 73 Figure 27. CIB1 shRNA cell clones are resistant to HIV-1 and HIV-2 infection ........... 74 Figure 28. mRNA knockdown in shRNA cell populations. .......................................... 74 Figure 29. shRNA cell populations are resistant to HIV infection................................ 75 Figure 30. EZH2 knockdown increases HIV-1 reactivation.. ....................................... 87 Figure 31. J-Lat 10.6 cells exhibit higher levels of EZH2 mRNA than Jurkat T cells.. . 88 Figure 32. Knockdown of EZH2 is efficient and viable in Jurkat T-cells ...................... 89 Figure 33. HIV-1 induces EZH2 mRNA levels in infected cells ................................... 90 XXXI.
(32) Figure 34. EZH2 is required for optimal HIV-1 and VSV-G infectivity .......................... 91 Figure 35. EZH2 depletion in Jurkat T cells does not influence Env-mediated virus entry............................................................................................................................ 91 Figure 36. Knockdown of EZH2 in primary CD4+ T lymphocytes. .............................. 93 Figure 37. EZH2 knockdown increases HIV-1 and VSV-G-envelope-mediated infectivity ................................................................................................................................... 94 Figure 38. Knockdown of CIB1 and CIB2 in Jurkat T-cells ....................................... 108 Figure 39. Depletion of CIB proteins impairs HIV-1 replication in the Jurkat T-cell line ................................................................................................................................. 109 Figure 40. Impairment of HIV replication is specific to down-modulation of CIB proteins ................................................................................................................................. 110 Figure 41. CIB1 and CIB2 are required for optimal HIV-1 envelope-mediated entry, but not VSV-G-mediated entry ........................................................................................ 111 Figure 42. Down-modulation of CIB proteins does not impair late steps in the HIV-1 replication cycle ........................................................................................................ 112 Figure 43. Knockdown of CIB proteins affects HIV-1 replication in primary CD4+ T-cells ................................................................................................................................. 114 Figure 44. HIV-1 induces CIB2 mRNA levels in infected cells. ................................. 115 Figure 45. Effect of knockdown of CIB proteins on HIV-1 replication in primary CD4+ Tcells is Env-dependent. ............................................................................................. 116 Figure 46. Depletion of CIB proteins in primary CD4+ T-cells does not influence viral attachment. ............................................................................................................... 117 Figure 47. Depletion of CIB proteins in primary CD4+ T-cells influences Env-mediated virus entry. ................................................................................................................ 118 Figure 48. Effect of down-modulation of CIB proteins in CD4+ T-lymphocytes on the expression of proteins implicated in HIV-1 entry........................................................ 120 Figure 49. Effect of down-modulation of CIB proteins in PM1 cells on the expression of HIV-1 co-receptors.. .................................................................................................. 121. XXXII.
(33) CHAPTER I General Introduction. 1. Human Immunodeficiency Virus. 1.1.. HIV as the etiological agent of AIDS. Acquired Immunodeficiency Syndrome (AIDS) was recognized in the early 1980s as the decline of immune system characterized by opportunistic diseases appearance in previously healthy individuals (Siegal et al. 1981). In 1983, the virus responsible for this syndrome was identified as a retrovirus capable of infecting CD4+ lymphocytes (Gallo et al. 1983; Barré-Sinoussi et al. 1983) and consequently induced their death with a half-life of less than two days (Turner and Summers 1999). Nowadays, the virus is known as Human Immunodeficiency Virus (HIV) and there are two types known: type 1 (HIV-1) (Barré-Sinoussi et al. 1983; Gallo et al. 1983) and type 2 (HIV-2) (Clavel et al. 1986). The former is more virulent and is more worldwide prevalent than type 2. Indeed, HIV‑2 is characterized by a slow disease progression and transmission, a lower plasma viral load and a low rate of CD4‑T cell decline (Jaffar et al. 1997; Whittle et al. 1994; Marlink et al. 1994). Also, HIV-2 has remained largely restricted to West Africa, with its highest prevalence rates recorded in Guinea-Bissau and Senegal (de Silva, Cotten, and RowlandJones 2008), although countries with West Africa ties also report HIV-2 infections such as Portugal (Soriano et al. 2000).. 1.
