DEPARTAMENTO DE QUÍMICA
PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
ANÁLISE QUÍMICA DE MATERIAIS CERÂMICOS:
DIGESTÃO POR FUSÃO E MEDIDAS POR ICP OES COM
CONFIGURAÇÃO AXIAL
Telma Blanco Matias
Tese apresentada como parte dos
requisitos para obtenção do título de
DOUTOR EM CIÊNCIAS, área de
concentração: QUÍMICA ANALÍTICA.
Orientador: Prof. Dr. Joaquim de Araújo Nóbrega
Ficha catalográfica elaborada pelo DePT da Biblioteca Comunitária/UFSCar
M433aq
Matias, Telma Blanco.
Análise química de materiais cerâmicos : digestão por fusão e medidas por ICP OES com configuração axial / Telma Blanco Matias. -- São Carlos : UFSCar, 2008. 110 f.
Tese (Doutorado) -- Universidade Federal de São Carlos, 2007.
1. Química analítica. 2. Materiais cerâmicos. 3. Análise química. 4. ICP-OES I. Título.
Declaramos para os devidos fins que TELMA BLANCO -MATrAS;-defendeu nesta d81asua tese~ de doutoraClO na -
áreã---de-Química Analítica, sob o título: "Análise Química de Materiais
Cerâmicos:
Digestão
por Fusão
e Medidas
por ICP OES com
Configuração
Axial", teve como banca examinadora
os Profs. Drs.
Joaquim de Araújo Nóbrega (Orientador), Edivaldo Egea Garcia (DQjUEM), Francisco José Krug (CENAjUSP-Piracicaba), Pedro Vitoriano de Oliveira (IQjUSP-SP) e Edenir Rodrigues Pereira Filho
(DQ jUFSCar) e foi considerada aprovada.
São Carlos, 26 de outubro de 2007.
~
~~
G
rP~
Prof. Dr. Joaquim de-hraújo Nóbrega
tn
Coordenador do PPGQ
.. Este
documento
é válido
por 3 meses.
UNIVERSIDADE FEDERAL DE SÃO CARLOS Via Washington Luiz, Km. 235
CEP 13.565-905 - São Car1os - SP - Brasil
CENTRO DE CIÊNCIAS EXATAS E DE TECNOLOGIA
DEPARTAMENTO DE QUÍMICA
“ Nunca ande pelo caminho traçado, pois ele conduz somente até onde os outros foram”.
Pais, pela estrutura sólida, amor e exemplo de vida
Ao meu filho, que desde sempre foi razão e inspiração para a vida, representando minha maior realização como ser humano.
À Deus
Ao Prof Joaquim de Araújo Nóbrega, mais que um orientador, um amigo com quem interagi tantos anos e com quem participei de desafios que me trouxeram cada vez mais experiência e amadurecimento é, sem dúvida, um professor no sentido profundo da palavra.
Ao CCDM pela oportunidade e apoio financeiro.
À Fapesp pelo auxílio financeiro ao desenvolvimento de projetos de pesquisas.
Ao Programa de Pós-graduação em Química da Universidade Federal de São Carlos pela oportunidade.
Às secretárias da Pós-graduação Ariane, Cristina e Luciani por estarem sempre dispostas a ajudar e pelos serviços prestados.
Aos meus companheiros de trabalho do CCDM Rosi, Fernando e Juninho que muito ajudaram na realização deste trabalho.
Aos meus amigos do GAIA e ex-GAIA pelo apoio e interesse manifestado.
Aos meus pais João e Verbena (in memoriam) pela estrutura familiar sólida, amor e exemplo de vida.
Ao meu filho querido por simplesmente existir e me fazer tão orgulhosa do homem e profissional que é.
Ao Abner pelo companheirismo, pelos cuidados e amor sempre presentes.
A todos, agradeço de coração.
• AAS: espectrometria de absorção atômica (absorption atomic spectrometry)
• BEC: concentração equivalente ao sinal de fundo (background equivalente concentration)
• CCD: dispositivo de carga acoplada (charge-coupled detector)
• CNR: condições não robustas
• CR: condições robustas
• DCP-AES: espectrômetro de emissão atômica com plasma de corrente
contínua (direct current plasma-atomic emission spectrometry)
• EIEs: elementos facilmente ionizáveis (easily ionizated elements)
• FAAS: espectrometria de absorção atômica com chama (flame atomic absorption spectroscopy)
• GFAAS:espectrometria de absorção atômica com forno de grafite (graphite furnace atomic absorption spectrometry)
• ICP OES: espectrometria de emissão óptica com plasma acoplado
indutivamente (inductively coupled plasma optical emission spectrometry)
• ICP-MS: espectrometria de massas com plasma acoplado indutivamente (inductively coupled plasma-mass spectrometry)
• LOD: limite de detecção (limit of detection)
• LOQ: limite de quantificação (limit of quantification)
• LTE: equilíbrio termodinâmico local (local thermal equilibrium)
• Mg II / Mg I: razão das intensidades de emissão de linha iônica (Mg 280 nm) / linha atômica (285 nm)
• NAA: análise por ativação neutrônica (analysis by neutronic activation)
• PTFE: politetrafluoretileno
• RSD: desvio padrão relativo (relative standard desviation)
• SBR: razão sinal analítico / sinal de fundo (signal background ratio)
• SRM: material de referência certificado (standard reference material)
• UV: ultravioleta
• VIS: visível
TABELA 2.0 - Pontos de fusão de compostos comumente utilizados para fusão de amostras. Adaptado de Anderson 16 e Sulcek.17...9
TABELA 2.1 - Aplicações do método de fusão na decomposição de materiais inorgânicos, adaptada de Krug.15...14
TABELA 2.2 - Categorias de amostras e exemplos, adaptada de Krug.15...15
TABELA 4.0 - Parâmetros instrumentais para as medidas por ICP OES com visão axial...39
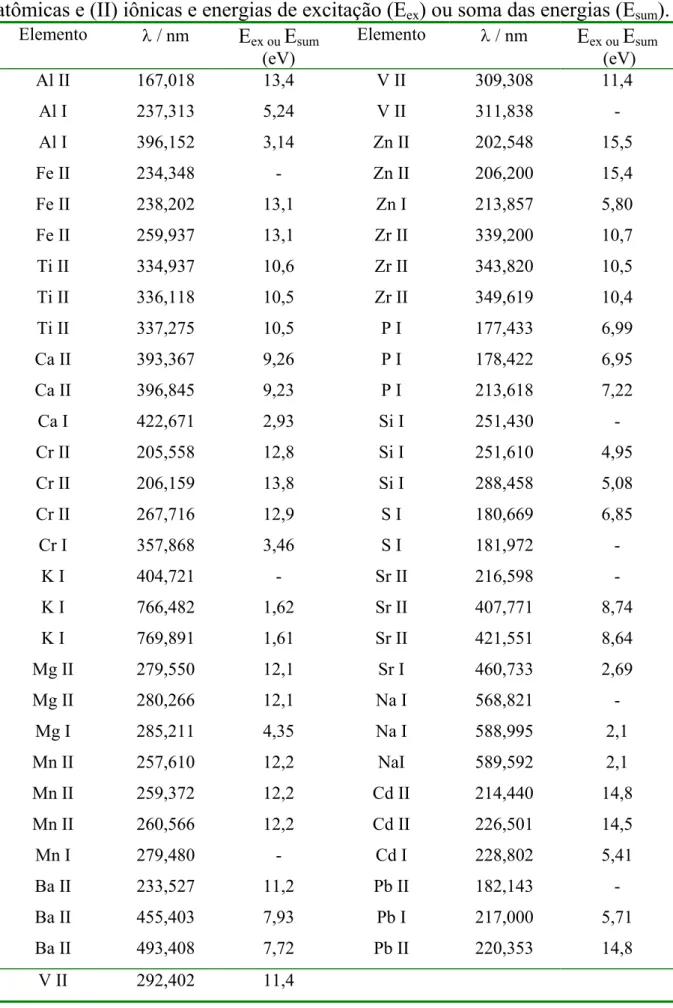
TABELA 4.1 - Elementos avaliados e suas respectivas linhas espectrais (I) atômicas e (II) iônicas e energias de excitação.8...40
TABELA 5.0 - Razão Mg II / Mg I em diferentes meios...53
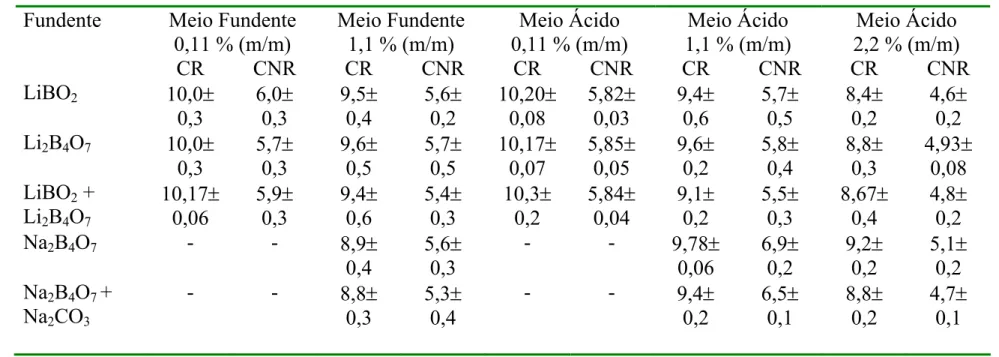
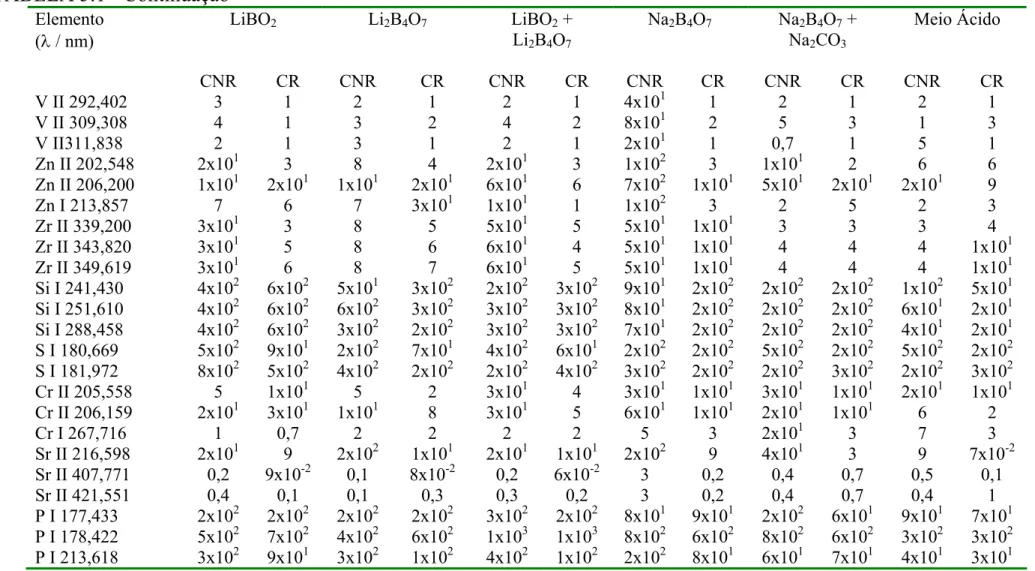
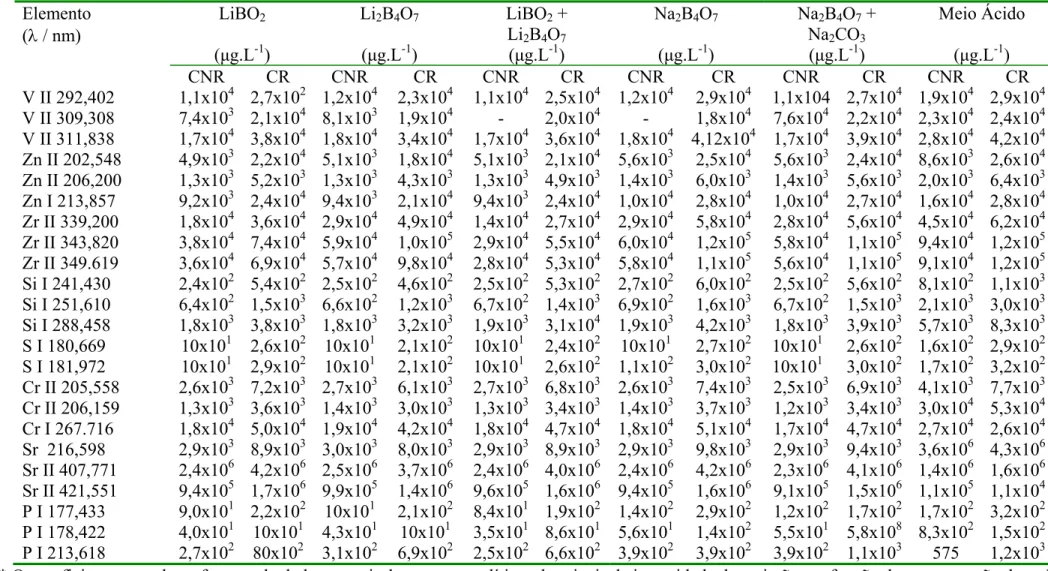
TABELA 5.1 - Limites de Detecção para todos os analitos nos diferentes meios estudados (1,1 % m/v de sólidos totais dissolvidos para os meios fundentes e HNO 5% v/v para o meio ácido)...56
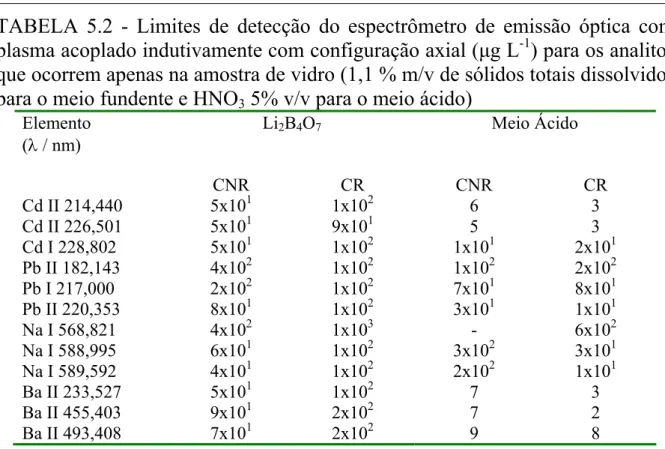
TABELA 5.2 - Limites de detecção para os analitos que ocorrem apenas na amostra de vidro. (1,1 % m/v de sólidos totais dissolvidos para o meio fundente e HNO 5% v/v para o meio acido)...58
TABELA 5.3 - Avaliação da linearidade em diferentes meios e condições de operação com curvas analíticas de calibração multielementares. Concentrações expressas em mg L-1...60
TABELA 5.4 - Avaliação da sensibilidade em diferentes meios (1,1 % m/v total de sólidos dissolvidos) e condições de operação...63
TABELA 5.7 – Análise química do refratário (SRM 77a) digerido com Li2B4O7 (1:10 m/m). Determinação dos constituintes com soluções de calibração em meio ácido e meio fundente para o fator de diluição de 1000 vezes (condições robustas de operação)...90
