Desenvolvimento de nova metodologia para avaliação da dissolução in vitro de
comprimidos de Besilato de Amlodipina comercializados em
Salvador/Bahia/Brasil, empregando planejamento experimental fatorial
Development of a new methodology for the evaluation of in vitro dissolution of
Amlodipine Besylate tablets sold in Salvador /Bahia /Brazil, using factorial
experimental design
DOI:10.34117/bjdv6n3-366
Recebimento dos originais: 14/02/2020 Aceitação para publicação: 24/03/2020
Fernanda de Souza Dias
Graduanda em Farmácia na Universidade do Estado da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
Gilmar Antônio de Carvalho Teles Júnior
Graduado em Farmácia pela Universidade do Estado da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
João Luís Silva de Oliveira
Graduando em Farmácia na Universidade do Estado da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
Desirée Aguiar Bonfim
Graduanda em Farmácia na Universidade do Estado da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
Jéssica Almeida Santos
Graduanda em Farmácia na Universidade do Estado da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
Laura Beatriz Souza e Souza
Graduanda em Farmácia na Universidade do Estado da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
Mestre em Química Aplicada pela Universidade do Estado da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
Fábio de Souza Dias
Doutor em Química pela Universidade Federal da Bahia Instituição: Universidade Federal da Bahia (UFBA) Endereço: Rua do Telégrafo, SN, Bomba, Camaçari - BA, Brasil
E-mail: [email protected]
Aníbal de Freitas Santos Júnior
Doutor em Química pela Universidade Federal da Bahia Instituição: Universidade do Estado da Bahia (UNEB)
Endereço: Rua Silveira Martins, 2555, Cabula, Salvador – BA, Brasil E-mail: [email protected]
RESUMO
Besilato de amlodipino (AB) é um vasodilatador (bloqueador dos canais de cálcio) usado no tratamento da hipertensão arterial. Na Farmacopeia Brasileira (2019) não existem metodologias para avaliar a dissolução de comprimidos contendo este fármaco. O objetivo deste estudo foi, com base em um planejamento fatorial completo de dois níveis 23, desenvolver e validar um método de dissolução utilizando espectrofotometria na região ultravioleta para determinar AB, em comprimidos (5 mg) disponíveis como referência (R), similares (S) e genérico (G) comercializados em Salvador, Bahia, Brasil. O planejamento experimental foi utilizado para analisar três variáveis: rotação (rpm), concentração de HCl (mol L-1) e volume do meio de dissolução (mL). Os dados foram submetidos à análise de variância (ANOVA) e o gráfico de Pareto foi construído para análise de variáveis e suas interações. As variáveis estudadas não afetaram significativamente a liberação (%) de AB. Considerando a concentração e o pH fisiológico do líquido gástrico (entre 1,5 e 2,0), os princípios da química verde e impactos ambientais, as condições mínimas (50 rpm e 500 mL de HCl 0,001 mol L-1) foram selecionadas. Todos os produtos liberaram AB satisfatoriamente, com pelo menos 75% do fármaco dissolvido em 30 minutos. O método proposto apresentou boa linearidade (r = 0,9997); precisão (desvio padrão relativo <1,0%) e exatidão (recuperação média de 99,89%); limites de quantificação e detecção de 5,46 e 1,80 µg mL-1, respectivamente. Essa nova metodologia contribui com a Farmacopeia Brasileira e com o controle de qualidade das formas farmacêuticas orais sólidas da AB.
Palavras-chave: perfis de dissolução, comprimidos de besilato de amlodipina, análise in vitro,
planejamento experimental.
ABSTRACT
Amlodipine Besylate (AB) is a vasodilator (calcium channel blocker) used to treat arterial hypertension. In the Brazilian Pharmacopoeia (2019) there are no methodologies for testing dissolution of tablets containing this drug. The aim of this study was, based on a Two-level full factorial design 23, to develop and validate a dissolution method using spectrophotometry in the ultraviolet region to determine AB, in tablets (5 mg) available as reference (R), similar (S) and generic (G) marketed in Salvador, Bahia, Brazil. Experimental factorial design was used to analyze three variables: rotation (rpm), HCl concentration (mol L-1) and volume of dissolution medium (mL). Data were subjected to analysis of variance (ANOVA) and the Pareto’s chart was constructed
for analysis of variables and their interactions. The studied variables had not significant effect on the release (%) of AB. Considering the concentration and physiological pH of gastric fluid (between 1.5 and 2.0), the principles of green technologies and environmental impacts, the minimum conditions (50 rpm and 500 mL of 0.001 mol L-1 HCl) were selected. All the products released AB satisfactorily, with at least 75% of the drug dissolved within 30 min. The proposed method presented good linearity (r = 0.9997); precision (relative standard deviation <1.0%) and accuracy (99.89% average recovery); quantitation and detection limits of 5.46 and 1.80 µg/mL, respectively. This new methodology to contribute with to Brazilian Pharmacopoeia and with quality control of AB solid oral dosage forms.
Keywords: dissolution profiles, amlodipine besylate tablets, in vitro analysis, experimental
factorial design.
1 INTRODUCTION
Amlodipine, a third-generation dihydropiridine, is a long-acting L-calcium channel blocker used worldwide, in the treatment of hypertension and angina pectoris [1]. It has reduced aqueous solubility and low permeability through the gastrointestinal tract, which affects its oral bioavailability and therapeutic targets. Therefore, it is commercially available in salt forms (besylate, mesylate, or maleate) to have better water solubility [2]. Amlodipine besylate – AB, is the most commonly used and their chemical name is 3-ethyl-5-methyl(±)-2-[(2-aminoethoxy) methyl]-4-(2-chlorophenyl)-1,4-dihydro-6-methyl-3,5-pyridinedicarboxylate, monobenzene sulphonate [3-5]. AB (Figure 1) belongs to Class 1 (high solubility and high permeability) of the Biopharmaceutical Classification System – BCS [6].