(34) Approximately 36.7 million of people live with HIV, with 2.1 million new cases of HIV infection described in 2015 (UNAIDS, 2015). Highly active anti‑retroviral therapy (HAART) is the current antiviral therapy and consists in the administration of a combination of antiviral drugs that act against HIV proteins or viral RNA (De Clercq 2009). Despite the progress in the development of antiretroviral drugs and their substantiated efficiency in reducing AIDS-related deaths, eradication of HIV‑1 and consequently the cure of AIDS has still not been achieved (Flexner 2007; Richman et al. 2009; Barouch 2008). The uncertainty of an effective vaccine or a curative treatment advocates AIDS as a significant public health threat for years to come.. 1.2.. HIV taxonomy. HIV is a member of Lentivirus genus from the Retroviridae family (Turner and Summers 1999). Lentiviruses have long incubation periods before clinical manifestations and cause chronic persistent infections in various mammalians, including bovines, horses, sheep, felines, and primates (Gifford 2012). HIV-1 comprises four distinct lineages, termed groups M (Major), O (Outlier), N (non-M, non-O) and P, each of which resulted from an independent cross-species transmission event. Group M represents the pandemic form of HIV-1 as it has infected millions of people worldwide (approximately 90% of HIV/AIDS cases) and it is further subdivided into subtypes (from A to K) (Mourez, Simon, and Plantier 2013; Sharp and Hahn 2011). Although these subtypes differ vastly in their distribution within the human population, all are capable of causing CD4+ T-cell depletion and, consequently, AIDS (Sharp and Hahn 2011). HIV-2 also comprises distinct lineages, termed groups A–H. Each lineage appears to represent an independent host transfer, and only groups A and B have spread within humans to a considerable degree (Sharp and Hahn 2011). 2.
(35) 1.3.. HIV Characterization. 1.3.1. Structure and Genome of HIV-1. HIV‑1 mature virions are spherical shaped with 100nm of diameter approximately. Its outer viral membrane is derived from the host cell membrane, consisting of a lipid bilayer where the surface glycoprotein (gp120 or SU) is. anchored. via. interaction. with. the. transmembrane glycoprotein (gp41 or TM) forming a trimeric complex (Turner and Summers 1999; Freed 2001). Cell surface. Figure 1. HIV-1 virion. Adapted from National Institute of Allergy and Infectious Diseases (NIAID) (https://www.niaid.nih.gov/).. molecules such as heparan-sulfate moieties, mannose receptor, histocompatibility antigens or DC-SIGN and other lectins can also be present in the lipid membrane (Anzinger, Olinger, and Spear 2008). Matrix proteins (p17 or MA) composes the protein scaffold underneath the viral inner membrane, and encloses the conical capsid core, formed by capsid proteins (p24 or CA). The core of the virion comprises the viral genome – two single strand RNA molecules of approximately 9 Kb – associated to nucleocapsid proteins (p7 or NC). Along with the two genomic-length RNA copies, the viral enzymes protease (p11 or PR), reverse transcriptase (p66/p51 or RT), and integrase (p31 or IN), are also incorporated in the particle (Figure 2), Three of six additional proteins, often called accessory proteins, are found in the viral particle: negative regulator factor (Nef), virion infectivity factor (Vif) and viral protein R (Vpr).. HIV genome organization is generally depicted in the form of viral DNA after integration into cellular DNA (i.e., provirus). Provirus representation places viral promoter, RNA start site and polyadenylation site in the positions typically found in host genes (Coffin, Hughes, 3.