TABELA 5.8 - Parâmetros instrumentais para medidas por ICP OES com visão axial...92
TABELA 5.9 - Análise química do carbeto de silício (SRM 1412) digerido com a mistura 1Na2B4O7 + 1Na2CO3 (1:10 m/m). Determinação dos constituintes com soluções de calibração em meio fundente e fator de diluição de 1000 vezes e condições não robustas...93
TABELA 5.10 - Parâmetros instrumentais para medidas por ICP OES com visão axial...95
TABELA 5.11 - Análise química do vidro (SRM 1412) digerido com Li2B4O7 (1:10 m/m). Determinações realizadas em condições não robustas de operação e soluções analíticas de calibração em meio fundente para o fator de diluição de 1000 vezes (condições robustas de operação)...97
TABELA 1 - Percentuais de recuperação para a bauxita (SRM 69b) utilizando calibração do instrumento com soluções padrão em meio ácido em condições não robustas - fator de diluição 500 vezes (n=3 ± coeficiente de variação)...1
TABELA 2 - Percentuais de recuperação para a bauxita (SRM 69b) utilizando calibração do instrumento com soluções padrão em meio ácido em condições robustas - fator de diluição 500 vezes (n=3 ± coeficiente de variação)...2
TABELA 3 - Percentuais de recuperação para a bauxita (SRM 69b) utilizando calibração do instrumento com soluções padrão em meio ácido em condições não robustas - fator de diluição 1000 vezes (n=3 ± coeficiente de variação)...3
TABELA 4 - Percentuais de recuperação para a bauxita (SRM 69b) utilizando calibração do instrumento com soluções padrão em meio ácido em condições robustas - fator de diluição 1000 vezes (n=3 ± coeficiente de variação)...4
TABELA 5 - Percentuais de recuperação para a bauxita (SRM 69b) utilizando calibração do instrumento com soluções padrão em meio ácido em condições robustas e não robustas - fator de diluição de 10.000 vezes (n=3 ± coeficiente de variação)...5
TABELA 6 - Percentuais de recuperação para a bauxita (SRM 69b) utilizando calibração do instrumento com soluções padrão em meio fundente em condições não robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...6
TABELA 7 - Percentuais de recuperação para a bauxita (SRM 69b) utilizando calibração do instrumento com soluções padrão em meio fundente condições robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...7
TABELA 8 - Balanço geral de todas as condições estudadas para a bauxita (SRM 69b) utilizando Li2B4O7 como fundente, apresentando os melhores resultados (n=3
± coeficiente de variação)...8
robustas - fator de diluição de 500 vezes (n=3 ± coeficiente de variação)...10
TABELA 11 - Percentuais de recuperação para o refratário (SRM 77a) utilizando calibração do instrumento com soluções padrão em meio ácido e condições não robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...11
TABELA 12 - Percentuais de recuperação para o refratário (SRM 77a) utilizando calibração do instrumento com soluções padrão em meio ácido e condições robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...12
TABELA 13 - Percentuais de recuperação para o refratário (SRM 77a) utilizando calibração do instrumento com soluções padrão em meio ácido em condições robustas e não robustas - fator de diluição de 10.000 vezes (n=3 ± coeficiente de variação)...13
TABELA 14 - Percentuais de recuperação para o refratário (SRM 77a) utilizando calibração do instrumento com soluções padrão em meio fundente e condições não robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...14
TABELA 15 - Percentuais de recuperação para o refratário (SRM 77a) utilizando calibração do instrumento com soluções padrão em meio fundente e condições robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...15
TABELA 16 - Percentuais de recuperação para o refratário (SRM 77a) utilizando calibração do instrumento com soluções padrão em meio fundente em condições robustas e não robusta - fator de diluição de 10.000 vezes (n=3 ± coeficiente de variação)...16
TABELA 17 - Percentuais de recuperação para o carbeto de silício (SRM 112B) utilizando calibração do instrumento com soluções padrão em meio ácido e condições não robustas - fator de diluição de 500 vezes (n=3 ± coeficiente de variação)...17
condições não robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...18
TABELA 20 - Percentuais de recuperação para o carbeto de silício (SRM 112B) utilizando calibração do instrumento com soluções padrão em meio ácido e condições robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...18
TABELA 21 - Percentuais de recuperação para o carbeto de silício (SRM 112B) utilizando calibração do instrumento com soluções padrão em meio fundente e condições não robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...19
TABELA 22 - Percentuais de recuperação para o carbeto de silício (SRM 112B) utilizando calibração do instrumento com soluções padrão em meio fundente e condições robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...19
TABELA 23 - Percentuais de recuperação para o vidro (SRM 112B) utilizando calibração do instrumento com soluções padrão em meio ácido e condições robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...20
TABELA 24 - Percentuais de recuperação para o vidro (SRM 112B) utilizando calibração do instrumento com soluções padrão em meio ácido e condições não robustas - fator de diluição de 1000 vezes (n=3 ± coeficiente de variação)...21
TABELA 25 - Percentuais de recuperação para o vidro (SRM 1412) utilizando calibração do instrumento com soluções padrão em meio ácido em condições robustas e não robustas - fator de diluição de 10.000 vezes (n=3 ± coeficiente de variação)...22
TABELA 26 - Percentuais de recuperação para o vidro (SRM 1412) utilizando calibração do instrumento com soluções padrão em meio fundente (Li2B4O7) em condições robustas e não robustas e diluições 1000 e 10.000 vezes (n=3 ± coeficiente de variação)...23
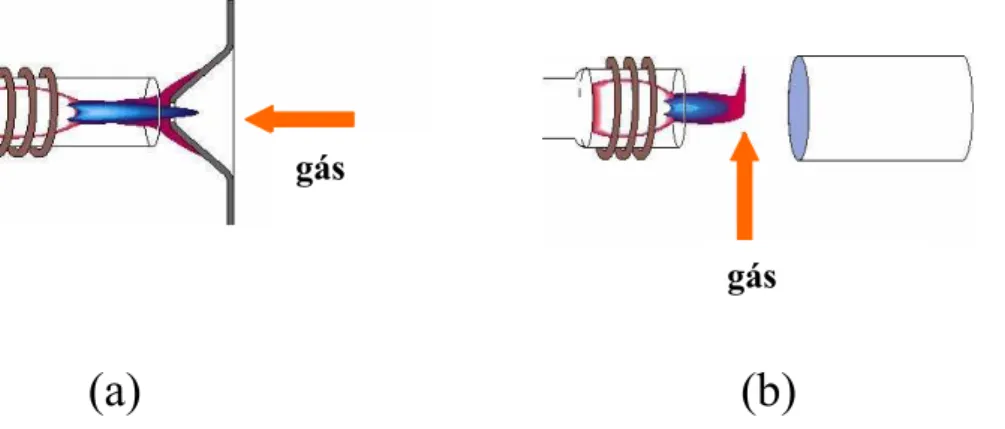
FIGURA 2.0 - Interfaces de remoção da zona fria do plasma: (a) end-on gas; (b) shear-gas...5
FIGURA 4.0 - (a) ICP OES Vista AX; (b) detalhe da configuração axial...37
