Estudo do Espectro Vibracional de Nanotubos de
Carbono por Simula¸c˜
oes de Dinˆ
amica Molecular
Jessiara Garcia Pereira
Ouro Preto - MG
Estudo do espectro vibracional de nanotubos de
carbono por simula¸c˜
oes de dinˆ
amica molecular
Disserta¸c˜ao apresentada ao Programa de P´os-Gradua¸c˜ao em Ciˆencias: F´ısica de Materiais da UFOP, como requisito parcial para a obten¸c˜ao do t´ıtulo de Mestre em Ciˆencias.
Orientador: Alan Barros de Oliveira
Ouro Preto - MG
Catalogação: [email protected]
P436e Pereira, Jessiara Garcia.
Estudo do espectro vibracional de nanotubos de carbono por simulação de dinâmica molecular [manuscrito] / Jessiara Garcia Pereira. – 2014.
71 f.: il. color., grafs., tabs.
Orientador: Prof. Dr. Alan Barros de Oliveira e Ronaldo Junio Campos Batista (Co-orientador).
Dissertação (Mestrado) - Universidade Federal de Ouro Preto. Instituto de Ciências Exatas e Biológicas. Departamento de Física.
Área de concentração:Física de materiais
1. Nanotubos de carbono - Teses. 2. Dinâmica molecular - Teses. I. Universidade Federal de Ouro Preto. II. Título.
e competˆencia ao longo do desenvolvimento deste trabalho. Ao meu coorientador Ronaldo
Junio Campos Batista pelas contribui¸c˜oes. `A UFOP e ao Programa de P´os-Gradua¸c˜ao em
Ciˆencias com ˆEnfase em F´ısica de Materiais pela oportunidade de realizar o mestrado. Aos
professores do programa, `a secretaria do mestrado e do departamento de f´ısica, bem como
aos meus colegas pela paciˆencia e pelo apoio. Agrade¸co tamb´em aos meus pais e ao meu
irm˜ao por estarem ao meu lado durante os momentos dif´ıceis. A todos que colaboraram
direta ou indiretamente para a elabora¸c˜ao deste trabalho, o meu sincero reconhecimento.
Por fim, agrade¸co `as agˆencias de fomento pelo apoio financeiro durante a realiza¸c˜ao das
bracionais de nanotubos de carbono, bem como aplica¸c˜oes de espectroscopia Raman. Al´em
disso, apresentamos as principais caracter´ısticas da t´ecnica de simula¸c˜ao computacional e
dos potenciais emp´ıricos utilizados. Finalmente, mostramos os resultados obtidos nas
sim-ula¸c˜oes empregando diferentes condi¸c˜oes de adsor¸c˜ao de ´atomos de hidrogˆenio aos nanotubos.
Atrav´es das simula¸c˜oes verificamos que se mais de 104 ´atomos de hidrogˆenio forem absorvidos
nos nanotubos, essas estruturas tornam-se mecanicamente inst´aveis `a temperatura de 300
K. No entanto, a 100 K, os tubos s˜ao est´aveis, mesmo que totalmente cobertos com ´atomos
de hidrogˆenio. Os espectros vibracionais dos nanotubos foram estudados por meio da
inves-tiga¸c˜ao da banda-G. Os resultados obtidos com AIREBO, em compara¸c˜ao com ReaxFF, s˜ao
significativamente diferentes. Embora os resultados obtidos com AIREBO n˜ao mostrem
nen-huma separa¸c˜ao dos modos de vibra¸c˜ao ´optica transversais horizontais e verticais, o m´etodo
utilizando ReaxFF foi capaz de detectar tal efeito. Al´em disso, as simula¸c˜oes indicaram
que a absor¸c˜ao de hidrogˆenios nas laterais dos nanotubos de carbono causa deforma¸c˜oes
of single-wall carbon nanotubes. Two empirical models were used, namely, AIREBO and
ReaxFF. We have reviewed the literature about spectral and vibrational properties of carbon
nanotubes as well as Raman spectroscopy applications. Also, we present the main
character-istics of the computational simulation technique and the empirical potentials used. Finally,
we show the results obtained in the simulations employing different conditions of adsorption
of hydrogen atoms in the nanotubes. We find that if more than 104 hydrogen atoms are
adsorbed in the tube these structures become mechanically unstable at temperature of 300
K. However, at 100 K, the tubes are stable even if completely covered with hydrogen. The
vibrational spectra of the nanotubes was studied by investigating the G-band. The results
obtained with AIREBO compared to ReaxFF are significantly different. While the results
obtained with AIREBO show no separation of the horizontal and vertical transverse optical
vibrational modes, the method using ReaxFF was able to detect such an effect. In
addi-tion, the simulations indicate that the absorption of hydrogen on the sides of the carbon
1.1 Nanotubos de Carbono . . . 5
1.1.1 M´etodos de S´ıntese . . . 5
1.1.2 Estrutura e Geometria . . . 9
1.1.3 Dispers˜ao de Fˆonons . . . 13
1.1.4 Espectroscopia Raman . . . 15
1.1.5 Espectroscopia Raman em nanotubos . . . 17
2 M´etodos 20 2.1 Mecˆanica Estat´ıstica . . . 21
2.1.1 Ensemble Microcanˆonico . . . 21
2.1.2 Ensemble Canˆonico . . . 24
2.1.3 Ensemble das Press˜oes . . . 28
2.2 Dinˆamica Molecular . . . 30
2.2.1 Algoritmo de Verlet . . . 31
2.3 O Potencial de Intera¸c˜ao . . . 32
3 Resultados 51
3.1 Potencial AIREBO . . . 51
3.2 Potencial ReaxFF . . . 55
3.3 Considera¸c˜oes sobre os resultados: AIREBO vs. ReaxFF . . . 57
4 Modelo Te´orico 59
4.1 Tubo comprimido . . . 59
4.2 Tubo hidrogenado . . . 65
5 Conclus˜oes 68
1.2 NTCMs obtidos por Iijima. a) 5 folhas de grafeno com 6,7 nm de diˆametro; b) 2 folhas de grafeno com diˆametro externo de 5,5nm; c) 7 folhas de grafeno com diˆametro externo de 6,5 nm e diˆametro interno de 2,2nm[14]. . . 3