(36) Figure 2. HIV-1 genome. Adapted from from Ayinde et al. 2010.. and Varmus 1997). The 5’-LTR is the viral promoter containing functional regions essential for HIV‑1 transcription regulation, such as the transactivation response (TAR) element, the basal promoter and the core enhancer (Whitcomb and Hughes 1992; W. K. Wang et al. 2000; Frankel and Young 1998). TAR is a regulatory element for transcription, located downstream from the initiation site, and is only functional when it is placed in the correct position and orientation (Muesing, Smith, and Capon 1987). The expression of HIV‑1 genes is mediated by viral proteins through regulatory mechanisms with overlapping reading frames and through alternative mRNA splicing (Wang et al. 2000).. HIV‑1 genome encodes three genes common to all retroviruses, gag, pol, and env, which are translated into polyprotein precursors, subsequently cleaved into individual proteins (Frankel and Young 1998). The group specific antigen (gag) gene encodes the internal structural proteins under the form of a precursor protein of 55 kDa (p55Gag), which is proteolytically processed into the mature proteins MA or p17 (matrix), CA or p24 (capsid), NC or p7 (nucleocapsid), p6 domain, as well as two spacer peptides SP1 and SP2 (Campbell and Hope 2015). MA, CA and NC form consecutive shells to protect the viral RNA, while p6 is involved in viral particle release. The pol gene encodes the viral enzymes in the context of a precursor protein of 160 kDa which results from a shift in the ribosome reading frame (-1 nucleotide to the 5 ') at the end of Gag (Qiao et al. 2016). The relative proportion of polyproteins Gag and Gag-Pol (20 to 1) is finely regulated (Kobayashi et al. 2010). The viral enzymes are reverse transcriptase (RT), integrase (IN), and protease (PR). Reverse Transcriptase (RT) is a heterodimer. 4.
(37) composed of subunits p66 and p51. This enzyme catalyzes the reverse transcription step which consists on DNA synthesis from viral RNA. Integrase (IN) catalyzes the provirus integration into the DNA of the infected cell (Chiu et al.2004), and Protease (PR) participates in virion morphology change which leads to their maturation during or immediately after the release of the virion (Loeb et al., 1989). The Env precursor envelope glycoprotein or gp160 is cleaved by the cellular furin protease to give rise to the envelope glycoprotein subunits gp120 (or SU) and gp41 (or TM) (Checkley, Luttge, and Freed 2011). These two subunits interact non-covalently and a trimeric complex allows the virus to bind to receptor and coreceptors when attaching to cell surface.. Two other proteins, Tat and Rev, provide essential gene regulatory functions. Tat is able to increase the transcription efficiency from the LTR promoter (Bannwarth and Gatignol 2005), while Rev is essential to export unspliced and incompletely spliced viral mRNA to the cytoplasm allowing their translation (Chang and Sharp 1990). HIV-1 genome also encodes for four additional proteins, often called accessory proteins: negative regulator factor (Nef), virion infectivity factor (Vif), viral protein R (Vpr) and viral protein U (Vpu). While Vif has a specific role in assisting reverse transcription by counteracting restriction factors from APOBEC proteins family, the other proteins perform multiple functions. Nef induces downregulation of several molecules including CD4 from the surface of infected cells, enhances virion infectivity, and modulates T cell activation state of its cell host (reviewed in Jere et al. 2010). The Vpr protein is involved in various processes to improve HIV-1 efficiency, such as cell cycle arrest in G2/M phase (Li et al. 2010; Laguette et al. 2014), suppression of immune activation and induction of apoptosis (reviewed in Romani and Engelbrecht 2009). Vpu has also been shown to downregulate CD4 surface expression and induce apoptosis (reviewed in Nomaguchi, Fujita, and Adachi 2008), although its most proclaimed role is assisting virion release by counteracting the action of cellular protein Tetherin (Neil, Zang, and Bieniasz 2008). 5.
