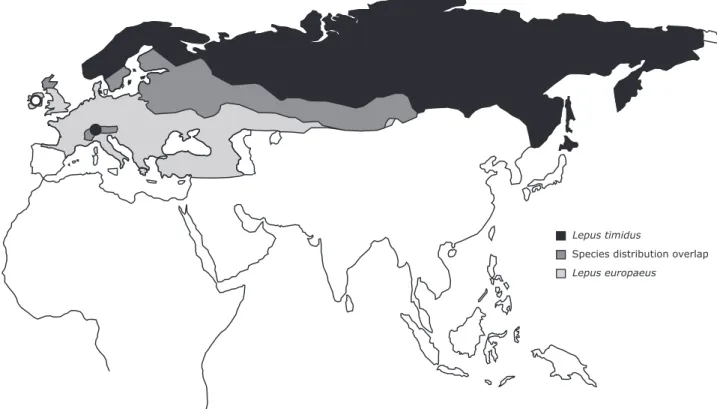
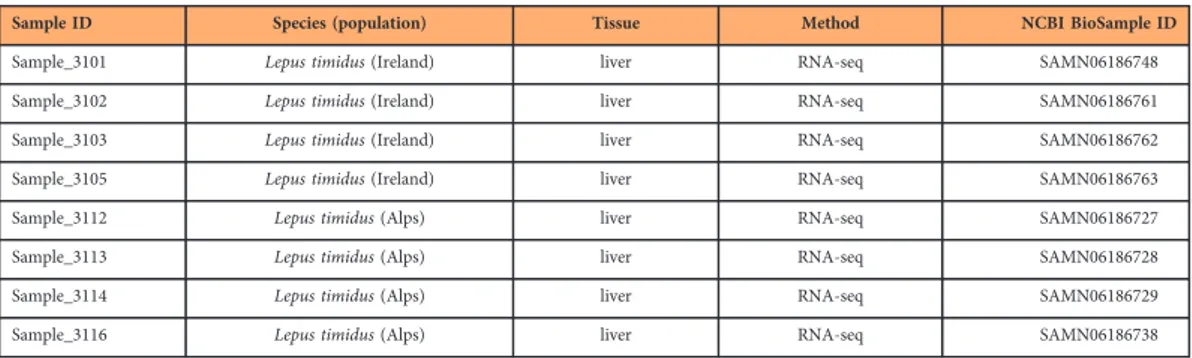
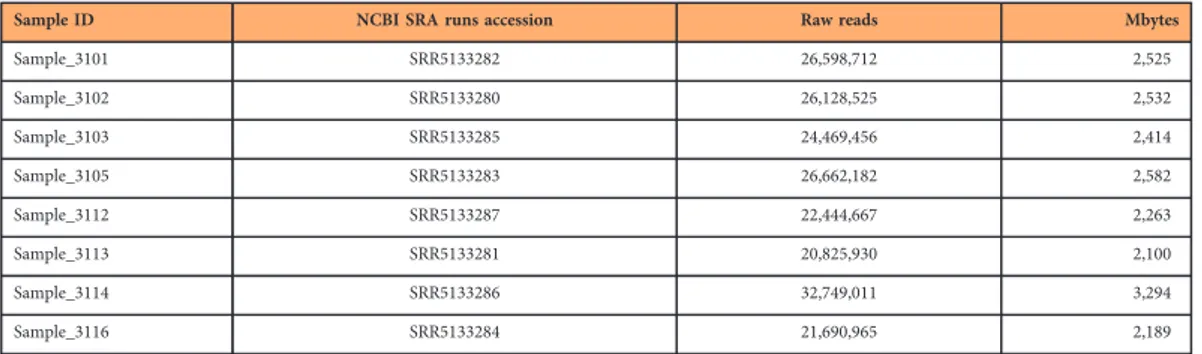
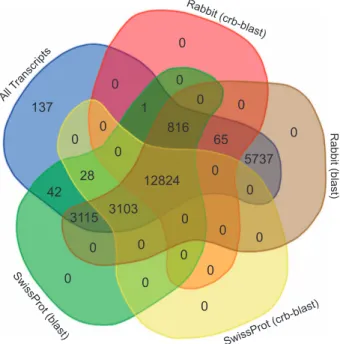
Data Descriptor: Mountain hare transcriptome and diagnostic markers as resources to monitor hybridization with European hares
Texto
Imagem

Documentos relacionados
Ao considerar que as actividades realizadas nas creches enriquecem as experiências infantis e possuem um significado para a vida das crianças, Wiggers (2008)
Para evitar o colapso da estrutura, as forças e des- locamentos aplicados não se aproximaram de valores críticos, embora fosse notória a perda de resistência da parede de fachada.
A ciência sedentária, por outro lado, existe somente no espaço estriado, fechado, limitado pelo horizonte ao sistema métrico e dimensional, o qual é extensivo
Durante a realização do estudo, observou-se que o conceito decisão de preço de venda é o mais predominante para uma empresa porque através desse conceito
To mimic at best the human disease which initiate in a bone microenvironment and spontaneously spread mainly to the lungs, we established different in vivo orthotopic models, either
Mesmo uma embalagem, cuja função é a proteção do alimento, pode estar externamente contaminada e apresentar traços de matéria orgânica, suficientes para servirem de substrato
Avaliação de inseticidas sobre a biologia e embriologia de Spodoptera frugiperda (J.E. Smith) (Lepidoptera: Noctuidae) e o efeito em Trichogramma pretiosum Riley