1.3 Representa¸c˜ao esquem´atica de uma camada de grafite que ao ser enrolada
origina um nanotubo. . . 4
1.4 Nanotubos de carbono com diferentes graus de deforma¸c˜ao [9] . . . 4
1.5 NTCs produzidos pela t´ecnica de Ablas˜ao por Laser [14]. . . 6
1.6 Os NTCs formam um emaranhado de filamentos de 10 a 20 nm de diˆametro
e 100µm de comprimento [14]. . . 7
1.7 Vista transversal de um dispositivo de descarga a arco para produ¸c˜ao de NTCs
[14]. . . 8
1.8 a) Representa¸c˜ao do sistema de crescimento de nanotubos de carbono por
CVD. b) e c) Fotos de um sistema comercial [16]. . . 8
1.9 a) Estrutura cristalina do grafeno. A c´elula unit´aria est´a delimitada pelas
linhas pontilhadas. b) Rede rec´ıproca do grafeno. A regi˜ao hachurada
corre-sponde `a primeira zona de Brillouin [14]. . . 9
1.10 Classifica¸c˜ao dos nanotubos: a) armchair, b) zigzag, c) quiral [14]. . . 10
vetores s˜ao descritos em termos de a1 ea2 que s˜ao os vetores unit´arios usados
para descrever a c´elula unit´aria do grafeno [14]. . . 11
1.12 (a) Diagrama de dispers˜ao de fˆonons para o grafeno. (b) Densidade de estados
de fˆonons para o grafeno. (c) Diagrama de dispers˜ao de fˆonons para um
nan-otubo armchair com (n,m) = (10, 10) com 72 ramos distintos. (d) Densidade
de estados de fˆonons para o nanotubo (10, 10) [16]. . . 14
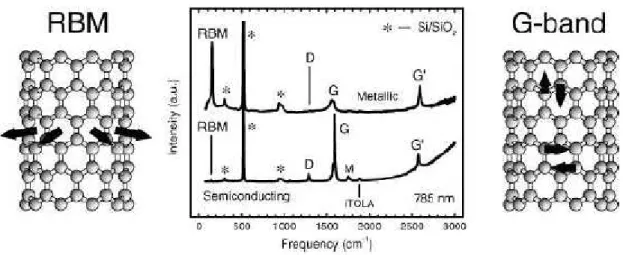
1.13 Espectro Raman de um NTCPS met´alico (acima) e semicondutor (abaixo),
mostrando o RBM, as bandas D, G e G’. Os nanotubos de carbono est˜ao
de-positados sobre um substrado de ´oxido de sil´ıcio que fornece contribui¸c˜ao para
o espectro Raman denotado por∗, que s˜ao utilizados para fins de calibra¸c˜ao [4]. 19
2.1 Dois fluidos separados por uma parede adiab´atica fixa e imperme´avel [13]. . 22
2.2 Sistema S em contato com o reservat´orio R (a temperatura T) [13]. . . 24
2.3 Dependˆencia da distˆancia interatˆomica da ordem liga¸c˜ao carbono-carbono. . 40
2.4 a) Efeito da corre¸c˜ao na ordem de liga¸c˜ao na Equa¸c˜ao 2 na liga¸c˜ao C-C e C-H
em uma mol´ecula de eteno em que a liga¸c˜ao C-C ´e encurtada para 1 ˚A com
o restante da geometria fixa. b) Efeito do encurtamento do comprimento da
liga¸c˜ao C-C no etano para 1 ˚A no comprimento liga¸c˜ao do par C-H calculado
por DFT e ReaxFF [6]. . . 42
2.5 Dependˆencia da distˆancia interatˆomica das liga¸c˜oes carbono-carbono,
carbono-hidrogˆenio e hidrogˆenio-hidrogˆenio e energia de van der Waals dos
sistemas C-C, C-H e H-H. Contribui¸c˜oes de energia para as intera¸c˜oes de
Coulomb e efeitos de energia relatados para sub e sobre-coordena¸c˜ao s˜ao
ig-norados na curva total de energia [6]. . . 49
3.1 Vis˜ao lateral (plano xz) do nanotubo de carbono hidrogenado (20 H) quando simulado com o ReaxFF a T = 300 K e P = 1 bar. . . 52
H’s. . . 54
3.4 Posi¸c˜oes dos m´aximos dos picos da banda G, TOh e TOv mostrados na Fig.
3.3, para adsor¸c˜oes de H desde 0 at´e 20 . . . 55
3.5 Densidade de estados de fˆonons [do inglˆes, phonon density of states (DOS)]
como fun¸c˜ao da frequˆencia de vibra¸c˜ao dos ´atomos obtidos usando o potencial
ReaxFF para hidrogna¸c˜oes do tubo com 0 (tubo perfeito), 4, 8, 12, 16 e 20 H’s. 56
3.6 Posi¸c˜oes dos m´aximos dos picos da banda G, TOh e TOv mostrados na Fig.
3.5, para adsor¸c˜oes de H desde 0 at´e 20 . . . 57
4.1 Modelo com oito massas e oito molas antes e depois de ser comprimido pela
ponta de AFM. Quando comprimido, as constantes de mola mudam para
κ+λ1 (superior e inferior) e κ−λ2 (laterais). . . 60
4.2 Deslocamentos arbit´arios uj com j = 0, . . . ,3. . . 61
4.3 Modos antisim´etricos de acordo com o modelo te´orico, Eqs. (4.10) e (4.13). . 65
4.4 Modos sim´etricos de acordo com o modelo te´orico, Eq. (4.7). . . 66
4.5 Modelo massa-mola com massas de valores diferentes, m1 e m2. Este modelo
imita o nanotubo de carbono hidrogenado. Sendo assim, neste trabalho, m1
´e a massa do carbono, mC enquanto quem2 ´e a soma das massas do carbono
e do hidrogˆenio, mH. . . 67
Introdu¸c˜
ao
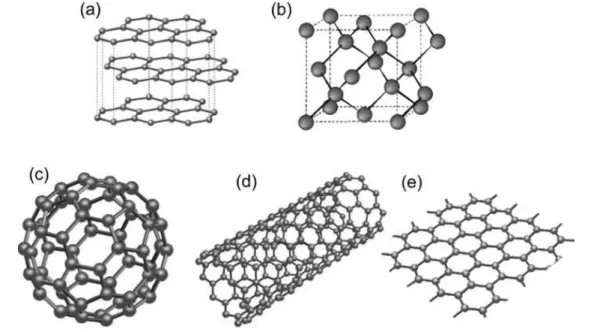
O carbono ´e um dos elementos mais abundandantes do universo, sua versatilidade deve-se
principalmente `as diversas maneiras nas quais seus quatro orbitais de valˆencia podem se
hibridizar, formando estruturas t˜ao distintas quanto o grafite e o diamante. A hibridiza¸c˜ao
ocorre entre os orbitais s e p da banda de valˆencia. No estado fundamental, o carbono apresenta a configura¸c˜ao eletrˆonica 1s2 2s2 2p2. Ao ser excitado, ele assume a configura¸c˜ao
1s22s12p3, de modo que os orbitais da banda de valˆencia podem combinar-se formando
orbitais h´ıbridos dos tipossp,sp2 e sp3.
Na hibridiza¸c˜aosp3, os ´atomos fazem liga¸c˜oes covalentes dando origem `a estrutura tetra´edrica
do diamante. Na hibridiza¸c˜aosp, s˜ao formadas cadeias lineares de carbono altamente reati-vas, as poliinas, estruturas dificilmente observadas em sua forma isolada. J´a na hibridiza¸c˜ao
sp2, os ´atomos formam liga¸c˜oes covalentes em uma rede hexagonal bidimensional, formando
o grafeno, cuja comprova¸c˜ao experimental foi apresentada em 2004 por Novoselov et all [10].