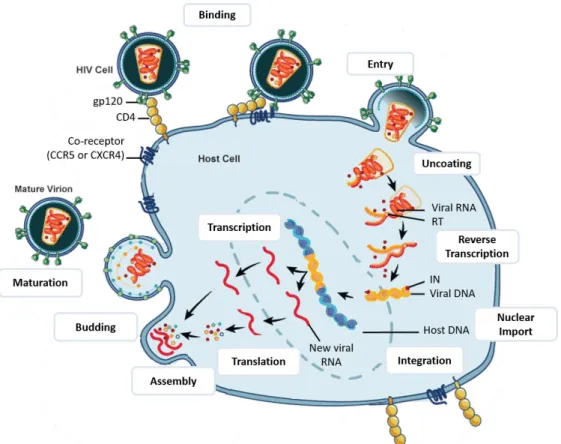
(38) 1.3.2. Structure and Genome of HIV-2. Less consideration has been given to HIV type 2 comparing to HIV-1, due to its restricted endemicity. HIV-2 is closely related to HIV-1, presenting ~60% similarity to HIV-1 at the aminoacid level in Gag and Pol, and 30-40% in the regions encoding the Env (de Silva, Cotten, and Rowland-Jones 2008). Besides these structural genes, HIV-2 also comprises the regulatory and accessory proteins Tat, Rev, Nef, Vif, Vpr and Vpx (instead of Vpu as in HIV-1) (Figure 3). In HIV-2, the molecular weight of some proteins varies from those on HIV-1. This is particularly true for surface glycoprotein gp125 (gp120 for HIV-1) and transmembrane glycoprotein gp32 (gp41 in HIV-1) (Bron et al. 1997; de Silva, Cotten, and Rowland-Jones 2008; Reeves and Doms 2002).. Figure 3. HIV-2 genome. Adapted from Ayinde et al. 2010.. 1.4.. HIV replication cycle. In addition to genomic and structural similarity, HIV-1 and HIV-2 have very similar replication cycles. The main difference concerns the early phase, more precisely the entry step, since HIV-2 can often use a wider repertoire of both CC and CXC chemokine receptors including CCR1 to CCR5, CXCR2, and CXCR4 (Bron et al. 1997). Henceforth, only HIV-1 will be referred in this section, since every aspect of HIV-2, otherwise indicated, is considered to be analogous to HIV-1.. 6.
(39) Figure 4. HIV-1 replication cycle. After fusion with the host membrane, HIV-1 undergoes uncoating, followed by reverse transcription of viral ssRNA genome into dsDNA by reverse transcriptase (RT). This DNA is delivered into the nucleus and is then inserted into the host genome by the viral integrase (IN) to form the integrated provirus. Afterwards, viral DNA is transcribed and viral RNAs are processed and exported out to the cytoplasm. The new viral RNA is then used as genomic RNA and to be transcribed into viral proteins. Both viral RNA and proteins are translocated to the cell surface and a new virion is formed. The HIV virion suffers maturation through conformational changes upon the action of viral protease. Adapted from National Institute of Allergy and Infectious Diseases (NIAID) (https://www.niaid.nih.gov/).. 1.4.1. Early phase of HIV-1 cycle. The early phase of HIV-1 life cycle comprises several steps such as viral attachment, viral entry, uncoating, reverse transcription, nuclear import and integration. These steps will be overviewed in the sections below.. Viral attachment In order to enter the target cell, the virus needs to adhere to the cell surface before receptors engagement. This attachment can be relatively nonspecific via cellular lectins such as Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin. 7.
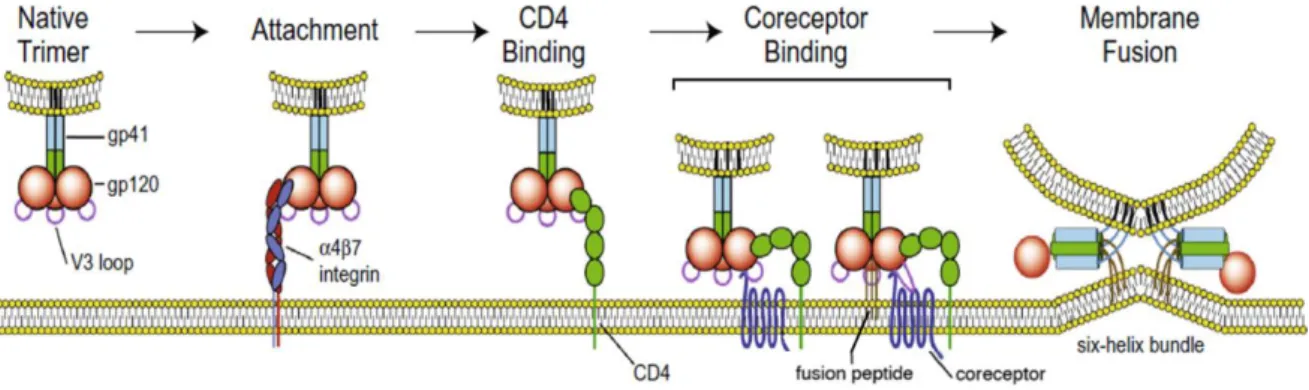
(40) (DC-SIGN) or Sialic acid binding Ig like lectin 1 (Siglec1), heparan sulphates, or the mannose receptor (reviewed in Clapham and McKnight 2002). Adhesion molecules can also play a role during this attachment process. For instance, Leukocyte Antigen Function1 (LFA-1) protein is expressed in target cells and can interact with Inter Cellular Adhesion Molecule 1 (ICAM1) proteins present on the virions surface (Fortin et al. 1999). Viral attachment can also result from specific interactions between Env and α4β7 integrin (Arthos et al. 2008; Cicala et al. 2009). HIV attachment to the host cell through any of these factors likely conveys Env into close proximity with the receptor CD4 and coreceptor, increasing