Figure 1. Molecular structure of Amlodipine besylate.
Font: Anumolu et al. (2014) [7]
In Brazil, AB (5 mg) can be found in reference, generic and similar medicines. Generic drug has the same active substance, pharmaceutical form, dosage and indication as the original reference drug. The country's health agencies require comparative studies of pharmaceutical and therapeutic (bio)equivalences with the reference product, by in vitro and in vivo tests. The same tests are required for similar drugs, which are produced after the expiration of the patent for
manufacturing reference medicines [8-10]. However, these medicines (reference, similar and generic) differ in characteristics such as size and formulation, with a variety of excipients which may influence the dissolution kinetics of the solid oral dosage forms (tablets and capsules, specially).
The Brazilian Pharmacopoeia (BP) defines quality control as the set of measures designed to guarantee, at any time, the production of lots of medicines and other products that meet the standards of identity, activity, content, purity, efficacy and safety [11]. The dissolution of the drug (assessed by the in vitro dissolution test), dissolution in the physiological medium and permeability through the gastrointestinal tract, after administration of solid oral dosage forms, are conditions determined by BP. However, no methodologies for testing dissolution of tablets containing BA, in the latest issue of BP, in 2019.
The dissolution test indicates a punctual analysis of the process. Therefore, a multipoint analysis is necessary to obtain detail of drug release through dissolution profiles [12]. In pharmaceutical industry, dissolution profiles are used for formulation development and optimization, drug quality and stability assessment, formulation and manufacturing process changes [13-15].
The experimental variables, independently and their possible interactions, can provide statistically significant parameters about the dissolution test, with the aid of the factorial experimental design method. The behavior of the system and optimal experiment conditions can be defined based on the design of few experiments, so that the main effects and interactions can be statistically determined [16,17]. Factorial design enables more efficiency and economy, since the determination of the most significant variables in a process can promote reduction in operating cost, improvement in productivity, reduction of analysis time, as well as better agreement between the obtained and expected values [18].
The dissolution studies of amlodipine besylate is unexplored, requiring further investigation. In the present study, a UV spectrophotometric method, based on an factorial experimental design 23, was developed and validated to study and compare the in vitro dissolution profiles, in different reaction media, of AB 5-mg tablets (reference, generic and similar drugs) produced by Brazilian national pharmaceutical companies. In addition, consequently to contribute to the expansion of the methodologies established by the Brazilian pharmacopoeia for AB solid oral dosage forms.
2 MATERIAL AND METHODS
All chemicals used, in this study, showed of analytical reagent grade and purchased from Quimex® (Merck, Brazil). Distilled water was obtained from a Q341 Quimis distiller (São Paulo, SP, Brazil) and used throughout the experiments. For to prepare all standard and sample solutions, in all analyzes, ultrapure water (with resistivity 18 MΩ cm–1) was obtained from a Milli-Q Pluswater purification system (Millipore Molsheim, France). All labware was soaked in a 10% (v v -1) HNO
3 solution bath for 24 h, rinsed with high-purity water and dried at ambient temperature. Hydrochloric acid (HCl), boric acid (H3BO3), potassium chloride (KCl), glacial acetic acid (CH3COOH) and sodium hydroxide (NaOH) were obtained from Neon Comercial (São Paulo, SP, Brazil).
The reference chemicals of Amlodipine besylate was obtained from Sigma-Aldrich (São Paulo, SP, Brazil) and Bioethica® Pharmacy (Salvador, BA, Brazil), respectively. Amlodipine besylate 5 mg tablets (reference, generic and similar) were purchased from local pharmacies in Salvador/Bahia/Brazil. All tests were performed on products within their expiration date. The characteristics of the three products as described in their drug labels (Table 1).
Table 1. Composition of drugs containing AB (5 mg).
Product Excipients
Reference (R)
Microcrystalline cellulose, anhydrous dibasic calcium phosphate, sodium starch glycolate and magnesium stearate.
Generic (G)
Sodium starch glycolate, dibasic calcium phosphate, microcrystalline cellulose and magnesium stearate.
Similar (S)
Microcrystalline cellulose, sodium starch glycolate, silicon dioxide, dibasic calcium phosphate and magnesium stearate.
Font: Drug package inserts/instructions for use
2.2 INSTRUMENTS
AB tablets were submitted to average weight and disintegration tests following BP [11]. An electronic analytical balance M164-AI (Mark®, Piracicaba, SP, Brazil)) and a disintegration test apparatus Model 301/AC 01 (Ethik®, Vargem Grande Paulista, Brazil) were used for tests. As the BP does not report methodology for AB dissolution test, a factorial experimental design was performed based on The United States Pharmacopeia – USP 41 [19]. A Two-level full factorial design (23) with central point replication for three variables was delineated for detect the influence of the variables on dissolution test. A dissolution test apparatus Model 299 (Ethik®, Vargem Grande Paulista, Brazil) multi-bath (n = 6) was used. A PG 2000 pH meter (Gehaka®, São Paulo,
Brazil) was used to determine the pH values of the dissolution media. All spectrophotometric measurements were carried out using an UV-Vis spectrophotometer (λ = 190-1100 nm) 1240 model (Shimadzu®, Kyoto, Japan) equipped with a diode array detector (DAD). The detector was set at λ = 238 nm. The absorbances of reference and sample solutions were read in 1 cm quartz cells, in triplicate.