FIGURA 4.1 - Câmara de nebulização Sturman-Masters: (1) Tubulação de transporte do aerossol para a tocha; (2) suporte; (3) orifício para introdução de amostra; (4) descarte...38
FIGURA 4.2 - Nebulizador com ranhura em V...38
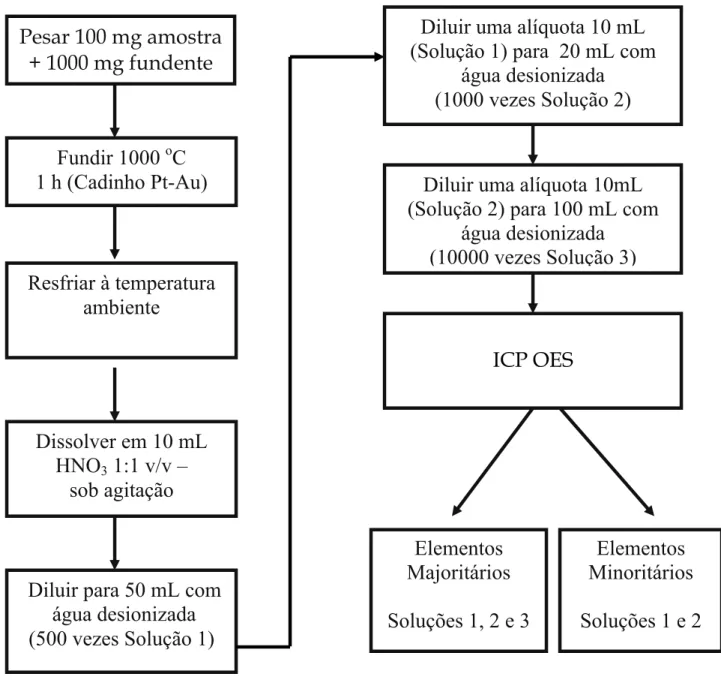
FIGURA 4.3 - Esquema da seqüência analítica adotada para o preparo das amostras ...44
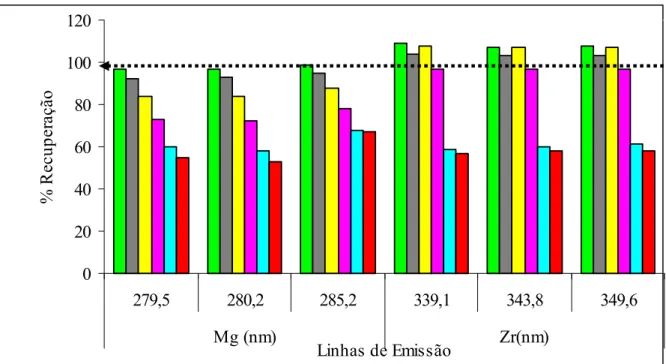
FIGURA 5.0 - Efeito da adição de concentrações crescentes do íon Na+ sobre as linhas de emissão de Mg e Zr em condições não robustas de operação (0,7 kW e 0,7 L min-1) Mg I / Mg II = 5,09...67
FIGURA 5.1- Efeito da adição de concentrações crescentes do íon Na+ sobre linhas de emissão de Mg Zr em condições robustas de operação (1,4 kW e 0,7 L min-1) Mg I / Mg II = 11,5...67
FIGURA 5.2 - Efeito da adição de concentrações crescentes do íon Li+ sobre as linhas de emissão de Mg e Zr em condições não robustas de operação (0,7 kW e 0,7 L min-1) Mg I / Mg II = 5,09...68
FIGURA 5.3 - Efeito da adição de concentrações crescentes do íon Li+ sobre as linhas de emissão de Mg e Zr em condições robustas de operação (1,4 kW e 0,7 L min-1) Mg I / Mg II = 11,5...68
Li2B4O7 como fundentes. As barras representam os teores médios de recuperação e os respectivos desvios padrão...74
FIGURA 5.6 - Percentuais de recuperação para Al I 237,313 nm (5,24 eV) nas diversas condições estudadas, utilizando (1) LiBO2, (2) Li2B4O7 e (3) LiBO2 + Li2B4O7 como fundentes. As barras representam os teores médios de recuperação e os respectivos desvios padrão...75
FIGURA 5.7 - Percentuais de recuperação para Fe II 238,202 nm (13,1 eV) nas diversas condições estudadas, utilizando (1) LiBO2, (2) Li2B4O7 e (3) LiBO2 + Li2B4O7 como fundentes. As barras representam os teores médios de recuperação e os respectivos desvios padrão...76
FIGURA 5.8 - Percentuais de recuperação para Ti II 334,937 nm (10,6 eV) nas diversas condições estudadas, utilizando (1) LiBO2, (2) Li2B4O7 e (3) LiBO2 + Li2B4O7 como fundentes. As barras representam os teores médios de recuperação e os respectivos desvios padrão...77
FIGURA 5.9 - Percentuais de recuperação para P I 177,433 nm (6,99 eV) nas diversas condições estudadas, utilizando (1) LiBO2, (2) Li2B4O7 e (3) LiBO2 + Li2B4O7, (4)Na2B4O7 e (5) Na2B4O7 + Na2CO3 como fundentes. As barras representam os teores médios de recuperação e os respectivos desvios padrão...79
FIGURA 5.10 - Percentuais de recuperação para Cr II 267,716 nm (12,9 eV) nas diversas condições estudadas, utilizando (1) LiBO2, (2) Li2B4O7 e (3) LiBO2 + Li2B4O7, (4) Na2B4O7 e (5) Na2B4O7 + Na2CO3 como fundentes. As barras representam os teores médios de recuperação e os respectivos desvios padrão...80
FIGURA 5.11 - Percentuais de recuperação para K I 766,482 nm (1,62 eV) nas diversas condições estudadas, utilizando (1) LiBO2, (2) Li2B4O7 e (3) LiBO2 + Li2B4O7, (4) Na2B4O7 e (5) Na2B4O7 + Na2CO3 como fundentes. As barras representam os teores médios de recuperação e os respectivos desvios padrão...81
ANÁLISE QUÍMICA DE MATERIAIS CERÂMICOS:
DIGESTÃO POR FUSÃO E MEDIDAS POR ICP OES COM
CHEMICAL ANALYSIS OF CERAMIC MATERIALS: FUSION AND
AXIALLY VIEWED ICP OES MEASUREMENTS. Ceramic samples are
1.0. INTRODUÇÃO...2
2.0. REVISÃO BIBLIOGRÁFICA...4
2.1. Aspectos históricos de plasmas com configuração radial e axial...4
2.2. Preparo de amostras para espectrometria de emissão óptica com plasma induzido – considerações gerais...5
2.3. Fusão...6
2.4. Figuras de Mérito – Como avaliar o desempenho de um método analítico em ICP OES...17
2.5. Aplicações do método de fusão para digestão de amostras de sólidos inorgânicos com determinação dos constituintes de interesse por ICP OES...22
2.5.1 Espectrômetros de emissão óptica com plasma acoplado indutivamente radiais e axiais………...…….23
3.0. OBJETIVO...36
4.0.PARTE EXPERIMENTAL...37
4.1. Instrumental...37
4.1.1. ICP OES...37
4.1.2. Balança Analítica...41
4.1.3. Forno Mufla...41
4.1.4. Purificador de Água...41
4.2. Reagentes, soluções e materiais...41
4.3. Amostras...42
4.4. Preparo das amostras...43
4.4.1 Fusão e diluição das amostras...43
4.5.3. Condições de operação do plasma...47
4.5.3.1. Razão Mg II / Mg I...47
4.5.3.2. Seleção dos comprimentos de onda...48
4.5.4. Limites de Detecção...48
4.5.5. Efeitos causados por elementos facilmente ionizáveis...49
4.5.6. Faixa Linear de Calibração...49
4.5.7. Sensibilidade...50
5.0. RESULTADOS DISCUSSÃO...51
5.1. Razão Mg II / Mg I...51
5.2. Figuras de mérito para soluções obtidas por fusão...54
5.2.1. Limites de Detecção...54
5.2.2. Linearidade...58
5.2.3. Sensibilidade...61
5.3. Efeitos causados por elementos facilmente ionizáveis...65
5.4. Análise química das amostras...69
5.4.1. Bauxita...70
5.4.1.1. Procedimento de Análise...86
5.4.2. Refratário...88
5.4.2.1. Procedimento de Análise...90
5.4.3. Carbeto de Silício...92
5.4.3.1. Procedimento de Análise...94
5.4.4. Vidro...96
5.4.4.1. Procedimento de Análise...97
6.0. CONCLUSÃO...101
1.0 - INTRODUÇÃO
O rápido e constante desenvolvimento das atividades humanas
aumenta a necessidade e a importância da análise química, que possibilita a
tomada de decisões cotidianas em diferentes campos, como o industrial,
ambiental ou da saúde. A espectrometria de emissão óptica com plasma
acoplado indutivamente (ICP OES) do inglês inductively coupled plasma optical
emission spectrometry, é uma técnica instrumental que apresenta numerosas
aplicações na caracterização de materiais. A grande aceitação dessa técnica pode
ser explicada por suas excelentes características analíticas, tais como alta
precisão e exatidão, alta seletividade e sensibilidade, baixos limites de detecção
e ampla faixa linear de calibração. Contudo, a ocorrência de indesejáveis efeitos
matriciais pode afetar a exatidão e precisão dos resultados analíticos. Uma etapa
crítica na caracterização de materiais por ICP OES é a escolha de um
procedimento adequado para a digestão das amostras, que podem ser divididas
entre aquelas que já estão na forma de uma solução aquosa (amostras de água,
sangue, urina, entre outras), em outras formas de líquidos (óleos, combustíveis,
solventes orgânicos, entre outras) ou na forma sólida (solos, cerâmicas,
refratários, plantas, tecidos animais, metais, plásticos, entre outros).