the efficiency of infection.. Viral entry The cellular entry starts with the specific-binding of HIV-1 viral particles to cells bearing CD4 receptor via specific interactions between the viral envelope glycoprotein gp120 and the amino-terminal immunoglobulin domain of CD4. The gp120 subunit contains five relatively conserved domains (C1–C5) and five variable loops (V1–V5), named for their relative genetic heterogeneity. The variable loops are predominantly at the surface of gp120 and play critical roles in immune evasion and coreceptor binding, particularly the V3 loop (reviewed in Wilen et al. 2012). The binding of Env to CD4 causes rearrangements of V1/V2 and subsequently V3. The gp120-CD4 interaction is sufficient for binding but not for infection. Infection requires the binding of Env to chemokine receptors, in particular CCR5 and CXCR4, which are relevant for the natural course of infection due to their role in viral tropism (R5 and X4-tropic viruses, respectively). These surface receptors, responsible for mobilizing intracellular calcium and inducing leukocyte chemotaxis, serve as essential viral coreceptors to trigger membrane fusion. After the engagement of gp120 V3 loop to the extracellular domains of HIV coreceptor, gp41 undergoes conformational changes exposing a region of the viral transmembrane protein, the “fusion peptide”. Each gp41 in the trimer then folds at a hinge region, bringing an amino-terminal helical region. 8.
(41) (HR-N) and a carboxy-terminal helical region (HR-C) from each gp41 subunit together to form a six-helix bundle (6HB) (Chan et al. 1997; Weissenhorn et al. 1997) (Figure 5). The formation and enlargement of the fusion pore is accomplished through the formation of multiple 6HB, and allow the viral core to gain access to the cytoplasm (reviewed in Melikyan 2008; Klasse 2012). Although the direct fusion at the cell surface is the “classical route” of HIV-1 entry, the virus can also enter cells by endocytosis or by the formation of virological synapses. In the last years, numerous evidences have provided insights into the role of endocytic virus entry. Firstly, blocking the acidification of endosomal compartments to avoid the viral degradation in lysosomes can increase HIV infection (Schaeffer, Soros, and Greene 2004; Fredericksen et al. 2002). Secondly, coreceptor independent internalization of HIV occurs through an endocytic mechanism, leading to the detection of vesicular structures that contain intact particles (Blanco et al. 2004). Moreover, endocytosed viral particles can remain infectious and are able to endure until an appropriate environment allows for productive infection (Blanco et al. 2004; Bosch et al. 2008; Miyauchi et al. 2009). Furthermore, it has been shown that HIV-1 can enter lymphoid cells through an endocytic pathway (Miyauchi et al. 2009). Finally, after a sequential CD4 and coreceptor binding, HIV-1 undergoes endocytosis in a clathrin-dynamin-dependent mechanism (Bosch et al. 2008; Miyauchi et al. 2009). The clathrin-dependent endocytosis has also been associated. Figure 5. The HIV entry process. HIV-1 gp120 binds to its primary cellular receptor CD4 leading to conformational changes that allow binding of gp120 to the coreceptor—either CCR5 or CXCR4. This Env-coreceptor interaction allows the exposure of the gp41 “fusion peptide” and formation of the sixhelix bundle required to drive fusion of the viral and host cell membranes. Figure adapted from Haqqani and Tilton 2013.. 9.