No grafeno, os orbitais 2s, 2px e 2py se hibridizam para formar os orbitais σ. J´a o orbital
de valˆencia 2pz n˜ao se hibridiza, dando origem ao orbital π. Os orbitais σ s˜ao respons´aveis
pelas fortes liga¸c˜oes covalentes entre os ´atomos de carbono no plano de grafeno, enquanto
os el´etrons presentes no orbital π atuam na condu¸c˜ao el´etrica e transi¸c˜oes ´oticas na regi˜ao do vis´ıvel. O grafite surge devido ao empilhamento de v´arias camadas de grafeno, unidas
atrav´es de intera¸c˜oes do tipo van der Waals. Al´em disso, na hibridiza¸c˜ao sp2 podem ser
encontrados materiais de diferentes dimensionalidades, cuja estrutura prim´aria ´e o plano
Figura 1.1: (a) grafite, (b) diamante, (c) fulereno, (d) nanotubo de carbono, (e) grafeno.
conforme est´a representado na Figura 1.1.
Historicamente, as substˆancias compostas por ´atomos de carbono tiveram grande
im-portˆancia no desenvolvimento tecnol´ogico. Os filamentos utilizados por Thomas Edison
para fabricar as primeiras lˆampadas el´etricas eram compostor por fibras de carbono. Na
Se-gunda Guerra Mundial, a exigˆencia por equipamentos mais leves e resistentes determinou a
busca por novos materiais. Neste per´ıodo as fibras de carbono se constitu´ıram como um dos
elementos mais promissores da ind´ustria aeroespacial. O progresso da produ¸c˜ao de fibras de
carbono levou `a sistematiza¸c˜ao e ao controle dos processos produtivos, estimulando o estudo
de filamentros de carbono com diˆametros cada vez menores. Estes estudos levaram `a
de-scoberta do fulereno (C60) por Kroto e Smalley em 1990. A partir da´ı surgiram expecula¸c˜oes
sobre a existˆencia de nanotubos de carbono com dimens˜oes comparadas `a do fulereno [14].
A primeira evidˆencia experimental dos nanotubos de carbono (NTCs) ocorreu em 1991 por
Sumio Iijima [8]. An´alises feitas por ele em um Microsc´opio Eletˆonico de Transmiss˜ao (TEM)
indicaram tubos formados por 2 a 50 folhas enroladas umas sobre as outras, cujo diˆametro
chegava a 2,2 nm. A descoberta de Iijima se referia aos nanotubos de paredes m´ultiplas (NCPMs), como mostrado na Figura 1.2. Dois anos depois, ele, em parceria com Toshinari
Ichihashi, publicaram um artigo relatando a obten¸c˜ao de nanotubos de parede ´unica (NCPSs)
de 1nm de diˆametro [8]. Apesar de outros autores terem relatado ind´ıcios da presen¸ca dos
Figura 1.2: NTCMs obtidos por Iijima. a) 5 folhas de grafeno com 6,7 nm de diˆametro; b) 2 folhas de grafeno com diˆametro externo de 5,5 nm; c) 7 folhas de grafeno com diˆametro externo de 6,5nm e diˆametro interno de 2,2 nm[14].
precis˜ao.
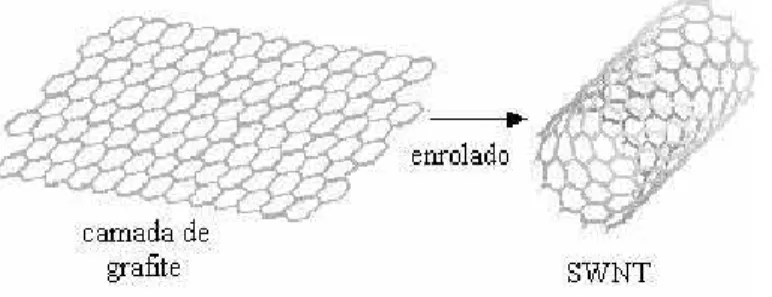
Os NTCs s˜ao representados por uma folha de grafeno enrolada em forma de cilindro e, em
condi¸c˜oes ideais, apresentam simetria esf´erica (Figura 1.3).
Apesar de ser formado por grafeno, um material extremamente r´ıgido, os nanotubos de
car-bono s˜ao bastante suscet´ıveis aos fatores ambientais, podendo sofrer deforma¸c˜ao. Estudos
recentes tˆem mostrado que os NCPSs podem interagir com os substratos s´olidos nos quais s˜ao
depositados, destruindo a forma idealizada de tubos livres (Figura 1.4 -b, c) e,
consequente-mente, suas propriedades eletrˆonicas [17]. As altera¸c˜oes na simetria dos nanotubos devido `as
deforma¸c˜oes provocam mudan¸cas nas propriedades eletrˆonicas, pois elas est˜ao relacionadas
com sua estrutura.
Al´em das deforma¸c˜oes causadas pela intera¸c˜ao do nanotubo com o substrato, a aplica¸c˜ao
de for¸cas externas tamb´em pode acarretar altera¸c˜oes permanentes em sua simetria. Um
estudo recente realizado por Araujo et all [2] constatou altera¸c˜oes permanentes na
-Figura 1.3: Representa¸c˜ao esquem´atica de uma camada de grafite que ao ser enrolada origina
um nanotubo.
Figura 1.4: Nanotubos de carbono com diferentes graus de deforma¸c˜ao [9]
Atomic Force Microscopy) com ponta de ouro.
As altera¸c˜oes estruturais induzidas pelo ambiente externo em NTCs podem ser medidas
ex-perimentalmente por Espectroscopia Raman. Em particular, a banda G do espectro Raman
informa os modos de vibra¸c˜ao tangenciais (TO) e longitudinais (LO) de NTCs, podendo
ser utilizada para estudar as altera¸c˜oes estruturais induzidas pelo ambiente externo [5]. Do
ponto de vista computacional, as altera¸c˜oes na simetria nanotubos podem ser estudadas por
t´ecnicas como a Dinˆamica Molecular (DM). Uma das vantagens da simula¸c˜ao computacional
´e a possibilidade de esclarecer fenˆomenos que n˜ao podem ser totalmente compreendidos
experimentalmente. Um destes casos ´e o estudo das deforma¸c˜oes mecˆanicas em NTCPSs.
Em um estudo realizado por Araujo et all [2] foram constatadas altera¸c˜oes permanentes na
simetria de NTCs ap´os serem comprimidos pela ponta de ouro de um Microsc´opio de For¸ca
Atˆomica. Embora a separa¸c˜ao permanente da banda G do espectro Raman dos NTCs tivesse
sido verificada, as causas deste fenˆomeno n˜ao foram completamente elucidadas.