2.3 CALIBRATION STANDARDS
A reference stock solution of 200 µg/mL was prepared with the reference chemical. Calibration standards with concentrations ranging from 0.5 to 20.0 μg mL-1 were prepared daily from the stock standard solution by appropriate dilution, stored in polyethylene bottles decontaminated and analyzed in triplicate by UV spectrophotometry at λ = 238 nm.
2.4 WEIGHT VARIATION AND DISINTEGRATION TESTS
For each brand, 20 tablets were randomly selected and weighed individually in the analytical balance. Then, the average weight, standard deviation and individual deviation of each tablet from the average for the three specialties (reference, generic and similar) were determined. BP [11] establish a maximum variation of ± 7.5%, and no more than two units outside the specified limits may be tolerated in relation to the average weight, but none may be above or below twice the indicated percentages.
In the disintegration test apparatus, six units of each sample (tablets) of each brand were used under the following conditions: distilled water as disintegration medium at 37 ± 1 °C and time of 30 minutes.
2.5 FULL FACTORIAL DESIGN AND DISSOLUTION TEST CONDITIONS
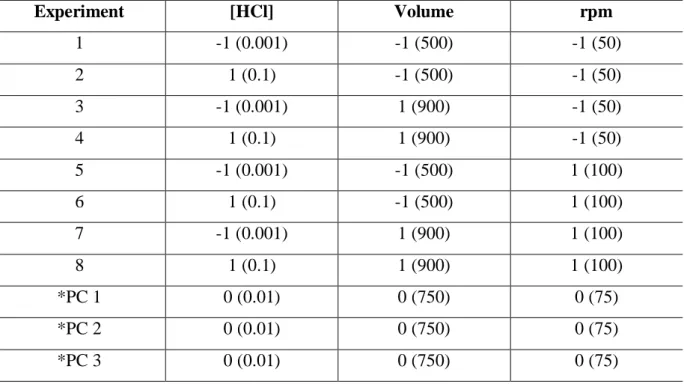
Factorial design with three variables (23) with central point replication was performed to detect the influence of the variables on dissolution test. Thus, 11 experiments were generated (Table 2). The variables analyzed were: rotation (rpm), HCl concentration (moL L-1) and volume of dissolution medium (mL). HCl concentration was based on physiological pH of gastric fluid (between 1.5 and 2.0). The fixed variables were temperature (37 ± 0.5 ° C) and type of apparatus (2, paddle), for each dissolution test, in accordance with USP 41 [19].
Table 2. Factorial design 23, with triplicate at central point, for dissolution test of AB tablets. Experiment [HCl] Volume rpm 1 -1 (0.001) -1 (500) -1 (50) 2 1 (0.1) -1 (500) -1 (50) 3 -1 (0.001) 1 (900) -1 (50) 4 1 (0.1) 1 (900) -1 (50) 5 -1 (0.001) -1 (500) 1 (100) 6 1 (0.1) -1 (500) 1 (100) 7 -1 (0.001) 1 (900) 1 (100) 8 1 (0.1) 1 (900) 1 (100) *PC 1 0 (0.01) 0 (750) 0 (75) *PC 2 0 (0.01) 0 (750) 0 (75) *PC 3 0 (0.01) 0 (750) 0 (75) *PC: central point
In all experiments, 10 mL sample aliquots were withdrawn at 30 minutes and replaced with an equal volume of fresh medium to maintain a constant total volume. After the filtration of the dissolution samples using 0.44-μm membrane filters, the concentrations of AB were determined simultaneously by the proposed spectrophotometric method. Absorbances were submitted to analysis of variance (ANOVA) and Pareto’s chart by Statistica 7.0 software. Thus, it is possible to determine the conditions of the variables for the test and dissolution profiles.
2.6 DISSOLUTION STUDIES
The dissolution tests was performed on the dissolution tester with 6 tablets with the dissolution medium at a temperature of 37 ± 0.5 °C and apparatus 2 (paddle). The rotation conditions (rpm), medium volume (mL) and HCl concentration (moL L-1) were fixed after the factorial experimental design was performed. For to obtain dissolution profiles, 10 mL sample aliquots were withdrawn at predetermined time intervals (1, 3, 5, 10, 15, 20, 25, 30, 45, and 60 min) and replaced with an equal volume of fresh medium to maintain a constant total volume. The collected aliquots were filtered (0.44-μm membrane filters) and transferred to amber vials for subsequent UV spectrophotometry reading at λ = 238 nm. Absorbances were converted to concentrations obtained from the equation on the standard curve. The calculations were performed considering the amount of drug removed in each aliquot. The results were expressed in percentage as a function of time.
From the obtained dissolution profiles were calculated the dissolution efficiency (DE), which is a parameter in the evaluation of pharmaceutical equivalence between pharmaceuticals products. This parameter was calculated from the area under the drug dissolution curve to the time (t) in minutes (ASC0-t), relative to 100% value label product (ASCTR) [20]. DE was expressed as a percentage and can be defined by the following equation: ED = ASC0-t / ASCTR × 100, where t is time (min) and TR is product label value.
In addition, to complement the dissolution profiles studies, tests were conducted to verified the influence of pH on the solubility of AB, in generic tablets, due to the greater availability of the samples. Thus, dissolution profiles were performed in the following media: hydrochloric acid (pH = 1.0), distilled water (pH = 6.03), sodium acetate at pH = 4.5 (glacial acetic acid + hydroxide sodium) and borate at pH = 8.0 (boric acid + potassium chloride + sodium hydroxide) buffers, under the same conditions and collection times mentioned above.