As amostras sólidas analisadas por ICP OES, geralmente são
convertidas em uma solução representativa empregando um método de digestão
apropriado1. Para quase todas as técnicas de digestão existentes são utilizados
reagentes químicos que podem ser ácidos diluídos e concentrados, misturas de
ácidos, mistura de ácidos com outros reagentes ou sais, entre outros. No caso de
amostras inorgânicas complexas, como materiais cerâmicos, geológicos etc., a
dificuldade em obter uma solução representativa é maior, sendo que os métodos
mais comumente usados são: dissolução via ataque ácido envolvendo o uso de
ácido fluorídrico ou fusão.2,3 Os métodos baseados em fusão são eficientes e
aplicáveis a uma ampla faixa de materiais e composições, pois sua eficiência
determinação de elementos em concentrações maiores a traços. No entanto, o
alto conteúdo de sólidos dissolvidos presentes nas soluções resultantes pode
causar interferências de transporte e espectrais, afetar os brancos analíticos e
deteriorar os valores para as concentrações equivalentes aos sinais de fundo e,
conseqüentemente, os limites de detecção. Esses efeitos podem ser agravados
quando as medidas são efetuadas em um espectrômetro de emissão óptica com
plasma acoplado indutivamente com configuração axial, que, desde a sua
concepção, é tido como inadequado para amostras complexas.5 A presença de
elementos facilmente ionizáveis em altas concentrações também pode causar
perturbações nos processos do plasma. Dessa forma, amostras sólidas complexas
podem ser adequadamente convertidas em soluções por fusão e, por sua vez,
essas poderiam ter sua composição química determinada por ICP OES. Essa
combinação atrativa de estratégias pode ser considerada rotineira quando se
emprega um espectrômetro de emissão óptica com plasma acoplado
indutivamente com configuração radial, porém pode afetar criticamente o
desempenho de um espectrômetro de emissão óptica com plasma acoplado
indutivamente com configuração axial. Esse último aspecto será enfocado no
2.0 - REVISÃO BIBLIOGRÁFICA
2.1 - Aspectos históricos de plasmas com configuração radial e
axial
Na década de 60, dois grupos de pesquisadores, o de Stanley
Greenfield na Inglaterra e Velmer A. Fassel nos Estados Unidos, utilizaram pela
primeira vez uma fonte de plasma com acoplamento indutivo para fins
analíticos.6,7 Desde então, a espectrometria de emissão óptica com plasma
acoplado indutivamente tem mostrado grande potencial para a determinação
multi-elementares nas mais diferentes áreas de pesquisa científica. Muitos
estudos foram realizados ao longo dessas décadas levando à aperfeiçoamentos
na instrumentação, sempre objetivando ampliar cada vez mais o campo de
aplicações dessa técnica.8 Um dos marcos da evolução dessa técnica, desde a sua
introdução em 1969, foi a proposição de equipamentos com configuração axial
em 1975, que propiciaram uma maior sensibilidade às determinações analíticas.
A idéia geralmente difundida é que equipamentos com visão axial (geralmente
com tocha de quartzo posicionada horizontalmente) se caracterizam por
possibilitarem medidas com maior sensibilidade, porém com maior ocorrência
de interferências espectrais quando comparados aos equipamentos com visão
radial (geralmente com tocha de quartzo posicionada verticalmente). Portanto,
apesar do significativo ganho em sensibilidade alcançado com a configuração
axial, essa idéia apresentou baixa aceitação devido ao limitado desempenho do
equipamento frente às interferências espectrais observadas. Somente nos anos
90, com o desenvolvimento de interfaces adequadas denominadas shear-gas e
end-on-gas (FIGURA 2.0), é que foi possível minimizar as interferências
associadas à região mais fria do plasma na qual predominam os processos de
recombinação.8 A interface shear-gas utiliza um fluxo de gás (15 a 20 L min-1)
perpendicular ao gás de formação e manutenção do plasma, enquanto que
end-on-gas, um fluxo de gás (1 a 2 L min-1) em contracorrente. Desde então, a
destacando, apesar de acreditar-se ainda hoje, que a configuração axial é
inadequada para aplicações nas quais a complexidade matricial é elevada.
Entende-se que para esse tipo de amostra a configuração radial ainda é a melhor
alternativa, como estabelecido em diversos trabalhos publicados na literatura.9-14
(a) (b)
FIGURA 2.0 - Interfaces de remoção da zona fria do plasma: (a) end-on gas; (b) shear-gas
2.2 - Preparo de amostras para espectrometria de emissão óptica
com plasma induzido – considerações gerais
O desenvolvimento de um procedimento de preparo de amostras
que resulte em completa solubilização, sem a volatilização ou perda de
elementos voláteis, redução de contaminações causadas pelo frasco e pela
atmosfera, baixos valores de branco dos reagentes e rapidez, é um dos pontos
mais críticos de uma análise química empregando ICP OES. Partindo do
princípio que não existe uma técnica de preparo de amostras que seja universal,
devem ser observados, prioritariamente, os requerimentos do elemento a ser
determinado, da amostra e das condições da solução final (concentração ácida,
viscosidade, entre outras).3
Dentre as estratégias empregadas para o preparo de amostras
sólidas para determinação por ICP OES podem ser citadas a dissolução direta
gás
gás
em água sem mudança química e a dissolução em ácido, mistura de ácidos ou
fusão, com mudança química.
A decomposição por via úmida pode ser promovida em sistemas
abertos e sistemas fechados, com aquecimento convencional ou assistido por
microondas. Já o preparo de amostras via combustão pode ocorrer em sistemas
abertos denominados via seca, ou sistemas fechados em frascos de combustão.
Outra técnica de digestão de amostras sólidas é a fusão que envolve uma reação
heterogênea executada em altas temperaturas entre um fundente e o material da
amostra. Como resultado desse procedimento, um mineral original ou fases
refratárias são convertidos em formas sólidas diferentes que são facilmente
dissolvidas em ácidos, bases ou até mesmo em água.15
Um procedimento ideal para digestão de sólidos inorgânicos deve
ser eficiente e rápido, apresentar ausência de interferências entre reagentes e
analitos, sendo que os reagentes devem estar disponíveis em elevado grau de
pureza. Perdas por volatilização e contaminações devem ser desprezíveis e tanto
os reagentes como a amostra não devem atacar o recipiente no qual será
conduzida a reação. Além disso, o método de digestão deve apresentar baixa
insalubridade e periculosidade, sendo a solução final representativa do sólido
original e compatível com a técnica instrumental de medida.16
2.3 - Fusão
O método de fusão pode ser utilizado no preparo de amostras tanto
para as técnicas espectrométricas que utilizam a amostra na forma sólida (XRF)
do inglês X-ray fluorescence, como para aquelas que utilizam a amostra na
forma de uma solução representativa, como é o caso da espectrometria de
emissão óptica com plasma acoplado indutivamente. Esse método é empregado
quente, ou são atacados lentamente e/ou dissolvidos parcialmente. A fusão é
adequada ainda àqueles materiais que dão origem a soluções ácidas instáveis
apresentando componentes com tendência para precipitar, como a sílica. Como
exemplos de materiais de difícil dissolução em ácidos podem ser citados:
cimento, aluminatos, silicatos, minérios de Ti e Zr; minerais mistos de Be, Si,
Al, resíduos insolúveis de minérios de ferro, óxidos de cromo, silício e ferro,
óxidos mistos de tungstênio, silício e alumínio. O ácido comumente utilizado na
digestão desses materiais é o HF, que forma compostos voláteis com Si (SiF4) e
B (BF3)16, tornando-se uma limitação do método quando se pretende determinar
tais elementos. O procedimento típico da fusão envolve o aquecimento da
mistura da amostra finamente moída (preferencialmente abaixo de 200 mesh –
75 µm) com o fundente sob elevadas temperaturas até fusão completa. Os
fundentes podem ser hidróxidos de metais alcalinos, carbonatos, dissulfatos,
boratos, peróxidos, fluoretos ou óxido bórico, e a proporção entre as massas de
amostra e fundente varia de 1:2 a 1:50 m/m.16 A escolha do fundente depende do
tipo de amostra a ser dissolvida e da sua basicidade, sendo que um fundente
ácido é adequado para digestão de amostras contendo altos teores de óxidos
básicos e carbonatos, enquanto que amostras ácidas requerem o uso de fundentes
básicos.17 Geralmente a mistura é colocada em cadinho de Ni ou Pt-Au e o
líquido fundido se solidifica quando resfriado à temperatura ambiente, que se
desprende das paredes do cadinho quando quebrado em movimentos leves. Um
reagente químico (do inglês non-wetting) pode ser utilizado para facilitar a
remoção desse bolo fundente das paredes do cadinho (NaBr, LiBr, KI, CsI, entre
outros), sendo que sua massa, em geral, deve ser limitada a no máximo 100 mg.