O trabalho realizado por Araujo et all [2] foi o ponto de partida para a realiza¸c˜ao da nossa
pesquisa. Nela tivemos como principal objetivo investigar as poss´ıveis causas da separa¸c˜ao
permanente da banda G e a consequente deforma¸c˜ao nos NTCPSs. A ideia fundamental
que norteou o estudo consistiu no fato de que a introdu¸c˜ao de ´atomos de hidrogˆenio (H)
simula¸c˜oes computacionais de Dinˆamica Molecular. Durante as simula¸c˜oes foram
introduzi-dos ´atomos de H na parede do NTCPS a fim de verificar o comportamento do seu espectro
vibracional e, consequentemente, verificar se houve deforma¸c˜ao do tubo. Este processo foi
realizado atrav´es de dois potenciais emp´ıricos: o Adaptive Intermolecular Reactive Empirical
Bond Order Potential (AIREBO) e o Reactive Force Field (ReaxFF), que ser˜ao descritos
com detalhes no pr´oximo cap´ıtulo.
Antes de apresentar os resultados obtidos nas simula¸c˜oes, segue um referencial te´orico
so-bre os NTCs a fim de apresentar processos de s´ıntese, a estrutura e propriedades dos NTCs,
bem como suas propriedades vibracionais. No Cap´ıtulo 2 ser˜ao apresentados os fundamentos
te´oricos da metodologia de pesquisa empregada no trabalho. Posteriormente, no Cap´ıtulo 3,
ser˜ao apresentados os resultados obtidos no estudo sobre o espectro vibracional dos NTCPSs
atrav´es das simula¸c˜oes computacionais de DM. No Cap´ıtulo 4 ser˜ao apresentadas as
consid-era¸c˜oes finais sobre o trabalho.
1.1
Nanotubos de Carbono
1.1.1
M´
etodos de S´ıntese
Ablas˜ao por Laser
Na Ablas˜ao por Laser um alvo de grafite ´e vaporizado pela irradia¸c˜ao de um feixe de laser
pulsado na presen¸ca de um g´as inerte. O grafite ´e colocado no meio de um tubo de quartzo
que ´e evacuado e submetido a temperatura de 1200 ◦
C. Posteriormente, o tubo ´e preenchido
com o g´as inerte e o laser ´e posicionado sobre o alvo de grafite. O fluxo de g´as inerte arrasta
as part´ıculas de carbono geradas na zona de alta temperatura e as deposita em um coletor
de cobre, resfriado por ´agua, localizado na extremidade oposta do tubo de quartzo [14]. A
Figura 1.5 mostra a representa¸c˜ao de um aparato experimental para produ¸c˜ao de NTCs pela
t´ecnica de ablas˜ao por laser.
Atrav´es desta t´ecnica podem ser produzidos nanotubos de carbono de paredes m´ultiplas
Figura 1.5: NTCs produzidos pela t´ecnica de Ablas˜ao por Laser [14].
´e submetido `a abla¸c˜ao. J´a os NCPSs s˜ao produzidos introduzindo ao alvo de grafite uma
pequena quantidade de metal de transi¸c˜ao, geralmente Co, Ni, Fe, e Y, ou uma combina¸c˜ao
deles. Os NCPSs apresentam uniformidade de diˆametro e uma grande tendˆencia a formar
feixes. Durante a produ¸c˜ao podem ser encontradas impurezas tais como part´ıculas de grafite,
carbono amorfo, fulerenos e part´ıculas met´alicas, necessitando uma etapa de purifica¸c˜ao
para remover essas esp´ecies. O material produzido pode ser observado em um Microsc´opio
Eletrˆonico de Varredura (SEM - Scanning Electron Microscopy) como um emaranhado de
filamentos de 10 a 20 nm de diˆametro e 100 µm de comprimento (Figura 1.6) [14].
Descarga por Arco
Na Descarga por Arco (Figura 1.7), dois eletrodos de grafite (c´atodo e ˆanodo) s˜ao mantidos
a uma distˆancia de aproximadamente 1 mm para que a corrente passe ao ser aberto o arco voltaico. Na regi˜ao entre os dois eletrodos ´e gerado um plasma cuja temperatura pode
atingir entre 3000 e 4000 ◦
C. As altas temperaturas aquecem os eletrodos, vaporizando o
grafite. Em seguida o grafite vaporizado ´e sublimado na forma de uma fuligem que cont´em
Figura 1.6: Os NTCs formam um emaranhado de filamentos de 10 a 20 nm de diˆametro e
100µm de comprimento [14].
e carbono amorfo nas proximidades dos nanotubos demandando uma etapa de purifica¸c˜ao.
Assim como na t´ecnica de Ablas˜ao por Laser, a produ¸c˜ao de NCPSs demanda uma mistura
de metais de transi¸c˜ao, geralmente Fe, Co, Ni, Y, ou uma combina¸c˜ao deles [14].
Deposi¸c˜ao Qu´ımica a Vapor (CVD)
Nas d´ecadas de 1960 e 1970, a necessidade de sintetizar fibras de carbono em larga
escala demandou a elabora¸c˜ao de t´ecnicas como a Deposi¸c˜ao Qu´ımica a Vapor (CVD
-Chemical Vapour Deposition). Atualmente esta t´ecnica ´e empregada para a s´ıntese de NTCs
[14]. O processo de CVD ocorre atrav´es da decomposi¸c˜ao de compostos de carbono como
metano, etileno, acetileno, etc., que funcionam como agentes precursores de carbono e s˜ao
catalisados por nanopart´ıculas met´alicas, que tamb´em servem como s´ıtios de nuclea¸c˜ao para
iniciar o crescimento dos NTCs. Um sistema de CVD comercial, como o mostrado nas
fotos da Figura 1.8 b-c, consiste de um tubo de quartzo posicionado dentro de um forno
com controle automatizado da temperatura e do fluxo de gases dentro do reator (tubo de
quartzo), como ilustrado esquematicamente na Figura 1.8-a. Este sistema ´e considerado
vers´atil, simples e eficiente, pois permite a produ¸c˜ao de amostras de ´otima qualidade e com
Figura 1.7: Vista transversal de um dispositivo de descarga a arco para produ¸c˜ao de NTCs
[14].
Figura 1.8: a) Representa¸c˜ao do sistema de crescimento de nanotubos de carbono por CVD.
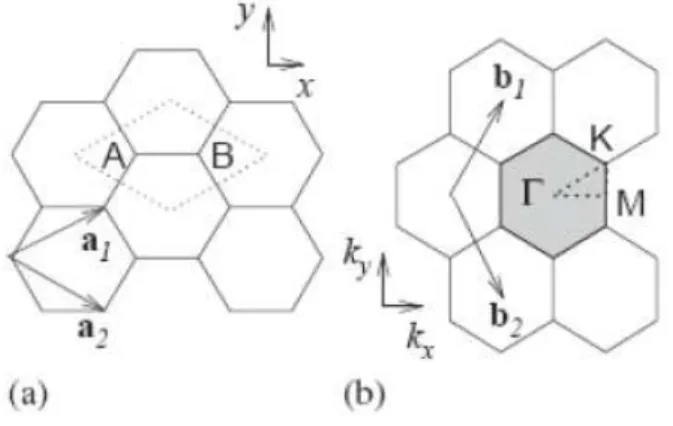
Figura 1.9: a) Estrutura cristalina do grafeno. A c´elula unit´aria est´a delimitada pelas linhas
pontilhadas. b) Rede rec´ıproca do grafeno. A regi˜ao hachurada corresponde `a primeira zona
de Brillouin [14].