2.7 ANALYTICAL METHOD VALIDATION
In order to demonstrate whether the dissolution test method was adequate, it was validated following Brazilian Resolution 166/2017 [21] and International Conference on Harmonisation – ICH [22]. Thus, analysis of selectivity, linearity, precision (repeatability and intermediate precision), accuracy, limit of detection (LOD) and quantification (LOQ) were performed. Possible interferences were evaluated by testing the components of the formulations (excipients) of the formulations and comparing them with a placebo.
The linearity was obtained through the correlation coefficient, evaluated by linear regression analysis using the least squares regression method, of three analytical curves with ten different concentrations ranging from 0.5 to 20.0 µg mL-1 for AB. Repeatability and intermediate precision were carried out to assess the precision of the method. Relative standard deviation (RSD) was used to calculate the precision of quantification of three known concentrations of AB (1.0; 5.0, and, 10.0 µg mL-1) on the same and on alternate days. Accuracy was expressed as the agreement between the set reference value and the measured value of each concentration above, in triplicate, by UV-VIS spectrophotometer.
2.8 STATISTICAL ANALYSIS
A Student’s t-test for paired samples and ANOVA One-way by Tukey’s Multiple Comparison Test for p-value < 0.05 were used to compare the dissolving ability of AB and factorial design 23. Pareto’s chart were constructed for analysis of variables and their interactions. DE values were statistically analyzed using the Student’s t-test with a significance level of p ≤ 0.05.
3 RESULTS AND DISCUSSION
3.1 ANALYTICAL METHOD VALIDATION
The UV spectrophotometry method proposed in this study confirmed the capability for rapid analysis with good repeatability of this analytical technique for Amlodipine determination in pharmaceutical formulation [23]. There was no interference from matrix components (excipients). Linearity of concentration range AB standard calibration curve was obtained with a correlation coefficient (r) over (0.99) in the studied (5.0 to 25.0 μg mL-1), and then used to calculate the amount of the drug dissolved in each sample. The method was validated to demonstrate selectivity, linearity, LOD, LOQ, precision and accuracy (Table 3). Attimarad et al [23] developed and validated a method for quantification of amlodipine by manipulation of ratio spectra in pure and pharmaceutical formulation. Theirs results showed showed agreement with the validation parameters obtained in this study.
Table 3. Evaluation of Analytical parameters for AB determinations.
Analytical parameter Results
Regression equation y = 0.0342x – 0.0004
Correlation coefficient (r) 0.9995
Limit of detection (LOD), in µg mL-1 1.6
Limit of quantification (LOQ), µg mL-1 5.4
Precision* (%) 0.04 – 0.31
Accuracy* (%) 93.40 – 104.09
* precision and accuracy of the method were evaluated by calculating relative standard deviation (RSD %) and relative error percentage (RE %), respectively, for six determinations of A (1,0, 5.0 and, 10.0 µg/mL) over 2 days, under the same experimental conditions.
3.2 PHYSICAL TESTS (WEIGHT UNIFORMITY AND DISINTEGRATION)
The test for weight variation is applicable for solid oral dosage forms (hard capsules, uncoated tablets and film-coated). Uniformity of weight is a test that allows to evaluate the homogeneity of the weight of the units of each lot and, can be evaluated either by measuring the content uniformity or the weight of the tested units [11, 24]. AB tablets (5 mg) basically had the same excipients. The results showed that drugs R, G, and, S have satisfactory average weight values (0.2042; 0.1608 and, 0.1843 mg, respectively) according to the BP (for tablets with more than 150
and less than 300 mg of the drug, the allowable variation range is ± 7.5 %) [11]. For the similar medicine (S) a greater variation in the weight of the tablets was verified, which may be justified by the uniformity of granulated powder (irregular diameter).
All formulations met the official compendium requirements for tablets (disintegration time less than 30 min for tablets). Reference, generic and similar products disintegrated in 13, 35 and 24 seconds, respectively. The production process and the compression force employed may influence on disintegration. This rapid disintegration may be related to the presence of sodium starch glycolate and microcrystalline cellulose in formulations. Sodium starch glycolate (usual concentration between 2% and 8%) is widely used in oral pharmaceuticals as a disintegrant in tablet formulations prepared by either direct-compression or wet-granulation processes. Microcrystalline cellulose is widely used in pharmaceuticals, primarily as a binder/diluent in oral tablet and capsule formulations. In addition to its use as a binder/diluent, microcrystalline cellulose also has some lubricant and disintegrant properties that make it useful in tableting [25]. In presence of these excipients, disintegration occurs by rapid uptake of water followed by rapid and enormous swelling.
3.3 FULL FACTORIAL DESIGN AND DISSOLUTION TEST CONDITIONS
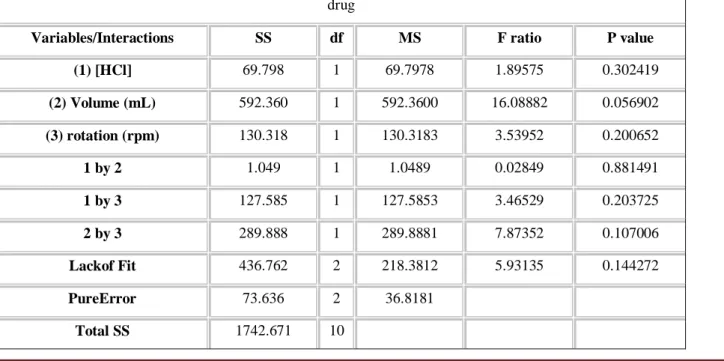
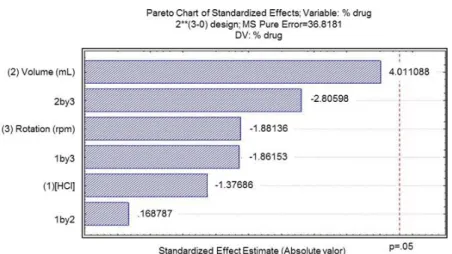
From the results of dissolution tests performed by factorial design 23 and central point replication, a statistical treatment (ANOVA) was applied (Table 4). Also, the Pareto’s chart was plotted (Figure 2). None of the variables under study were significant (p> 0.05) for the tested systems.