(42) with virus transfer during cell-cell contacts (Bosch et al. 2008), implicating HIV endocytosis as a relevant pathway not only in cell-free virus entry but also in cell-to-cell transmission. Viral entry can be modulated by different cellular components. For instance, this step can be disrupted by family of proteins IFITM (Interferon Induced Transmembrane Proteins), either in target cells by retaining incoming particles in endosomes, or in virus-producing cells by leading decreased infectivity of produced virus (J. Yu et al. 2015; Lu et al. 2010; Tartour et al. 2014). Also, it has been reported that the lipid composition of the target membrane affects HIV-1 Env-mediated fusion and entry, being the HIV-1 entry in target cells associated to lipid rafts (Nisole, Krust, and Hovanessian 2002; Popik, Alce, and Au 2002). Lipid rafts are small but concentrated platforms in the plasma membrane, rich in glycosphingolipids, cholesterol and several proteins, including CD4, CCR5 and CXCR4 (Kozak, Heard, and Kabat 2002; Wysoczynski et al. 2005; Popik, Alce, and Au 2002).. Uncoating Once inside the cell, the HIV-1 core endures the uncoating process that involves the core disassembly by the dissociation of the capsid monomers (CA). It is known that CA proteins have functions beyond the simple encapsidation of the viral genome, and it has been described to have roles in reverse transcription and nuclear import (Yamashita et al. 2007; Dismuke and Aiken 2006; Iordanskiy et al. 2006; Forshey et al. 2002). For instance, CA seems to confer to HIV-1 the ability to infect non-dividing cells, separating lentivirus from other retrovirus (Yamashita and Emerman 2004; Yamashita et al. 2007). Lentivirus genomes penetrate the nucleus through an active nuclear import process. The field remains divided concerning the precise moment and location for uncoating, and several models have been proposed along the years (see Figure 6). Of note, these models may not be mutually exclusive, and factors such as cell type or activation state of target cells at the moment of infection may govern the uncoating process. The first model proposes. 10.
(43) that core disassembly occurs rapidly after virion fusion into the cytoplasm and in proximity to the plasma membrane, where most CA is dissociated before the formation of reverse transcriptional complex (RTC) (Suzuki and Craigie 2007; M. Bukrinsky 2004; LehmannChe and Saïb 2004). In the second model, supported by imaging-based approaches, the capsid remains intact for some time until the initiation of reverse transcription, suggesting. Figure 6. Models of HIV-1 uncoating. A) Uncoating occurs at the plasma membrane and is required to trigger viral reverse transcription. B) Some core disassembly occurs in the cytoplasm but a measurable amount of the capsid (CA) protein remains associated with the reverse transcription complex (RTC), suggesting that uncoating occurs gradually along the transport towards the nucleus. C) The successful completion of reverse transcription at the nuclear pore is the trigger for the uncoating process. Adapted from Campbell and Hope 2014.. 11.
(44) that uncoating occurs gradually along the transport towards the nucleus (Xu et al. 2013; Hulme, Perez, and Hope 2011). A third model has been proposed where the capsid remains intact until it arrives at the nuclear membrane where uncoating occurs at the nuclear pore upon completion of reverse transcription (Rasaiyaah et al. 2013; Lahaye et al. 2013). Both the second and the third models can support the finding that some level of CA remains associated with the pre-integration complex (PIC) in the nucleus (Peng et al. 2014; Matreyek et al. 2013; L. Zhou et al. 2011). Despite the different models, there are evidences for the involvement of several cellular factors in this step (reviewed in Ambrose and Aiken 2014). For example, Tripartite Motif 5 (TRIM5) proteins have been described to block HIV-1 infection by binding to its capsid and inducing premature disassembly before reverse transcription can occur (Stremlau et al. 2004). Another protein that binds to CA cores is cyclophilin A protein (CypA), a peptidylprolyl isomerase that is also packaged into HIV-1 virions (Luban et al. 1993). Although its role has been controversial, one study demonstrated that the binding of CypA either stabilizes or destabilizes the HIV-1 capsid depending on the target cell type (Li, Kar, and Sodroski 2009). Regarding cytoplasmic trafficking, HIV-1 virions hijack microtubule- and dynein-dependent trafficking to move towards the nucleus (McDonald et al. 2002; Lukic et al. 2014; Arhel et al. 2006).. Reverse Transcription Reverse transcription, although being a complex process, is well established (reviewed in Hu and Hughes 2012). The activation of reverse transcriptase (RT) to copy the singlestranded positive-sense viral RNA genome into double-stranded linear DNA is triggered by the exposure of the viral complex to non-limiting deoxyribonucleotides in the cytoplasm. While the role of p51 subunit of RT is mainly structural, p66 subunit contains both polymerase and RiboNuclease enzyme H (RNase H) enzymatic activities, essential for the reverse transcription process (Kohlstaedt et al., 1992). This process starts with the cellular. 12.