1.1.2
Estrutura e Geometria
Os NTCs s˜ao representados por uma folha de grafeno enrolada em forma de cilindro. Por
isso, antes de falar dos detalhes sobre a estrutura e geometria dos nanotubos ´e importante
apresentar alguns pontos importantes sobre o grafeno, cuja c´elula unit´aria e primeira zona
de Brillouin est˜ao mostradas na Figura 1.9.
O grafeno possui dois ´atomos de carbono por c´elula unit´aria, cujos vetores de rede s˜ao dados
pela Equa¸c˜ao 1.1:
~ a1 =
√
3 2 a,
a
2
! , ~a2 =
√
3 2 a,
−a
2
!
(1.1)
onde a=|a~1 |=|a~2 |=aC−C
√
3, sendo que aC−C = 1,42 ˚A corresponde ao comprimento de
liga¸c˜ao entre dois ´atomos de carbono.
Os vetores b~1 e b~2 da rede rec´ıproca do grafeno s˜ao obtidos a partir da Equa¸c˜ao 1.2:
~ai~bj = 2πδij, (1.2)
Figura 1.10: Classifica¸c˜ao dos nanotubos: a) armchair, b) zigzag, c) quiral [14].
~b1 =
2π
√
3a,
2π a
,~b2 =
2π
√
3a,
−2π a
. (1.3)
A primeira zona de Brillouin do grafeno corresponde `a regi˜ao hachurada da Figura 1.9 -b, onde est˜ao destacados os pontos de simetria Γ, K e M.
As altera¸c˜oes na orienta¸c˜ao da folha de grafeno, bem como no diˆametro do tubo, modificam
suas propriedades de transporte eletrˆonico fazendo com que eles sejam met´alicos ou
semi-condutores. Levando em considera¸c˜ao a orienta¸c˜ao da folha de grafeno, os nanotubos podem
ser classificados em quirais ou aquirais.
O nanotubo quiral possui simetria espiral e n˜ao pode ter sua imagem sobreposta `a original
quando colocada diante de um espelho. J´a o nantubos aquiral ´e aquele cuja imagem no
espelho produz a mesma estrutura original. Este ´e o caso dos nanotubos zigzag e armchair
(Figura 1.10).
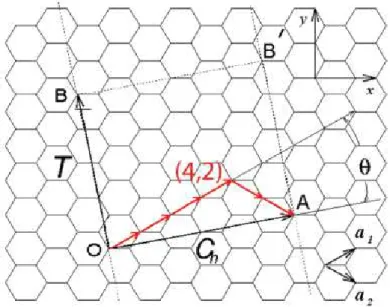
A c´elula unit´aria de um NTC pode ser determinada a partir de dois vetores perpendiculares
entre si no espa¸co real do grafeno: o vetor quiral (C~h) e o vetor transla¸c˜ao (T~).
A seguir, ser˜ao apresentadas as caracter´ısticas dos vetoresC~h eT~ de acordo com a referˆencia
Figura 1.11: O retˆangulo pontilhado representa a c´elula unit´aria de um nanotubo de carbono
(4, 2). S˜ao necess´arios dois vetores para descrever esta c´elula e, portanto, a estrutura do
nanotubo: o vetor quiral Ch e o vetor de transla¸c˜ao T. Ambos vetores s˜ao descritos em
termos de a1 e a2 que s˜ao os vetores unit´arios usados para descrever a c´elula unit´aria do
grafeno [14].
Vetor Chiral: C~h
O vetor chiral (C~h) fornece a dire¸c˜ao na qual a folha de grafeno ´e enrolada para formar o
nanotubo. A origem deste vetor est´a em um ´atomo de carbono e sua extremidade se localiza
na posi¸c˜ao de outro ´atomo idˆentico a ele na rede do grafeno. Qualquer ponto da rede onde
um ´atomo de carbono se localiza pode ser escrito em termos de uma combina¸c˜ao linear dos
vetores~a1 e~a2. Em termos destes vetores, C~h ´e dado pela Equa¸c˜ao 1.4:
~
Ch =n ~a1+m ~a2 ≡(n, m), (1.4)
onde n e m s˜ao n´umeros inteiros que devem satisfazer a rela¸c˜ao: 0≤m≤n.
O diˆametro, dt, de um NTC ´e dado por Lπ, em que L ´e o comprimento do nanotubo e
corresponde ao comprimento do vetor quiral, isto ´e,L=Ch,:
dt=
L π =
~ Ch
π = a π
√
variam entre 0 e 30 graus por causa da simetria da rede hexagonal do grafeno e ´e definido
pelo produto vetorial entreCh e a1. O ˆangulo θ pode ser obtido atrav´es da Equa¸c˜ao 1.6 que
relaciona θ com os n´umeros inteiros (n, m) da Equa¸c˜ao 1.4:
cosθ = Ch·a1
|Ch ||a1 |
= 2n+m
2√n2 +m2+nm, (1.6)
Os nanotubos zigzag e armchair correspondem a θ = 0◦
e θ= 30◦
, respectivamente.
Vetor Transla¸c˜ao: T~
O vetor transla¸c˜ao, T~, definido a partir do mesmo ponto de origem de C~h, ´e perpendicular a
este e define o eixo de rota¸c˜ao do nanotubo. A c´elula unit´aria pode ser constru´ıda
enrolando-se o retˆangulo de grafeno cuja ´area ´e delimitada pelos vetores C~h e T~ em torno do eixo de
rota¸c˜ao.
~
T =t1a1+t2a2 (1.7)
onde t1 e t2 s˜ao n´umeros inteiros e podem ser obtidos a partir das express˜oes:
t1 =
2m+n dR
, t2 = −
2m+n dR
(1.8)
onde dR ´e o m´aximo divisor comum de (2m+n) e (2n+m). Atrav´es da introdu¸c˜ao de d
como o maior divisor comum de n e m, dR pode ser relacionada com d. Assim, se n - m n˜ao
´e m´ultiplo de 3ddR ter´a o valor d; se n - m ´e m´ultiplo de 3d temos que dR= 3d.
No caso da Figura 1.11, onde Ch = (4,2), d = dR e T = (4,−5). O comprimento do vetor
T =|T |=√3L/dR, (1.9)
onde a circunferˆencia (L) do nanotubo ´e dada pela Equa¸c˜ao 1.5.