Se a fusão for bem sucedida, o sólido resultante é facilmente dissolvido em água
ou ácido diluído.18
A velocidade de decomposição depende da área superficial das
substâncias a serem dissolvidas. Um fator limitante é a alta viscosidade dos
e a formação de produtos insolúveis na interface plasma/espectrômetro. O
processo é também afetado pelas características químicas do solvente e
propriedades das substâncias a serem decompostas, como por exemplo, energia
de ligação dos átomos e íons na estrutura. Os produtos da fusão nem sempre são
prontamente dissolvidas e freqüentemente, várias novas fases são obtidas
durante a fusão, as quais são mais facilmente solúveis em ácidos que as fases
originais. A formação de compostos com solubilidades variadas pode ser
vantajosa na separação de grupos de substâncias.17
Os métodos de fusão em geral são bastante eficientes e isso se deve
ao fato dos eletrólitos inorgânicos serem solventes poderosos, nas elevadas
temperaturas alcançadas durante o processo de fusão (atingindo a 1200 oC
dependendo do fundente utilizado) e o fato do eletrólito fundido agir como ácido
ou base de Lewis.16
As reações heterogêneas que ocorrem nas fusões podem ser
divididas em 2 grupos maiores: (1) reações de ácido-base e (2) reações de
óxido-redução. As reações de ácido-base podem ocorrer em fusões alcalinas
(carbonatos, boratos e hidróxidos) e fusões ácidas (dissulfatos, fluoretos e óxido
de boro). Por sua vez, as reações de óxido-redução podem ocorrer em fusões
oxidantes (agentes de fusões alcalinas + oxidantes, peróxidos) e fusões redutoras
(agentes de fusões alcalinas + redutores, fusões com enxofre e bases)17.
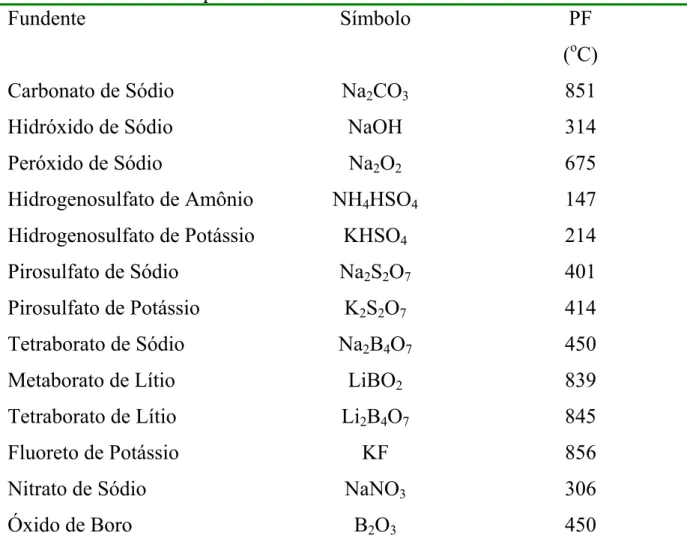
Na TABELA 2.0 estão listados alguns fundentes utilizados em
decomposições por fusão.
Muitas aplicações da utilização de fusão na digestão de materiais
inorgânicos podem ser encontradas na literatura, tanto nas décadas de 1950 e
19702,19-20 quanto em artigos de 2003 e 2005.21-23Dessa forma, alguns trabalhos
foram selecionados e serão discutidos com o intuito de ressaltar a aplicabilidade
de fundentes desde os mais comuns como Na2CO3, LiBO2, Li2B4O7, Na2B4O7 e
mistura deles, até aqueles mais específicos como por exemplo (NH4)2SO4, entre
TABELA 2.0 - Pontos de fusão de compostos comumente utilizados para a fusão de amostras. Adaptado de Anderson16 e Sulcek.17
Fundente Símbolo PF
(oC)
Carbonato de Sódio Na2CO3 851
Hidróxido de Sódio NaOH 314
Peróxido de Sódio Na2O2 675
Hidrogenosulfato de Amônio NH4HSO4 147
Hidrogenosulfato de Potássio KHSO4 214
Pirosulfato de Sódio Na2S2O7 401
Pirosulfato de Potássio K2S2O7 414
Tetraborato de Sódio Na2B4O7 450
Metaborato de Lítio LiBO2 839
Tetraborato de Lítio Li2B4O7 845
Fluoreto de Potássio KF 856
Nitrato de Sódio NaNO3 306
Óxido de Boro B2O3 450
O agente fundente mais comum é o carbonato de sódio que é
encontrado comercialmente em pureza adequada para a determinação de
constituintes maiores e menores. Os recipientes utilizados para fusão com
carbonatos são usualmente de Pt ou suas ligas, sendo que o tempo de vida útil do
cadinho dependerá da textura do metal, temperatura de fusão e a composição do
material decomposto. Como os carbonatos são usados principalmente na
decomposição de silicatos (tais como rochas e vidros), quando se utiliza uma
chama como fonte de calor, pode ocorrer perda acentuada de ferro da amostra
para o cadinho de platina, causando perdas do analito e prejuízos ao cadinho. No
Durante a fusão, ligações de polissilicatos e aluminosilicatos são
dissociadas formando silicatos alcalinos, solúveis em água ou em ácidos
minerais.
É sabido que o Na2CO3 é uma excelente fonte de íons O
:
CO32- CO2 + O2-
(base de Lewis)
SiO2 + O
SiO3 2-
(ácido de Lewis)
O sal sódico do SiO3
é solúvel em água, promovendo assim a
dissolução da sílica.16
Carbonatos alcalinos são bastante eficientes como agentes
fundentes na determinação de impurezas em carbetos de silício, apesar de alguns
compostos serem volatilizados durante o processo de fusão como As e Se
parcialmente, Tl e Hg completamente, conforme TABELA 2.1. No entanto, a
determinação de Re em amostras de interesse geológico por ICP-MS (do inglês
inductively coupled plasma-mass spectrometry) foi possível a partir da digestão
via fusão com Na2CO3 em cadinho de platina. Os resultados foram concordantes
com àqueles obtidos a partir da digestão ácida das amostras.21
A utilização de combinações de Na2CO3 com agentes oxidantes
como KNO3, KClO3 ou Na2O2 16
pode melhorar a eficiência da fusão, para
materiais de elevada resistência química. Bem como misturas com outros
agentes fundentes como o K2CO3 utilizado na decomposição de pós de
cerâmicas avançadas, Si3N4 e BN. A mistura 1:1 m/m de Na2CO3 e K2CO3 foi
adicionada às amostras e a fusão efetuada em cadinho de platina.11
O Na2B4O7 (bórax) é um fundente que pode ser utilizado
isoladamente ou em combinação com o Na2CO3, sendo que as temperaturas de
fusão podem variar de 1000 – 1200 oC. A mistura pode ser utilizada em
procedimentos de decomposição de solos como proposto por Medved et al..24
proporção de 1:20 m/m (amostra:fundente) em cadinho de platina. A mistura é
eficiente apesar de TiN ser um material com elevada dureza, resistência à
abrasão, alto ponto de fusão, alta estabilidade química à elevadas temperaturas e
condução metálica.22 Da mesma forma, o nitreto de alumínio (AlN) pode ser
digerido utilizando essa mistura devido à sua baixa solubilidade em meio ácido,
mesmo em sistemas de aquecimento assistido por microondas. A mesma
proporção amostra:fundente utilizada para TiN foi requerida no caso do AlN. A
solução resultante após dissolução do bolo fundente em água, foi utilizada para
comparar os resultados obtidos na determinação de impurezas a partir da
introdução de suspensões em espectrômetro de emissão óptica com plasma
acoplado indutivamente.25
Ácido bórico quando misturado ao Na2CO3 (2:3 m/m) origina uma
mistura capaz de digerir óxido de zircônio, quando submetido à temperatura de
1000 oC durante 15 min em cadinho de Pt-Au (5 % m/m).26
O metaborato de lítio (LiBO2) é um fundente freqüentemente
utilizado, pois além de estar disponível comercialmente em elevada pureza,
apresenta excelente solubilidade para muitos elementos. Apesar de ser
susceptível à cristalização, é o fundente preferido para o preparo de soluções
devido à sua baixa viscosidade.15 Diversas aplicações utilizando LiBO2 podem
ser encontradas na literatura, como a decomposição de silicatos em cadinho de
grafite na proporção 1: 1 m/m (amostra/fundente). A fusão foi realizada à 1000
o
C durante 30 min. A dissolução do fundido foi feita em HNO3 8 % v/v, sendo
que o procedimento total foi finalizado num período de 3 - 4 h.20 Outras
amostras de interesse geológico foram digeridas via fusão com LiBO2, sendo
que em alguns casos, o fundido foi dissolvido em HNO3 diluído
23,27,28
e HCl
também diluído.29
Amostras de materiais cerâmicos também podem ser digeridas
utilizando fusão com LiBO2em cadinho de platina sob temperaturas em torno de
digestão de materiais é o caso da determinação de Mn em sabão em pó por ICP
OES e ICP-MS. As amostras fundidas foram dissolvidas em HNO3 e limites de
detecção da ordem de sub µg g-1 foram obtidos para ambas as técnicas. Amostras altamente complexas como o sabão em pó, não são digeridas
facilmente. Os componentes orgânicos podem ser destruídos pela queima via
seca, mas os constituintes inorgânicos tais como silicato de sódio, são altamente
resistentes e insolúveis em ácidos concentrados. Por isso, a fusão alcalina se
apresenta como uma excelente opção para digestão dessas amostras,
principalmente devido ao tempo relativamente curto necessário para a execução
desse processo. Deve-se salientar que o método anteriormente utilizado se
baseava na extração do analito em HNO3 diluído à quente, resultando em várias
horas para o preparo de uma amostra.31
O tetraborato de lítio (Li2B4O7) é pouco higroscópico e pode ser
usado diretamente ou em combinação com LiBO2. O Li2B4O7 é um ácido fraco,
adequado principalmente para a fusão de amostras com características básicas.