Table 4. Significance test (ANOVA) for the variables studied.
ANOVA; Var.:%drug; R-sqr=.70712; Adj:.26779 (factorial) 2**(3-0) design; MS PureError=36.8181 DV: % drug
Variables/Interactions SS df MS F ratio P value (1) [HCl] 69.798 1 69.7978 1.89575 0.302419 (2) Volume (mL) 592.360 1 592.3600 16.08882 0.056902 (3) rotation (rpm) 130.318 1 130.3183 3.53952 0.200652 1 by 2 1.049 1 1.0489 0.02849 0.881491 1 by 3 127.585 1 127.5853 3.46529 0.203725 2 by 3 289.888 1 289.8881 7.87352 0.107006 Lackof Fit 436.762 2 218.3812 5.93135 0.144272 PureError 73.636 2 36.8181 Total SS 1742.671 10
Legend = SS: Sum-of-squares / df: Degrees of freedom / MS: Mean squares.
Figure 2. Pareto’s chart for the studied variables and their interactions.
None of the studied variables were statistically significant (p> 0.05) for the studied systems. A positive variation was observed for the variable “volume of dissolution medium”. Taking into account the maximum capacity of the dissolution recipient (up to 1000 mL) and the principles of green chemistry and pharmacy (minimization of residues and environmental impacts), the minimum conditions (0.001 moL L-1 HCl; 500 mL and 50 rpm) were established for dissolution test, as well as for the dissolution profiles of the amlodipine besylate tablets 5 mg (R, G and S).
3.4 DISSOLUTION TEST, DISSOLUTION PROFILES (DP) AND DISSOLUTION EFFICIENCY (DE)
The USP 41 [19] recommends that not less than 75% of the AB label amount (5 mg) must dissolve within 30 min. Several factors may affect dissolution results, such as the nature of the excipients used and the rate of disintegration. Dissolution test is an important tool to guarantee the quality control of solid oral dosage forms, however a multipoint study is necessary to study the solids dissolution kinetics, understanding the drug dissolution profile (DP) [26]. DP to evaluate formulation development and final products, for batch quality control and establishing the similarity between test formulations and reference products [27]. Multipoint analysis graphs were plotted to evaluate the amount of the drug dissolved versus time of AB tablets (reference, generic, and similar) as shown in Figure 3.
Figure 3. Comparative dissolution profiles of AB (5 mg) tablets of reference, generic, and similar products. (USP type 2 apparatus at 50 rpm with 500 mL HCl 0.001 mol L-1 at 37.0 ± 0.5 oC for 1 h.)
Products R, G and S showed comparable levels of AB (5 mg), in 30 minutes. AB, in all tablets (reference, generic and similar), have dissolved in accordance with the USP 41 (>75% of drug within 30 min), with AB release of 115.0, 97.2 and 91.7%, respectively. In Brazil, Martinez et al. [28] evaluated dissolution of reference (R), similar (S1, S2, and S3), and generic (G) products (tablets containing AB). In their comparative dissolution profile studies, the author founded that dissolution efficiency of products G and S2 was statistically different from product R, using a dissolution apparatus fitted with a paddle (type-II apparatus) rotated at 75 rpm for 30 minutes, in 500 mL of 0.01 mol L-1, at 37°C ± 1°C. The average amlodipine contents in the samples varied between 99.59% and 106.52%. The results obtained in this study are in agreement with these authors.
G and S products released less than 100% of the labeled drug. It was observed that R, G and S drugs have a dissolution rate faster; in less than 5 minutes, the samples had already released almost all the drug. This can be explained by the fact that Amlodipine is a class 1 drug by SCB and all tablets contains sodium starch glycolate and microcrystalline cellulose (disintegrants) in its formulations. As Amlodipine is a long-acting L-calcium channel blocker used in the treatment of angina pectoris, its release should be rapid.
Dissolution efficiency values (% DE) offers a suitable alternative for quality control of immediate release products due to calculation the current amount of drug dissolved [29]. The reference, generic, and similar drug products showed DE values of 73.64, 61.34, and 59.14%, respectively. The efficiency of dissolution is not a comparative parameter of dissolution kinetics, but it is a parameter which characterizes dissolution of the drug. Malesuik et al [30] developed, in
Brazil, a dissolution test and a comparative study between tablets and capsules containing amlodipine. The authors found that the average dissolved percentage for all products was greater than 95% in 30 minutes. They also calculated ED for capsules (73.2 to 93.7%) and tablets (75.3 to 91.1%). DE, the area under a dissolution curve between defined time points, and the fit factors (f1 and f2) have been important tools for characterisation of dissolution profiles. In this study, factors f1 and f2 were not calculated due to AB is a drug class I and, a very rapid dissolution (> 85% dissolution of the drug) was observed in 15 minutes, for according to the criteria in the literature [8].