O comprimento T ´e reduzido quando (n,m) tem um divisor comum ou quando (n −m) ´e um m´ultiplo de 3d. Para um nanotubo armchair Ch = (5,5) tem-se que dR = 3d = 5 e
T = (1,−1), enquanto para um nanotubo zigzag Ch = (9,0) tem-se que dR = d = 9 e
T = (1,−2).
A c´elula unit´aria do NTC da Figura 1.11 ´e o retˆangulo OAB’B, definido pelos vetores C~h e
~
T, enquanto os vetores~a1 e~a2 definem a ´area da c´elula unit´aria do grafeno. Quando a ´area
da c´elula unit´aria do nanotubo ´e dividida pela ´area de um hex´agono, o n´umero de hex´agonos
por c´elula unit´aria N ´e obtido como uma fun¸c˜ao de n e m como:
N = |Ch×T |
|a1×a2 |
= 2(m
2+n2+nm)
dR
= 2L
2
a2d
R
, (1.10)
onde L e dR est˜ao dados nas Equa¸c˜oes 1.5 e na rela¸c˜ao entredR e d indicada acima,
respec-tivamente, e cada hex´agono cont´em dois ´atomos de carbono. Portanto, h´a 2N ´atomos de
carbono ou 2pz orbitais em cada c´elula unit´aria do NTC.
1.1.3
Dispers˜
ao de Fˆ
onons
Os ´atomos que comp˜oem uma rede cristalina vibram naturalmente em torno de suas posi¸c˜oes
de equil´ıbrio no cristal. Essas vibra¸c˜oes s˜ao quantizadas e seu quantum de energia ´e chamado
de fˆonon. Propriedades t´ermicas, mecˆanicas e de transporte dos s´olidos s˜ao fortemente
afe-tadas pela presen¸ca de fˆonons no material. A energia dos fˆonons apresenta uma dependˆencia
com seu vetor de onda e o conhecimento da rela¸c˜ao de dispers˜ao de fˆonons ´e fundamental no
estudo das propriedades vibracionais dos s´olidos cristalinos. A rela¸c˜ao de dispers˜ao de fˆonons
em NTCs tamb´em pode ser obtida, em primeira aproxima¸c˜ao, a partir da rela¸c˜ao de
dis-pers˜ao de fˆonons para o grafeno. Estas rela¸c˜oes s˜ao calculadas resolvendo-se as equa¸c˜oes de
Figura 1.12: (a) Diagrama de dispers˜ao de fˆonons para o grafeno. (b) Densidade de estados
de fˆonons para o grafeno. (c) Diagrama de dispers˜ao de fˆonons para um nanotubo armchair
com (n,m) = (10, 10) com 72 ramos distintos. (d) Densidade de estados de fˆonons para o
nanotubo (10, 10) [16].
c´elula unit´aria. Na aproxima¸c˜ao harmˆonica, a equa¸c˜ao de movimento ´e escrita pela Equa¸c˜ao
1.11:
Mir˙i =
X
j
Kij(ri−rj)(i= 1, ..., N) (1.11)
onde Mi ´e a massa do ´atomo i, ˙ri ´e a derivada segunda da posi¸c˜ao do ´atomo i e Kij a
constante de for¸ca entre os ´atomos i e j. N ´e o n´umero de pares de ´atomos na c´elula unit´aria do nanotubo e a soma ´e feita sobre os n vizinhos mais pr´oximos do ´atomo i [16].
O grafeno tem dois ´atomos na sua c´elula unit´aria, o diagrama de dispers˜ao apresenta seis
ramos de dispers˜ao de fˆonons (Figura 1.12 (a)). Os trˆes ramos de dispers˜ao que tˆem
frequˆencias nulas no ponto Γ da zona de Brillouin (oTA, iTA e LA) s˜ao os ramos de fˆonons
ac´usticos e os outros trˆes (oTO, iTO e LO) correspondem aos ramos de fˆonons ´opticos[16].
Como os nanotubos de carbono podem ser considerados como uma folha de grafeno enrolada
na forma de um cilindro, em primeira aproxima¸c˜ao, sua dispers˜ao pode ser obtida atrav´es
do processo de dobramento de zonas, Figura 1.12-c. Ent˜ao, a rela¸c˜ao de dispers˜ao de fˆonons
para os nanotubos ´e obtida da rela¸c˜ao de dispers˜ao de fˆonons do grafeno a partir da Equa¸c˜ao
ωmµ1D(K) =ω m
2D k
K2
|K2 |
+µK1 (1.12)
onde ωm
2D representa a rela¸c˜ao de dispers˜ao de fˆonons para o grafeno, k ´e o vetor de onda
unidimensional, |K2 |´e a magnitude do vetorK2 e µ= 0,1, ..., n−1 e −π/|T |< k < π/|
T |. O ´ındice m = 1,2, ...,6 representa cada ramo do fˆonon do grafeno.
A Figura 1.12 mostra as dispers˜oes de fˆonons e a densidade de estados de fˆonons para o
grafeno e para o nanotubo (10,10). A grande quantidade de picos na densidade de fˆonons do tubo (10, 10) reflete os muitos ramos de fˆonons e a natureza unidimensional dos nanotubos
surgindo do confinamento quˆantico dos estados de fˆonons nas singularidades de van Hove[16].
O m´etodo de dobramento de zonas no diagrama de dispers˜ao do grafeno ´e aplic´avel a quase
todos os fˆonons dos nanotubos. Por´em, esse m´etodo nem sempre fornece um resultado
correto, principalmente para fˆonons de baixa frequˆencia. Por exemplo, o ramo ´optico
rela-cionado `as vibra¸c˜oes fora do plano vai a ω → 0 para q → 0 no grafeno. No nanotubo, esse modo d´a origem ao modo de respira¸c˜ao radial cuja frequˆencia emq = 0 (ponto Γ) ´e finita e diferente de zero, obedecendo a rela¸c˜ao ωRBMαd
−1
t , onde dt ´e o diˆametro do tubo. Ent˜ao,
para se chegar ao diagrama de dispers˜ao correto, deve-se encontrar o tensor de constantes
de for¸ca para os nanotubos e resolver as equa¸c˜oes de movimento [16].
Como j´a foi dito, as rela¸c˜oes de dispers˜ao s˜ao diferentes para cada tipo de nanotubo. ´E
mostrado, como exemplo, na Figura 1.12(c) o diagrama de dispers˜ao obtido para o nanotubo
armchair (10, 10). Esse nanotubo possui 40 ´atomos na c´elula unit´aria (N = 20) e, portanto,
120 graus de liberdade [16].
1.1.4
Espectroscopia Raman
Em 1928, o f´ısico indiano C. V. Raman descobriu que o comprimento de onda vis´ıvel de uma
pequena fra¸c˜ao da radia¸c˜ao espalhada por certas mol´eculas difere do feixe incidente e, al´em
disso, que os deslocamentos em comprimento de onda dependem das estruturas qu´ımicas
das mol´eculas respons´aveis pelo espalhamento. A seguir ser´a descrito o modelo ondulat´orio
para o espalhamento Raman, baseando-se na referˆencia [15].