O fundido é altamente viscoso, sendo que os vidros formados são dificilmente
dissolvidos em ácidos minerais. Tal como outros boratos fundidos, esse vidro
também adere fortemente à platina do cadinho, sendo aconselhável o uso de
cadinho de Pt-Au.17 Como citado anteriormente, o Li2B4O7 pode ser utilizado
isoladamente, como demonstrado por Paama et al.14 na digestão de amostras
arqueológicas e argilas, como também em mistura com LiBO2. Amostras de
rochas graníticas foram digeridas por fusão com mistura de LiBO2 + Li2B4O7 (1
+ 4 m/m) em cadinho de grafite, na proporção de 1:3 m/m (amostra/fundente). O
produto da fusão foi dissolvido em HCl diluído. A fusão foi realizada em forno
mufla sob temperatura de 1000 oC durante 20 min.32 A mesma mistura foi
utilizada na digestão de minério de niobio-tântalo para determinação de
diversos elementos via ICP OES.33
Em estudos anteriores, observou-se que o tetraborato de lítio é mais
mistura de 4 + 1 m/m de LiBO2 e Li2B4O7 corresponde a um eutético com ponto
de fusão de 832 oC, considerada o agente fundente universal para amostras com
elevadas concentrações de óxido de silício e alumínio.15
Outros fundentes menos comuns também podem ser úteis na
digestão de materiais inorgânicos, como a mistura de metaborato de potássio
(KBO2) e carbonato de potássio (K2CO3), utilizada para dissolução de silicatos.
Essa mistura binária (3:2 m/m) pode fundir amostras de rochas basálticas, areia
ou vidros em pó em cadinho de platina com aquecimento em forno mufla. A
vantagem desse procedimento é o curto tempo de fusão, cerca de 10 min e a
rápida dissolução do fundido em meio ácido, que pode ser HCl, HNO3 ou H2SO4
diluídos.34
Kohl et al.35 utilizaram (NH4)2SO4 na digestão de pós cerâmicos à
base de ZrO2. A proporção 1:10 m/m (amostra/fundente) foi fundida em
recipientes de quartzo sob temperaturas de até 450 oC em forno mufla, sendo o
produto resultante dissolvido em HNO3 2% v/v. A vantagem do emprego desse
fundente reside na baixa temperatura de fusão requerida (< 500 oC)
possibilitando o uso de recipiente de quartzo e minimizando a perda de
elementos potencialmente voláteis.35
Para a determinação de impurezas presentes em baixas
concentrações em ZrO2, outros fundentes podem ser utilizados como por
exemplo K2S2O7, LiBO2/H3BO3, Li2B4O7/Li2CO3 ou Na2B4O7. Esses métodos de
fusão demandam menos tempo que os processos de digestão ácida, porém
necessitam de elevadas temperaturas, podendo ocasionar contaminações e ou
perda de analitos.36
Das possíveis fontes de contaminação durante um processo de
fusão, o fundente e o cadinho podem ser considerados os agentes mais críticos.
Com a utilização de reagentes de grau analítico se minimiza a possibilidade de
contaminação proveniente do fundente. Já o cadinho pode causar contaminações
observada contaminação por Cu, Mn, Ni e Zn. Utilizando cadinho de Zr com o
mesmo fundente pode ser observada a contaminação do próprio Zr.37
TABELA 2.1 - Aplicações do método de fusão na decomposição de materiais inorgânicos, adaptada de Krug.15
Fundente T
(oC)
Material
do
Cadinho
Amostras Elementos
Voláteis
Na2CO3 850-1000 Pt, Ni, Zr silicatos, solos, óxidos
carbonatos, sulfatos,fosfatos,
fluoretos
Ag, As, Bi, Br, Cd, Cl, F, Ga, Hg, In, I, Os, Pb, Re, Ru, S, Sb, Se, Te, Tl, Zn Na2CO3 +
Na2B4O7
900-1000 Pt, Pt-Au silicatos, solos,
bauxita, óxidos de Al, Ti, Fe, Mn, Cr, Sn,
Zr, Nb, Ta
Ag, As, Bi, Br, Cd, Cl, F, Ga, Hg, In, I, Os, Pb, Re, Ru, S, Sb, Se, Te, Tl, Zn LiBO2 ou
Li2B4O7
900-950 Pt, Pt-Au,
Grafite
silicatos, solos, óxidos de Al, Cr, Mg, Ca, Fe,
materiais refratários
Ag, As, Bi, Br, Cd, Cl, F, Ga, Hg, In, I, Os, Pb, Re, Ru, S, Sb, Se, Te, Tl, Zn
KHSO4
ou K2S2O7
420-700 Pt sulfetos, óxidos de
Be, Cr, Fe, Nb, Ta, Ti, Zr e lantanídeos
Bi, Cd, Hg, Pb, S, Se, Tl, Zn
KOH 360-450 Ni, Fe, Ag,
Zr
silicatos óxidos de Fe, Ge, Nb, Sn, Te, Ti,
W, fosfatos
Bi, Cd, Hg, Se, Tl
Na2O2 450-1000 Ni, Fe, Ag,
Zr, carbono vítreo, Pt
(450 oC máx.)
concentrados de metais preciosos, refratários, silicatos,
solos, óxidos de Al, Ti, Fe, Mn, Cr, Sn,
Zr, Nb, Ta, ligas metálicas, materiais à
base de zinco, minérios
Cd, Hg
Constata-se que os hidróxidos alcalinos têm pontos de fusão
consideravelmente menores que os carbonatos alcalinos e são bastante reativos
limitante quando se pretende usar fundentes como KOH, NaOH ou LiOH.
Mesmo metais como platina ou paládio e suas ligas são relativamente corroídos,
principalmente sob elevadas temperaturas.17
A quantidade de fundente utilizada depende da basicidade da
amostra, sendo que um fundente ácido é adequado para digestão de amostras
contendo altos teores de óxidos básicos e carbonatos (um excesso de 3 a 5 vezes
comparativamente à massa de amostra é suficiente). Por outro lado, amostras
ácidas requerem o uso de fundentes básicos.17 A TABELA 2.2 apresenta
exemplos das categorias de amostras existentes:
TABELA 2.2 - Categorias de amostras e exemplos, adaptada de Krug.15
Amostras
Categorias Exemplos
ácidas SiO2 , P2O5
básicas MxOy
M= Na, Mg, Ca, Al
x = 1,2
y = 1,2,3
anfóteras Fe2O3
Quando analisamos criticamente uma técnica de digestão, vários
fatores devem ser considerados: (1) se digestão completa é requerida; (2)
possível perda do analito por volatilização; (3) a solução final deve ser adequada
à técnica instrumental escolhida e ou disponível (matriz da amostra, total de
sólidos dissolvidos e possíveis interferências); (4) exatidão e precisão para os
elementos determinados deverão ser adequadas e não deverão ser afetadas pelo
processo de diluição; (5) tempo de preparo da amostra não deve ser
exageradamente longo e (6) o procedimento deverá ter um custo razoável.9 Em
minerais resistentes ao ataque ácido, possibilitando a quantificação de elementos
refratários como exemplo Cr, Hf e Zr.9 No entanto, as soluções resultantes da
fusão apresentam alto conteúdo de sólidos dissolvidos, o que torna a análise por
espectrometria de emissão óptica com plasma acoplado indutivamente,
problemática. Uma vez que o total de sólidos dissolvidos deve ser menor que 2
% m/v, o que usualmente não é possível, pois para esse método é comum utilizar
uma massa de fundente 7-10 vezes maior que a massa da amostra.4 Esse
aumento de sais introduzidos pelo processo de fusão implica na necessidade de
um aumento da diluição da amostra, causando um aumento significativo dos
limites de detecção, prejudicando a determinação de muitos elementos traços.
Outra desvantagem é a possível perda dos analitos por volatilização devido às
elevadas temperaturas necessárias ao processo de fusão como por exemplo Pb,
Sb, Sn e Zn.9 Digestão com ácido fluorídrico também é muito utilizada quando
se trabalha com materiais de interesse geológico, cerâmicas, entre outros.
Porém, quando a digestão é promovida sob pressão ambiente e elevada
temperatura, o Si se perde por volatilização como SiF4. Além de que, amostras
digeridas com HF em sistemas fechados (tanto aquecimento condutivo como
aquecimento assistido por microondas) para determinação de constituintes de
interesse por espectrometria de emissão óptica com plasma acoplado
indutivamente, faz-se necessário a adição de 1,0 mL de ácido bórico 4% m/v
para cada 0,5 mL de HF utilizado no processo de digestão.38 O ácido bórico por
sua vez, complexa os íons fluoreto, evitando o desgaste da tocha de quartzo
quando desprovida de tubo central resistente a HF. Esse processo, da mesma
forma que a fusão alcalina, promove o aumento do conteúdo total de sólidos
dissolvidos na solução da amostra.