(45) Figure 7. HIV reverse transcription process. Reverse transcription is initiated by the synthesis of minus-strand DNA (in blue) at the PBS (Primer Binding Site) at the 5’ end of the RNA genome (in orange) by host cell tRNALys3. The minus-strand strong-stop DNA is then transferred to the 3’ end of the genome through complementarity with the R (Repeated) region of the LTR (Long-Terminal Repeats) thus allowing synthesis of the minus-strand DNA to be completed. Minusstrand DNA synthesis is accompanied by progressive degradation of the RNA matrix by the RNase H activity of reverse transcriptase (dashed orange line). In the HIV-1 genome, two polypurine tracts (PPT), the central PPT (cPPT) and 3’ PPT regions resist degradation by RNase H and serve as primers for synthesis of plus-strand DNA (green line). Plus-strand initiation in two distinct sites – cPPT and 3’ PPT – leads to a displacement of the downstream strand over 100 nucleotides, terminating at the central terminal sequence (CTS) and thus generating a discrete strand displacement called the central DNA Flap. Adapted from Esposito, Corona, and Tramontano 2012.. tRNA(Lys3) binding to the primer binding site (PBS) sequence of the viral RNA and continues with a series of steps involving cis-acting elements and RT enzymatic activity (see Figure 7). The final product of reverse transcription, full-length linear HIV-1 DNA, can be detected as early as 4h post-infection reaching the peak between 8h and 12h after infection (Barbosa et al. 1994; Kim et al. 1989). Reverse transcription can be altered by the action of the cellular apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3 family proteins (APOBEC3). The restriction factors APOBE3G and APOBEC3F are able to deaminate dC to dU in minus-strand viral cDNA during reverse transcription, leading to G-to-A hypermutation in the plus strand of viral genomes (Stopak et al. 2003; Mariani et al. 2003). HIV escapes this cellular restriction by expressing the viral infectivity factor (Vif), which induces proteasomal degradation of APOBEC3 proteins (Goila-Gaur and Strebel 2008; Sheehy et al. 2002). After reverse transcription is completed, the pre-integration complex (PIC) is formed.. 13.
(46) Nuclear Import The viral double-stranded DNA (dsDNA) is delivered into the nucleus in the form of a preintegration complex (PIC), which is composed by viral proteins, such as reverse transcriptase (RT), matrix (MA), Vpr, and integrase (IN) (Goff 2007; M. Bukrinsky 2004). The presence of CA in the PIC has been debated, as mentioned before. MA appears to hold two functional, yet rather weak, nuclear localization signals (NLS) (Haffar et al. 2000), Vpr presents one NLS (Popov et al. 1998), and IN seems to have several of these sequences (Ao et al. 2007). The HIV-1 DNA flap sequence has also been implicated in PIC nuclear import (Zennou et al. 2001; Arhel et al. 2007). These HIV-1 nucleoprotein complexes are believed to enter the nucleoplasm by passing through nuclear pore complexes (NPCs), which form stable channels through the nuclear envelope and gatekeep the trafficking of molecules between the cell nucleus and cytoplasm. It is known that HIV-1 hijacks several proteins for the active transport of viral dsDNA into the nucleus, such as nucleoporins Nup153, Nup93 and Nup358 (Di Nunzio et al. 2013), importins α, β (Suzuki and Craigie 2007) and transportin TNPO3/TRN-SR2 (Christ et al. 2008). PIC is also associated with many other cellular proteins, such as Ku70, Ku80, Ini1, PML, BAF, LEDGF/p75 and HMGA (Warrilow, Tachedjian, and Harrich 2009). All these factors may have an important role in the integration process. The modification of the viral DNA starts in the cytoplasm with the 3’-processing step (see Figure 8). In this catalytic process, IN multimerizes within the PIC to cleave the viral dsDNA genome in order to leave a recessed and chemically reactive hydroxyl group at both viral dsDNA 3’-ends (Li et al. 2006; Hare, Maertens, and Cherepanov 2012). HIV-1 PIC is then actively imported into the nucleus.. Integration After nuclear import of the PIC, the IN catalyzes the insertion of the linear viral dsDNA into the host cell chromosome, completing the early phase of HIV-1 replication cycle (Freed 2001; Goff 2007; Turner and Summers 1999). Besides the 3’-processing activity, IN also 14.