Drugs may have variable solubilities depending on pH and the dissolution rates of the drug may be varying in different regions of the gastrointestinal (GI) tract [31]. The pH of the GI fluids in the local region of the stomach and intestine will influence a drug’s dissolution rate and possibly its permeability. Therefore, the fraction of drug absorbed is a function of drug solubility, dose, and GI permeability [32]. Amlodipine is described as slightly soluble in water in different Pharmacopoeias [19, 33]. It is stable under ordinary conditions, and has a pKa value of 8.7 [34]. Within the gastrointestinal pH range, amlodipine is an ionized compound (weak base) [35].
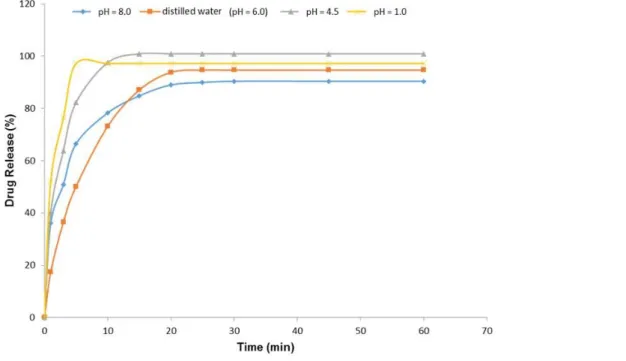
As BP [11] does not present methodology for dissolution tests of solid forms of AB, in this study, different reaction media with varying pH values were studied to evaluate the release of AB tablets. Influence of pH on the release of BA tablets (generic products) was studied in four different reaction media, based on the physiological pH range with different pH values (1.0; 4.5; 6.0 and 8.0). The results obtained showed that AB release is in agreed with the literature, indicating that this drug has a higher solubility in water, acidic medium and sodium acetate buffer (Figure 4).
Figure 4. Comparative dissolution profiles of AB (5 mg) tablets of generic product in distilled water and different pH solutions, (1.0; 4.5; 6.0; 8.0). (USP type 2 apparatus at 50 rpm with 500 mL HCl 0.001 mol L-1 at 37.0 ± 0.5 oC for 1 h.)
According to FDA, a drug product is considered to be very rapidly released if ≥ 85% of the drug is dissolved in 15 minutes, which corresponds to gastric emptying half-life (T50%) in fasting conditions [36]. In this study, after 15 minutes, more than 75% of drug was released in R, G and S products, regardless of the pH value of the medium.
In this study, the investigation of other means of dissolution was proposed, aiming to expand the studies about the behavior of the release of AB. Shohin et al [35] evaluated the dissolution characteristics (in different reaction media) of the reference and marketed products in Russia. The operational conditions were 500 mL of USP buffer solutions (pH 1.2, 4.5 and 6.8), apparatus 2 at 75 rpm. Samples were collected at 10, 15, 20, 30 and 45 minutes and quantified on the λ = 239 nm UV-visible spectrophotometer. Rapid dissolution was observed in both specialties, i. e. within 30 minutes more than 85% of the drug was dissolved in all media. In this study, in agreement with the aforementioned authors, in pH 6.8, the products rapidly dissolved and dissolution profiles were comparable. In Pakistan, Feroz et al [37] studied the release of AB 5 mg, in tablets (six brands) in 900 mL of different dissolution media (water, pH 1.2, 4.5 and 6.8), using apparatus 2 at 75 rpm. From the results obtained, the authors concluded that the products were considered pharmaceutical equivalents due to presented very fast dissolution.
Jung-Cook et al [38] studied the release of AB products in the different dissolution media (500 mL), in Mexico, using USP apparatus 2 at 75 rpm at 37 ± 0.5 °C. At pH 1.2 and 4.5, all the products fulfilled the criteria for very rapidly dissolving products (> 85 % disso lved within 15 min).
Nevertheless, when pH 6.8 was used, neither the reference nor the test products met the criteria for rapidly dissolving products. In Nigeria, Olusola et al [39] evaluated the dissolution profiles of two generic and reference products containing AB, using apparatus 2 at 50 rpm and 900 mL of buffers at pH 1.2, 4.5 and 6.8. Samples were collected at 5, 10, 15, 30 and 50 minutes and quantified on the UV-visible spectrophotometer at λ = 238 nm. In all media, dissolution was low, and hence none of the products complied the FDA criteria [36] for very rapidly or rapidly dissolving tablets. These data were in disagreement with the present and Shohin et al [35] studies. However, all drugs tested showed a release greater than 75% within 30 minutes of dissolution.
4 CONCLUSION
AB is a drug widely used in Brazil and worldwide to treat hypertension and angina. For the products tested (R, G and S), the weight uniformity and disintegration tests showed compliance with the requirements of the Brazilian Pharmacopeia. In Brazilian Pharmacopeia there are no methodologies for testing dissolution of tablets containing this drug. The application of factorial design 23 allowed us to evaluate the statistical significance of the factors investigated for the development of a dissolution test for AB tablets (5mg). The independent variables and their interactions were not statistically significant. Therefore, the factorial design made possible the reduction of generated residues, due to it was possible to perform the test and dissolution profile under the minimum conditions (50 rpm; 500 mL of 0.001 mol L-1 HCl). All tablets (reference, generic and similar) have dissolved in accordance with the USP 41 (>75% of drug within 30 min), with AB release of 115.0, 97.2 and 91.7%, respectively. In comparative dissolution profile studies, all products (R, G and S) have a dissolution rate faster (< 5 minutes) and G and S products released less than 100% of the labeled drug. Sodium starch glycolate and microcrystalline cellulose (disintegrants) were present in the formulations and may have contributed to the rapid release of AB. Influence of pH on the release of BA tablets (generic products) was studied. The results obtained showed that AB release is in agreed with the literature, indicating that this drug has a higher solubility in water, acidic medium and sodium acetate buffer.