Finalmente, apesar de avanços tecnológicos, como a introdução de
fornos de microondas com sistemas fechados (alta pressão) para digestão de
amostras, método de digestão por fusão em alguns casos se apresenta como uma
dependendo do processo de fabricação e moagem somente é digerido por fusão
alcalina. Assim, estabelecer as melhores condições para a determinação
elementar em soluções de amostras preparadas a partir de fusão via ICP OES
seja por configuração radial, como configuração axial é relevante principalmente
em laboratórios de análise de rotina nos quais a diversidade de amostras é
elevada. Deve-se ainda ressaltar que mesmo para aqueles materiais para os quais
já existe um método de digestão via ataque ácido estabelecido, muitas vezes,
dependendo da amostra, a dissolução não é adequada. Isso implica que em
muitas situações a fusão é o caminho mais adequado, senão o único, para essa
etapa tão crítica de uma análise química, que é a digestão da amostra.
2.4 - Figuras de mérito – Como avaliar o desempenho de um
método analítico em ICP OES
Para avaliar o desempenho de um método analítico em ICP OES,
foram estabelecidas algumas figuras de mérito como número de elementos,
seletividade, repetibilidade (precisão), estabilidade a longo período, robustez,
limites de detecção e exatidão.39 Essas figuras de mérito podem ser
determinadas a partir de experimentos simples e levam à caracterização de um
método analítico como é o caso da determinação do número de elementos, que
se refere diretamente à faixa de comprimentos de onda possível de ser medida
pelo sistema óptico. A seletividade, que está relacionada à resolução do sistema
dispersivo, pode ser determinada experimentalmente medindo-se a largura à
meia altura das linhas de emissão de Ba II em 233 e 405 nm para a região do UV
e VIS respectivamente, usando-se uma solução contendo 1 mg L-1 de Ba.
Quanto menor esse valor, maior a resolução do equipamento. A repetibilidade
pode ser expressa como desvio padrão relativo (RSD) do inglês relative
standard desviation da flutuação do sinal de emissão da linha de Mg I em 285
em condições normais de operação, esse valor é tipicamente inferior a 2 %40 e
dificilmente superior a 5 %.41 A estabilidade pode ser também expressa como o
RSD da flutuação do sinal, no entanto, considerando um período de várias horas.
Além disso, há também a estabilidade no início da operação do equipamento,
conhecida como warm up time, e definida como o tempo necessário a partir da
ignição do plasma para garantir que o sistema está pronto para análises
quantitativas, ou seja, que os sinais obtidos são aceitáveis (RSD < 1 % para as
linhas de Ar I 404 nm, Ba II 455 nm ou Zn II 206 nm).
Dentre as características de um método analítico, a exatidão,
precisão e estabilidade a longo período são figuras de mérito prioritárias. A
exatidão por sua vez, é a aproximação entre o valor medido experimentalmente
e o valor correto, podendo ser avaliada a partir do uso de materiais de referência
certificados.39
Outra figura de mérito importante é a robustez, que diz respeito às
condições de excitação do plasma. Mermet42, em trabalho publicado em 1991,
usou a razão das intensidades das linhas de Mg II 280,270 nm / Mg I 285,213
nm para avaliar e otimizar os processos de atomização, excitação e ionização no
plasma. O termo robustez é usado para descrever as condições de excitação e
atomização em um espectrômetro de emissão óptica com plasma acoplado
indutivamente, quando são obtidas razões Mg II / Mg I ≥10, correspondentes à condição de equilíbrio termodinâmico local em um plasma, conforme deduzido
teoricamente por Mermet et al..42 Um plasma robusto pode ser obtido usando
baixa vazão de nebulização, uma elevada potência de rádio-freqüência e um
tubo injetor da tocha de quartzo com diâmetro igual ou maior que 2 mm.43 No
entanto, outro fator importante para obtenção de exatidão e precisão nos
resultados em um espectrômetro de emissão óptica com plasma acoplado
indutivamente é o sistema de introdução de amostras.
Trevizan et al.44 avaliaram as influências da potência aplicada de
introdução de amostras em um espectrômetro de emissão óptica com plasma
acoplado indutivamente com configuração axial. Os resultados obtidos
indicaram que condições robustas são dependentes do tipo de nebulizador usado,
de forma que para um nebulizador concêntrico, com uma câmara de spray
ciclônica ou Sturman-Masters, condições robustas foram obtidas a uma alta
potência de rádio-frequëncia e a uma baixa vazão do gás de nebulização, como
já observado anteriormente. Entretanto, para um nebulizador V-groove com uma
câmara spray Sturman-Masters é necessário o uso de uma alta vazão de
nebulização (p. ex. > 1,0 L min-1) para garantir condições robustas de operação
e elevadas intensidades de emissão de Mg.44 Assim, esse plasma robusto é capaz
de suportar mudanças na natureza ou concentração da matriz da amostra, sem,
no entanto, apresentar variações significativas nos sinais analíticos.45 Outra
estratégia utilizada para minimizar interferências matriciais, além da utilização
de condições robustas, é a compatibilização de matriz (matrix matching). Esse
procedimento consiste no preparo de soluções analíticas de calibração contendo
concentração(ões) do(s) elemento(s) majoritário(s) e quantidade de ácido ou
agente fundente iguais ao da amostra. Entretanto, essa simulação requer um
perfeito conhecimento da matriz da amostra, o que nem sempre é possível. Para
amostras geológicas essa estratégia é muito utilizada, assim como a calibração
com materiais de referência certificados.46
A padronização interna também pode ser utilizada para melhorar a
repetibilidade e exatidão de um método analítico. O padrão interno é um
elemento que é adicionado em concentração constante em todas as amostras e
soluções analíticas de calibração, que será medido de tal forma a obter valores
da razão do sinal do analito sobre o sinal do padrão interno. Contudo, a escolha
do padrão interno é um fator crítico pois geralmente é um elemento não presente
entre os constituintes da amostra e que não seria usualmente determinado. A
correção de interferências de transporte e flutuações instrumentais que afetem a
repetibilidade dos sinais analíticos.41
Para espectrômetro de emissão óptica com plasma acoplado
indutivamente com configuração radial, a razão Mg II / Mg I apresenta valores
maiores que para configurações axiais, isso porque, quando o modo de
observação convencional é usado, as razões Mg II / Mg I são usualmente
medidas seguindo uma otimização da altura de observação correspondendo ao
ótimo da emissão da linha iônica. Em contraste, quando o modo de visão axial é
usado, ambas as zonas de emissão de linhas iônicas e atômicas são investigadas
pelo sistema de detecção.45 Portanto, resultados experimentais indicam que
plasmas radiais são considerados robustos quando a razão Mg II / Mg I > 10,
enquanto que para plasmas axiais a razão Mg II / Mg I > 8. Esses valores foram
obtidos multiplicando-se as razões de intensidade Mg II / Mg I por um fator de
correção para compensar a diferença na resposta do detector de estado sólido
para os dois comprimentos de onda do Mg. No caso do ICP OES Vista da
Varian, o fator foi estabelecido em 1,85:
No entanto, a possibilidade de trabalhar com plasmas em condições
não robustas de operação, não deve ser descartada. Mesmo porque, elevadas
potências de rádio-freqüência causam desgaste na tocha de quartzo, fonte de
potência e comprometimento da razão sinal analítico / sinal de fundo, mesmo
quando o modo de observação axial é utilizado. Além disso, a utilização de
condições robustas não garante a completa eliminação de efeitos matriciais.43
Diante de todas as considerações acima, a utilização ou não de
condições robustas de operação deve ser definida de acordo com as necessidades
analíticas. O analista deverá considerar ainda o tipo de configuração do
espectrômetro de emissão óptica com plasma acoplado indutivamente (radial ou
axial), a complexidade da matriz da amostra (total de sólidos dissolvidos,
(sensibilidade, linhas iônicas ou atômicas), sistema de introdução de amostra,
precisão e exatidão requeridas.
Um estudo comparando as duas configurações radial e axial quanto
ao desempenho analítico foi realizado por Silva et al.47 Os autores compararam
figuras de mérito para ambos os espectrômetros de emissão óptica com plasma
acoplado indutivamente quanto ao warm up time, estabilidade a curto e longo
prazo, resolução espectral das linhas UV e VIS e limites de detecção. Ambos os
equipamentos apresentavam sistema de detecção e gerador de rádio-freqüência
idênticos, diferindo apenas no modo de observação. Os resultados apontaram
que para um equipamento com configuração radial, o warm up time foi 2 vezes
menor que para aquele com configuração axial. A robustez, estabilidade a curto
e longo período e as resoluções espectrais nas regiões do UV e visível foram
similares para ambas configurações. Por outro lado, a sensibilidade alcançada
para o Ni com a configuração axial foi 20 vezes melhor que com a radial,
enquanto que os LOD´s do inglês limit of detection (limite de detecção) e
BEC´s do inglês background equivalente concentration (concentração
equivalente do sinal de fundo) foram similares para todas as concentrações de
carbono estudadas.47
Dessa forma, considerando-se a necessidade de emprego de fusão
para a digestão de amostras inorgânicas com alta inércia química e o efeito de
meios complexos sobre medidas em ICP OES, o próximo item discutirá a
literatura referente ao emprego de um espectrômetro de emissão óptica com
plasma acoplado indutivamente com configurações axial e radial para a análise
de digeridos obtidos por fusão e, conseqüentemente, contendo um alto teor de
2.5 - Aplicações do método de fusão para digestão de amostras de
sólidos inorgânicos com determinação dos constituintes de
interesse por ICP OES
Em revisão da literatura, constata-se que diversos trabalhos
envolvendo o método de fusão para digestão de amostras de sólidos inorgânicos
com determinação dos constituintes de interesse por ICP OES estão reportados.