(47) presents the capacity of DNA strand transfer to accomplish viral genome integration (Figure 8). Within the nucleus, IN binds the target DNA (tDNA) and uses the reactive viral dsDNA 3’-hydroxyl groups to cleave the tDNA phosphodiester backbone and insert the viral dsDNA molecule at the same time, through the process of strand transfer (reviewed in Serrao and Engelman 2015). After disassembly of the strand transfer complex, host-cell enzymes repair the DNA recombination intermediate to yield the integrated provirus through non-homologous end joining (NHEJ) DNA repair pathway (Daniel et al. 2004). HIV-1 integration occurs more frequently into chromatin that is in close proximity to the nuclear periphery, indicating that PIC transport through the NPC and integration may be mechanistically linked (Di Primio et al. 2013; Marini et al. 2015; Lelek et al. 2015). It seems that some fraction of HIV-1 CA remains associated with the PIC as it enters into the cell nucleus (Hulme et al. 2015; Peng et al. 2014), suggesting that CA-binding factors like CypA, TNPO3, Nup358 and Nup153 can also influence the distribution of HIV-1 integration (Craigie and Bushman 2014). Also, the cellular protein lens epithelium-derived growth factor and coactivator protein p75 (LEDGF/p75) has been shown to form a stable tetramer. Figure 8. Mechanism of retroviral integration. The formation of the intasome occurs in the cytoplasm, where multimerized integrase (IN) is bound to the viral LTRs. The integration starts with the 3’processing activity of IN leading to cleaved ends of viral genome leaving chemically reactive hydroxyl groups. After nuclear import of the PIC, IN uses the 3’hydroxyl groups to cleave the tDNA phosphodiester and insert the viral dsDNA through the process of strand transfer. After the dissociation of strand transfer complex, host cellular enzymes repair the gap to produce the integrated provirus flanked by host DNA. Figure from Serrao and Engelman 2015.. 15.
(48) structure with HIV-1 IN, enhancing the strand transfer activity of viral integrase (Cherepanov et al. 2003a) and participating in the selection of insertion site (Ferris et al. 2010). Another protein that facilitate HIV-1 integration is Emerin, an inner nuclear envelope protein that seems to improve viral dsDNA localization to the chromatin before integration in macrophages (Jacque and Stevenson 2006). Regarding the integration distribution, HIV-1 preferentially integrate in transcriptionally active regions with high gene density and high GC content (Mitchell et al. 2004; G. P. Wang et al. 2007; Elleder et al. 2002; Brady et al. 2009), although transcriptionally silent but replication-competent proviruses were also reported in regions with low level of transcription such as gene desert and alphoid regions in heterochromatin (Dieudonné et al. 2009; Carteau, Hoffmann, and Bushman 1998).. After integration, the viral DNA is referred to as the provirus, where the 5′ LTR operates like any eukaryotic promoter and the 3′ LTR acts as the polyadenylation and termination site. Of note, not all linear viral dsDNA is integrated into the host cell chromosomes. The unintegrated DNA can also become circularized, creating 1-LTR and 2-LTR circles, or degraded (Butler, Johnson, and Bushman 2002). Of note, these different forms of unintegrated viral DNA can constitute pre-integration latency (Coiras et al. 2009).. 1.4.2. Late phase of HIV-1 cycle. HIV-1 late phase encompasses the steps of transcription, translation, assembly, budding and maturation. These steps will be overviewed in the sections below.. Transcription After integration, the provirus persists in the host cell to either remain latent (post‑ integration latency) or become transcriptionally active. In the latter scenario, proviral DNA. 16.
Imagem


+7





Documentos relacionados