E =E0cos(2πνext), (1.13)
onde E0 ´e a amplitude da onda. Quando o campo el´etrico da radia¸c˜ao interage com uma
nuvem eletrˆonica de uma liga¸c˜ao, induz um momento de dipolo m na liga¸c˜ao que ´e dado
pela Equa¸c˜ao 1.14.
m=αE =αE0cos(2πνext), (1.14)
ondeα ´e uma constante de proporcionalidade chamada de polarizabilidade da liga¸c˜ao. Esta constante ´e uma medida do grau de deforma¸c˜ao da liga¸c˜ao em um campo el´etrico.
A polarizabilidadeαvaria em fun¸c˜ao da distˆancia entre os n´ucleos, de acordo com a Equa¸c˜ao 1.15:
α=α0+ (r−req)
∂α ∂r
, (1.15)
onde α0 ´e a polarizabilidade da liga¸c˜ao na distˆancia internuclear de equil´ıbrio req, e r ´e a
separa¸c˜ao internuclear em qualquer instante. A altera¸c˜ao na separa¸c˜ao internuclear varia
com a frequˆencia da vibra¸c˜aoνv e ´e dada por 1.16:
r−req =αrmcos(2πνvt), (1.16)
onde rm ´e a separa¸c˜ao internuclear m´axima com rela¸c˜ao `a posi¸c˜ao de equil´ıbrio.
Substituindo a equa¸c˜ao 1.16 na equa¸c˜ao 1.15, temos
α=α0+
∂α
∂r
1.17 na Equa¸c˜ao 1.14. Assim:
m=α0E0cos(2πνext) +E0rm
∂α ∂r
cos(2πνvt)cos(2πνext). (1.18)
Utilizando-se a identidade trigonom´etrica para o produto de dois cossenos indicada na
Equa¸c˜ao 1.19:
cosxcosy= [(cos(x+y) +cos(x−y)]/2, (1.19)
obtem-se da equa¸c˜ao 1.20:
m=α0E0cos(2πνext) +
E0
2 rm
∂α ∂r
cos[2π(vex−vv)t] +
E0
2 rm
∂α
∂r
cos[2π(vex+vv)t].
(1.20)
O primeiro termo nesta equa¸c˜ao representa o espalhamento Rayleigh (espalhamento el´astico),
o qual ocorre na frequˆencia de excita¸c˜ao v(ex). O segundo e o terceiro termos da Equa¸c˜ao
1.20 correspondem, respectivamente, `as frequncias stokes e anti-stokes devex−vv evex+vv.
Neste caso, a frequˆencia de excita¸c˜ao foi modulada pela frequˆencia vibracional da liga¸c˜ao.
O espalhamento Raman ocorrer´a somente se houver varia¸c˜ao da polarizabilidade durante a
vibra¸c˜ao, ou seja,∂α/∂r na Equa¸c˜ao 1.20 deve ser maior ou igual a zero para que as linhas Raman apare¸cam.
1.1.5
Espectroscopia Raman em nanotubos
Espectroscopia Raman tem se tornado uma das ferramentas mais valiosas para estudar
as propriedades eletrˆonicas e vibracionais de nanotubos de carbono. Os processos Raman
podem ser de primeira ordem (RBM e banda G) ou de segunda ordem (bandas D e G′
),
hexagonal), a banda G´e observada entre 1500 e 1600 cm e a banda G ´e observada em torno de 2700 cm−1
.
Modo de respira¸c˜ao radial
O modo de respira¸c˜aoo radial (RBM - radial breathing mode) est´a entre 50 e 500 cm−1
e
est´a associado `as vibra¸c˜oes dos ´atomos de carbono na dire¸c˜ao radial em rela¸c˜ao ao eixo do
nanotubo. A observa¸c˜ao do RBM no espectro Raman fornece a evidˆencia direta de que a
amostra cont´em nanotuos de carbono. A rela¸c˜ao entre a frequˆencia e o diˆametro do nanotubo
de carbono pode ser dada por:
ωRBM =
227
dt
(1 +Ce.d2t)
1
2, (1.21)
onde dt ´e o diˆametro do nanotubo e Ce ´e uma constante relacionada a fatores de ambiente,
tais como atmosfera, tipos diferentes de surfactantes em solu¸c˜oes de nanotubos ou intera¸c˜ao
entre o nanotubo e o substrato. Atrav´es do espectro Raman podem ser obtidas infoma¸c˜oes
sobre EiiS,M e ωRBM. O par (EiiS,M e ωRBM) ´e ´unico para cada NTCPS e com a ajuda do
gr´afico de Kataura, uma esp´ecie de mapa da energia de transi¸c˜aoEiiem fun¸c˜ao do diˆametro
do nanotubo (dt), que est´a ligado a ωRBM pela rela¸c˜ao dada na Equa¸c˜ao 1.21), ´e poss´ıvel
identificar cada ´ındice (n,m) que identifica um nanotubo de carbono. Mais informa¸c˜oes sobre
o gr´afico de Kataura e sobre a identifica¸c˜ao dos ´ındices (n,m) em nanotubos, bem como sua
rela¸c˜ao com o diˆametro dt podem ser obtidas na referˆencia [16].
Banda G
Os modos tangenciais, ou banda G, em nanotubos de carbono est˜ao diretamente relacionados
`a banda G em grafite, que ´e identificada com o movimento atˆomico no plano da folha de
grafeno, e ´e observado em torno de 1590 cm−1
. Em nanotubos esses modos correspondem
Figura 1.13: Espectro Raman de um NTCPS met´alico (acima) e semicondutor (abaixo),
mostrando o RBM, as bandas D, G e G’. Os nanotubos de carbono est˜ao depositados sobre
um substrado de ´oxido de sil´ıcio que fornece contribui¸c˜ao para o espectro Raman denotado
por∗, que s˜ao utilizados para fins de calibra¸c˜ao [4].
transversal ´optico (TO)). Geralmente, a banda G ´e dividida em duas componentes, uma de
maior e outra de menor frequˆencia, chamadas componentesG+eG−
. A an´alise da frequˆencia
do modo de estiramento tangencial pode ser usada para medir deforma¸c˜ao e dopagem, e para
distinguir entre tubos met´alicos e semicondutores por causa da presen¸ca de uma anomalia
de Kohn na dispers˜ao de fˆonon de NTCPS met´alico. A anomalia de Kohn est´a relacionada
ao comportamento anˆomalo na dispers˜ao de fˆonons do grafeno. Essa anomalia acontece para
determinados fˆonons com vetores de onda que conectam dois pontos da superf´ıcie de Fermi,
em que acontece uma mudan¸ca abrupta da blindagem eletrˆonica nas vibra¸c˜oes dos ´atomos.