The Dissolution test for AB was proposed using a validated UV spectrophotometric method with good linearity, precision and accuracy, in order to contribute to Brazilian Pharmacopeia.
ACKNOWLEDGEMENTS
The authors are grateful for the financial support received from Bahia State Research Support Foundation (FAPESB), Brazilian National Council for Scientific and Technological
Development (CNPq), collaborations of the State University of Bahia (UNEB) and Research Group: “Biopharmaceutics and Drugs”.
REFERENCES
1. Şen, S.; Demir, M.; Yiğit, Z.; Üresin, A. Y. Efficacy and safety of s-amlodipine 2.5 and 5 mg/d in hypertensive patients who were treatment-naive or previously received antihypertensive monotherapy. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 318–28. DOI: 10.1177/1074248418769054.
2. Sheraz, M. A.; Ahsan, S. F.; Khan, M. F.; Ahmed, S.; Ahmad, I. Formulations of amlodipine: a review. J. Pharm. (Cairo). 2016: 2016, 8961621. DOI: 10.1155/2016/8961621.
3. Rabhi, S.; Belkacemi, H.; Bououdina, M.; Kerrami, A.; Brahem, L. A.; Sakher, E. Effect of Ag doping of TiO2 nanoparticles on anatase-rutile phase transformation and excellent photodegradation of amlodipine besylate. Mater. Lett. 2019, 236, 640–43. DOI: 10.1016/j.matlet.2018.11.006.
4. Hussan, K. P. S.; Thayyil, M. S.; Rajan, V. K.; Muraleedharan, K. DFT studies on global parameters, antioxidant mechanism and molecular docking of amlodipine besylate. Comput. Biol.
Chem. 2019, 80, 46–53. DOI: 10.1016/j.compbiolchem.2019.03.006.
5. Kapoor, H.; Aqil, M.; Imam, S. S.; Sultana, Y.; Ali, A. Formulation of amlodipine nano lipid carrier: formulation design, physicochemical and transdermal absorption investigation. J. Drug
Deliv. Sci. Tec. 2019, 49, 209–18. DOI: 10.1016/j.jddst.2018.11.004.
6. Kaynak, M. S.; Bogacz, A.; Stelmasin´ski, M.; Şahin, S. Bioavailability file: amlodipine.
FABAD J. Pharm. Sci. 2011, 36, 207–22.
7. Anumolu, P. D.; Neeli, S.; Anuganti, H.; Ranganatham, S. B. P.; Satya, S. C. V. Development of dissolution test method for a telmisartan/amlodipine besylate combination using synchronous derivative spectrofluorimetry. Braz. J. Pharm. Sci. 2014, 50, 329–36. DOI: 10.1590/S1984-82502014000200012.
8. Resolution of the Board of Directors – RDC No. 31. Ministry of Health, Brazilian Health Surveillance Agency (ANVISA): Brasília, August 11, 2010. Accessed Jan 04, 2020.http://portal.anvisa.gov.br/documents/33880/2568070/res0031_11_08_2010.pdf/5e157d15-d3d5-4bb9-98db-5667e4d9e0c8.
9. Resolution of the Board of Directors – RDC No. 16. Ministry of Health, Brazilian Health Surveillance Agency (ANVISA): Brasília, March 02, 2007. Accessed Jan 04, 2020.http://bvsms.saude.gov.br/bvs/saudelegis/anvisa/2007/rdc0016_02_03_2007.html.
10. Resolution of the Board of Directors – RDC No. 17. Ministry of Health, Brazilian Health Surveillance Agency (ANVISA): Brasília, March 02, 2007. Accessed Jan 04, 2020.http://bvsms.saude.gov.br/bvs/saudelegis/anvisa/2007/rdc0017_02_03_2007.html.
11. Brazilian Pharmacopeia, 6th ed.; Brasília: Brazilian Health Surveillance Agency; 2019.
12. Jambhekar, S. S.; Breen, P. J. Drug dissolution: significance of physicochemical properties and physiological conditions. Drug Discov. Today 2013, 18: 1173–84. DOI: 10.1016/j.drudis.2013.08.013.
13. Nickerson, B.; Kong, A.; Gerst, P.; Kao, S. Correlation of dissolution and disintegration results for an immediate-release tablet. J. Pharmaceut. Biomed. 2018, 150, 333–40. DOI: 10.1016/j.jpba.2017.12.017.
14. Todeschini, V.; Sangoi, M. S.; Goelzer, G. K.; Machado, J. C.; Paim, C. S.; Araujo, B.V.; Volpato, N. M. Dissolution method for delapril and manidipine combination tablets based on an absorption profile of manidipine. J. Pharm. Anal. 2016, 6, 49–55. DOI: 10.1016/j.jpha.2015.10.002.
15. Conceição, A. P.; Sá, R. R.; Silva, V. C.; Ferreira, M. S.; Cazedey, E. C. L.; Magalhães, H. I. F.; Santos Junior, A. F. A comparative study of propranolol release by in vitro dissolution profiles in pharmaceutical formulations. Dissolut. Technol. 2018, 25, 54–61. DOI: 10.14227/DT250418P54.
16. Kumar, L.; Reddy, M. S.; Managuli, R. S.; Pai K., G. Full factorial design for optimization, development and validation of HPLC method to determine valsartan in nanoparticles. Saudi Pharm
17. Jain, A.; Jain, S. K. Formulation and optimization of temozolomide nanoparticles by 3 factor 2 level factorial design. Biomatter. 2013, 3, e25102. DOI: 10.4161/biom.25102.