A técnica de ICP OES é reconhecida como uma técnica multi-elementar
confiável, para determinação de elementos maiores, menores e traços em
diversos tipos de amostras. Porém, na grande maioria dos trabalhos, o
espectrômetro de emissão óptica com plasma acoplado indutivamente é
encarado apenas como uma ferramenta analítica. Poucos trabalhos avaliaram os
possíveis efeitos matriciais provocados pela presença de elementos facilmente
ionizáveis como Na e Ca5,48-50 na amostra, interferências de ácidos utilizados na
digestão das amostras51 ou avaliaram a eficiência da interface utilizada para
remover a zona fria indesejável do plasma no caso de plasmas axiais.8 Em geral,
esses trabalhos também não avaliaram sistematicamente as melhores condições
de operação52 ou até mesmo as figuras de mérito envolvidas como o número de
elementos, a sensibilidade, a seletividade, a repetibilidade, a estabilidade, a
exatidão, a robustez e os limites de detecção.39
Portanto, o desempenho do espectrômetro de emissão óptica com
plasma acoplado indutivamente foi em geral superficialmente avaliado e
discutido. Na discussão abaixo, exemplificaram-se aplicações da digestão por
fusão nas determinações de constituintes de interesse em espectrômetros de
emissão óptica com plasma acoplado indutivamente com configurações radiais e
axiais, considerando-se as condições experimentais utilizadas e quando possível
avaliar o desempenho do instrumento frente à introdução de soluções
2.5.1 Espectrômetros de emissão óptica com plasma acoplado
indutivamente radiais e axiais
Espectrômetros de emissão óptica com plasma acoplado
indutivamente com configuração radial, geralmente com tocha posicionada
verticalmente ao sistema óptico, foram os primeiros a serem introduzidos
comercialmente e, até recentemente, considerados como a configuração
convencional. Ao longo dos anos, diversos trabalhos foram desenvolvidos,
ampliando cada vez mais o campo de aplicações dessa técnica, como no caso da
caracterização de amostras de vidro borossilicato provenientes de
reprocessamento industrial de um “lixo de alta radioatividade” (combustível
utilizado em reatores nucleares). Mais uma vez a importância da análise
química é destacada, bem como a existência de métodos e técnicas analíticas
adequadas. Nesse estudo, os autores utilizaram Na2O2 como fundente, na
proporção de 1:15 m/m de amostra e fundente para determinação de Si e B. O
recipiente utilizado foi um cadinho de Zr a 700 oC. Como citado anteriormente,
a escolha do material do cadinho depende do fundente e, conseqüentemente, da
temperatura de fusão, do tipo de amostra e dos analitos a serem determinados.
Nesse caso, optou-se pelo cadinho de zircônio provavelmente devido à sua
elevada resistência química. Para a determinação dos demais analitos Li, Na,
Mg, Al, P, Cr, Fe, Ni, Sr, Mo, La, Ce, Nd, U e inclusive Zr, os autores
utilizaram a digestão com HF e HClO4. O resíduo insolúvel da digestão ácida foi
dissolvido posteriormente por fusão com Na2O2 em cadinho de níquel, uma vez
que o Zr era um elemento de interesse. As soluções resultantes foram analisadas
em um espectrômetro Shimadzu, modelo ICP 1000 II, com algumas
modificações que permitiram a introdução de amostras radioativas. As
condições de operação foram 1,2 kW de potência aplicada e 1,2 L min-1 de
vazão do gás de nebulização. Informações sobre o tipo de configuração radial ou
axial, e interferências encontradas não foram reportadas, mesmo porque o
emissão óptica com plasma acoplado indutivamente, e sim a viabilidade dos
métodos de caracterização. Os autores informaram apenas que os resultados
obtidos apresentaram boa concordância com os valores esperados, exceto para B
que apresentou valores menores, provavelmente devido à perda por volatilização
durante o processo de fusão sob elevada temperatura. Um material de referência
certificado foi utilizado (Vidro NIST SRM 1412) para avaliar a exatidão das
medidas.13
A fusão com boratos consiste em uma das técnicas mais limpas
para o preparo de soluções, sendo que os produtos de reação são isentos de gases
tóxicos e não requer a limpeza ácida dos recipientes de reação.18 A eficiência da
fusão com Li2B4O7 foi avaliada na dissolução de amostras de fibras de vidro, nas
quais tanto a capa da fibra como o preenchimento propriamente dito foram
analisados. Os analitos Zr, Ba, Y, La, Al e Na originalmente na forma de ZrF4,
BaF2, YF3, LaF3, AlF3 e NaF foram determinados a partir da dissolução por
fusão, ataque ácido e introdução de suspensões. Para o procedimento por fusão
utilizou-se uma proporção de amostra e fundente de 1:10 m/m, com uma
diluição de 1500 vezes para os analitos Y, La, Al e Na e de 15000 vezes para Zr
e Ba. As condições de operação do plasma foram 1,2 kW de potência aplicada
de rádio-freqüência e 1,0 L min-1 de vazão do gás de nebulização, que
provavelmente favoreceram condições robustas de operação apesar desse
aspecto não ter sido discutido. Os três métodos foram comparados e os
resultados foram concordantes.53
Um percentual significativo das aplicações da ICP OES utilizando
fusão para decomposição da amostra se refere à caracterização de materiais
geológicos. Esses materiais têm como característica marcante a complexidade
matricial e uma elevada resistência química devido as diferentes fases minerais
presentes. Por isso, a existência de técnicas analíticas para a caracterização
desses materiais é limitada. A fluorescência de raios-X (XRF) é uma técnica
com chama (FAAS) e a técnica de ICP OES. Outras técnicas mais sofisticadas
como análise por ativação neutrônica (NAA) do inglês analysis by neutronic
activation até espectrometria de massas também são ferramentas importantes
para essa tarefa.
Em trabalhos realizados anteriormente, observou-se que a precisão
analítica das medidas obtidas por ICP OES em análises de rotina, está em torno
de ± 2 – 3 % tanto para os elementos maiores quanto para os menores. No
entanto, quando se trata de XRF, essa precisão geralmente se situa entre ± 0,2 –
0,5 %.27 É claro, que existem outras limitações para essa técnica, como por
exemplo a elevada quantidade de amostra requerida para a análise de elementos-
traço,54 que tornam o espectrômetro de emissão óptica com plasma acoplado
indutivamente também atrativo para análises de materiais geológicos, apesar da
exatidão inferior. Por isso, uma avaliação objetiva da precisão analítica das duas
técnicas foi realizada por Ramsey et al..10 Nesse trabalho os autores compararam
as duas técnicas analíticas, sendo que para as determinações dos elementos-
traço por ICP OES, as amostras foram digeridas em meio ácido (diluição 100
vezes) e para os elementos maiores, por fusão com uma mistura de metaborato e
tetraborato de lítio, na razão 1:3 m/m (amostra/fundente). A solução preparada
por fusão (em meio de HNO3 diluído) foi diluída 1000 vezes. O espectrômetro
de emissão óptica com plasma acoplado indutivamente utilizado foi um ARL
34000 simultâneo operando em 1,25 kW de potência aplicada de rádio
freqüência e 1,0 L min-1 de vazão do gás de nebulização, com vazão de
bombeamento da amostra de 1,2 mL min-1. A estratégia utilizada pelos autores
para minimizar as interferências matriciais foi preparar soluções analíticas de
calibração monos-elementares para a determinação dos elementos maiores,
enquanto que os elementos presentes em baixas concentrações foram
determinados a partir da calibração do espectrômetro de emissão óptica com
plasma acoplado indutivamente com soluções multi-elementares . Após um
concluíram que a diferença entre eles foram relativamente baixos não devendo
afetar sua precisão analítica. Sendo < 1% para os elementos maiores e < 10%
para os elementos traços.
Uma avaliação de técnicas de dissolução para análises em ICP OES
foi realizada por Totland et al..9 Nesse estudo, os autores compararam três
técnicas de digestão: (1) fusão alcalina com LiBO2; (2) ácido fluorídrico e
perclórico em frascos de PTFE abertos e (3) mistura ácida (HF + HClO4) em
forno de microondas com sistema fechado. Os elementos de interesse foram
determinados por ICP OES e ICP-MS. Dos três métodos de digestão avaliados, a
fusão alcalina é o mais comumente usado para digerir amostras geológicas,
sendo que a sílica é retida em solução, permitindo a determinação de todos os
elementos a partir de uma única preparação. Entretanto, o conteúdo total de
sólidos dissolvidos na solução final compromete a exatidão e precisão das
medidas estabelecendo-se um total < 1- 2 % (ICP OES) e < 0,1 – 0,2%
(ICP-MS). Contudo, mesmo necessitando de diluições extras, a técnica é preferida
para determinação de elementos mais refratários como Ti, Zr, W e Cr.
Considerando-se que as informações referentes ao espectrômetro de emissão
óptica com plasma acoplado indutivamente são mais relevantes nesta discussão,
um equipamento com configuração radial da marca Jobin-Yvon JY70 Plus foi
utilizado com as seguintes condições de operação: 1,0 kW de potência aplicada,
0,35 L min-1 de vazão do gás de nebulização e 1,2 mL min-1 de vazão de
bombeamento da amostra. A altura de observação variou de 12 - 15 mm. No que
diz respeito à digestão da amostra por fusão com LiBO2, a razão
amostra/fundente foi de 1:5 m/m, sendo que a mistura foi colocada em cadinho
de grafite e submetida à temperatura de 1050 oC durante 20 min. Também foi
utilizado serpentinite UB-N (agente “non-wetting”) na razão de 1:7 m/m (UB-N
/ amostra+fundente) para prevenir que o fundido ficasse aderido às paredes do
cadinho. O produto da fusão foi dissolvido em HNO3 0,8 mol L-1, perfazendo