18. Calado, V.; Montgomery, D. C. Planejamento de experimentos usando Statistica, 1th ed.; Rio de Janeiro: E-Papers Serviços Editoriais; 2003, pp 1–260.
19. The United States Pharmacopeia and National Formulary USP 41–NF 3640; The United States Pharmacopeial Convention, Inc.: Rockville, MD, 2018.
20. Khan, K. A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. DOI: 10.1111/j.2042-7158.1975.tb09378.x.
21. Resolution of the Board of Directors – RDC No. 166. Ministry of Health, Brazilian Health Surveillance Agency (ANVISA): Brasília, July 24, 2017. Accessed Jan 04, 2020. https://www20.anvisa.gov.br/coifa/pdf/rdc166.pdf.
22. International Conference on Harmonisation (ICH). International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH Q14: Analytical Procedure
Development and Revision of Q2(R1)Analytical Validation; ICH Harmonised Tripartite Guideline:
Geneva, Switzerland, 2018. Accessed Jan 4, 2020. https://database.ich.org/sites/default/files/Q2R2-Q14_EWG_Concept_Paper.pdf.
23. Attimarad, M.; Narayanswamy, V. K.; Aldhubaib, B. E.; SreeHarsha, N.; Nair, A. B. Development of UV spectrophotometry methods for concurrent quantification of amlodipine and celecoxib by manipulation of ratio spectra in pure and pharmaceutical formulation. PLoS One.
2019, 14, e0222526. DOI: 10.1371/journal.pone.0222526.
24. Zaid, A. N.; Al-Ramahi, R. J.; Ghoush, A. A.; Qaddumi, A.; Zaaror, Y. A. Weight and content uniformity of lorazepam half-tablets: a study of correlation of a low drug content product. Saudi
Pharm. J. 2013, 21, 71–5. DOI: 10.1016/j.jsps.2011.12.009.
25. Rowe, R. C.; Sheskey, P. J.; Quinn, M. E. Handbook of pharmaceutical excipients, 6rd ed; London: Pharmaceutical Press, 2009, pp 1–917.
27. Storpirtis, S.; Gonçalves, J. E.; Chiann, C.; Gai, M. N. Biopharmacotechnics, Rio de Janeiro: Guanabara Koogan; 2011, pp 35.
28. Martinez, R. M.; Silva, J. F.; Jorge, L. R.; Ishikawa, R. L.; Novelli, A. P.; Cezar, T. L. C.; Georgetti, S. R.; Baracat, M. M.; Casagrande, R. Validation of methodology for assay, pharmaceutical equivalence, and comparative dissolution profile for tablets containing amlodipine besylate. J. Appl. Pharm. Sci. 2019, 9, 93–100. DOI: 10.7324/JAPS.2019.91112.
29. Simionato, L. D.; Petrone, L.; Baldut, M.; Bonafede, S. L.; Segall. A. I. Comparison between the dissolution profiles of nine meloxicam tablet brands commercially available in Buenos Aires, Argentina. Saudi Pharm. J. 2018, 26, 578–84. DOI: 10.1016/j.jsps.2018.01.015.
30. Malesuik, M. D.; Cardoso, S. G.; Lanzanova, F. A.; Bajerski, L.; Dorigoni, E. Development of dissolution test and comparative study of tablets and compounded capsules containing amlodipine.
Rev. Cienc. Farm. Basica Apl. 2006, 27, 37–49.
31. Kim, T. H.; Shin, S.; Jeong, S. W.; Lee, J. B.; Shin, B. S. Physiologically relevant in vitro-in vivo correlation (ivivc) approach for sildenafil with site-dependent dissolution. Pharmaceutics.
2019, 11, E251. DOI: 10.3390/pharmaceutics11060251.
32. Amidon, G. L.; Lennernas, H.; Shah, V. P.; Crison, J. R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995, 12, 413–20. DOI: 10.1023/a:1016212804288.
33. European Pharmacopoeia, 10th ed.; European Directorate for the Quality of Medicines & Healthcare, Council of Europe: Strasbourg, France, 2019.
34. Burges, R.; Moisey, D. Unique pharmacologic properties of amlodipine. Am. J. Cardiol. 1994,
73, 2A–9A. DOI: 10.1016/0002-9149(94)90268-2.
35. Shohin, I. E.; Ramenskaya, G. V.; Vasilenko, G. F.; Malashenko, E. A. In Vitro Dissolution Kinetics of Amlodipine Tablets Marketed in Russia Under Biowaiver Conditions. Dissolut.
36. Food and Drug Administration (FDA) Draft Guidance for Industry, Waiver of In vivo
bioavailability and bioequivalence studies for immediate-release solid oral dosage forms containing certain active moieties/active ingredients based on a biopharmaceutics classification system. February 1999, CDER/ FDA.
37. Feroz M.; Razvi, N.; Ghayas, S.; Anjum, F.; Ghazal, L.; Siddiqui, S. A. Assessment of pharmaceutical quality control and equivalence of various brands of amlodipine besylate (5 mg) tablets available in the Pakistani market under biowaiver conditions. Int. J. Pharm. Pharm. Sci.
2015, 6, 909–13.
38. Jung-Cook, H.; Mayet-Cruz, L.; Girard-Cuesy, M. E. Comparative in vitro dissolution and in vivo bioavailability of commercial amlodipine tablets. Trop. J. Pharm. Res. 2018, 17, 1685–91. DOI: 10.4314/tjpr.v17i9.1.
39. Olusola, A. M.; Olubukola, O. O.; Emeka, O. H.; Lilian, A. E. Equivalence of two generic brands of amlodipine besylate under biowaiver conditions. Int. J. Pharm. Pharm. Sci. 2012, 4, 265– 68.