Isso resulta em uma diminui¸c˜ao na frequˆencia dos fˆonons. Para o grafeno, a anomalia de
Kohn ocorre no ramo longitudinal ´optico (LO) no ponto Γ e no ramo TO no ponto K. No
caso dos nanotubos de carbono, devido `a sua baixa dimensionalidade, a anomalia de Kohn
´e ainda mais pronunciada no caso de nanotubos de carbono met´alicos [11].
Banda G’
Nos nanotubos de carbono, a banda G’ ´e o modo mais intenso de segunda ordem no espectro
Raman. A frequˆencia da banda G′
depende da energia de excita¸c˜ao do laser e aparece em
entre 2600 e 2700 cm−1
. Deslocamentos de frequˆencia neste modo podem ser usados para
O estudo do espectro vibracional dos NTCPS foi desenvolvido a partir de simula¸c˜ao
com-putacional. A simula¸c˜ao computacional (SC) tem como principal objetivo reproduzir o
comportamento de sistemas reais atrav´es de modelos usados em computadores. O emprego
de tais t´ecnicas se justifica nas situa¸c˜oes em que um experimento n˜ao pode ser realizado em
laborat´orio, seja pela complexidade do problema, sejam pelas limita¸c˜oes experimentais.
Existem v´arias t´ecnicas de SC, entre elas podem ser citados o M´etodo de Monte Carlo,
Autˆomatos Celulares e Dinˆamica Molecular. Neste trabalho, o estudo do espectro vibracional
dos NTC ser´a realizado via Dinˆamica Molecular (DM). A DM foi desenvolvida ao longo da
d´ecada de 1950 por Alder e Wainwright [1] e aperfei¸coada na d´ecada de 1960 por Rahman
[12], consagrando-se na solu¸c˜ao de problemas de f´ısica estat´ıstica computacional
expandindo-se, posteriormente, para outras ´areas. Atualmente a DM ´e bastante empregada no estudo
de sistemas f´ısicos e biol´ogicos.
Com esta t´ecnica se obt´em as posi¸c˜oes e velocidades das part´ıculas a partir da resolu¸c˜ao
das equa¸c˜oes cl´assicas do movimento. Para se obter as propriedades do sistema, calcula-se a
m´edia das propriedades das part´ıculas deste sistema utilizando a mecˆanica estat´ıstica. Por
ser fundamental para a compreens˜ao do formalismo que envolve os c´alculos computacionais
da Dinˆamica Molecular, o in´ıcio deste cap´ıtulo ser´a dedicado `a revis˜ao dos conceitos b´asicos
da Mecˆanica Estat´ıstica. Posteriormente, ser˜ao discutidos os temas relacionados `a Dinˆamica
A F´ısica Estat´ıstica explica as leis e os resultados da Termodinˆamica levando em considera¸c˜ao
o comportamento do grande n´umero de part´ıculas que comp˜oe os corpos macrosc´opicos. Sua
importˆancia deve-se ao fato de que apesar do movimento das part´ıculas microsc´opicas ser
governado pelas leis da mecˆanica, a resolu¸c˜ao das equa¸c˜oes do movimento para o grande
n´umero de part´ıculas que comp˜oem os sistemas, da ordem do n´umero de Avogadro (A ≃
6,02× 1023), torna-se impratic´avel. Nesse cen´ario, a Mecˆanica Estat´ıstica caracteriza-se
como uma ´area do conhecimento importante, uma vez que re´une elementos da mecˆanica
e os princ´ıpios da teoria das probabilidades para interpretar os resultados emp´ıricos da
Termodinˆamica [13].
Para se fazer uma an´alise mecˆanico-estat´ıstica ´e necess´ario estabelecer um postulado
es-tat´ıstico, fazer as conex˜oes com a Termodinˆamica, atrav´es das vari´aveis vis´ıveis do mundo
macrosc´opico e, por fim, definir um estado microsc´opico para o sistema. Este estado
microsc´opico pode ser definido como um conjunto de coordenadas de posi¸c˜ao e
veloci-dade de todas as N part´ıculas que comp˜oe o sistema, podendo ser representado por
xa = r1, ..., rN, v1, ...., vN. O conjunto de estados microsc´opicos, ou microestados acess´ıveis
ao sistema, constitui um conjunto (ensemble) estat´ıstico [13]. S˜ao comuns em simula¸c˜oes trˆes
ensembles estat´ısticos para descrever microscopicamente a mat´eria: microcanˆonico, canˆonico
e ensemble das press˜oes, que ser˜ao descritos a seguir.
2.1.1
Ensemble Microcanˆ
onico
O Ensemble Microcanˆonico refere-se a um sistema adiab´atico, imperme´avel e de tamanho
fixo. Muitas vezes ele ´e representado pela sigla NVE, que indica n´umero de part´ıculas,
volume e energia total fixos. A partir deste ensemble ´e poss´ıvel definir o n´umero de
microes-tados microsc´opicos (Ω) acess´ıveis do sistema para um dado valor de energia. Em Mecˆanica
Estat´ıstica, as vari´aveis termodinˆamicas do sistema, tais como a energia total, energia livre,
entropia e press˜ao, podem ser expressas em termos da fun¸c˜ao de parti¸c˜ao do sistema.
Para analisar o comportamento de um sistema termodinˆamico com E, N e V fixos, vamos
considerar um recipiente fechado composto por dois fluidos 1 e 2 separados por uma parede
Figura 2.1: Dois fluidos separados por uma parede adiab´atica fixa e imperme´avel [13].
O n´umero de estados microsc´opicos acess´ıveis ao Sistema 1 ´e dado por Ω1(E1, V1, N1) e
ao Sistema 2 por Ω2(E2, V2, N2). O n´umero de estados microsc´opicos acess´ıveis ao sistema
constitu´ıdo pelos dois subsistemas independentes ´e dado pelo produto:
Ω = Ω1(E1, V1, N1)Ω2(E2, V2, N2). (2.1)
Supondo que em um determinado instante a parede divis´oria interna se torna diat´ermica. A
energia total permanece constante, mas a energia de cada subsistema E1 e E2 pode variar,
contanto que E1 +E2 = E0, onde E0 ´e a energia total constante. Os outros parˆametros
macrosc´opicos dos dois sistemas (V1, V2, N1, N2) permanecem constantes. Eliminando os
parˆametros constantes para simplificar a nota¸c˜ao, o n´umero de estados microsc´opicos do
sistema composto, numa situa¸c˜ao em que o subsistema 1 tenha energia E1 e o subsistema 2
tenha energia E2 = (E0−E1) ´e dado por:
Ω(E1;E0) = Ω1(E1)Ω2(E0−E1). (2.2)
De acordo com o postulado fundamental da mecˆanica estat´ıstica, a probabilidade de
encon-trar o sistema composto em um estado microsc´opico em que a energia do subsistema 1 seja
dada porE1 e a energia do sistema 2 dada por E2 =E0−E1, pode ser descrita como:
composto pelos dois subsistemas:
1
c = Ωc =
E0
X
E1=0
Ω1(E1)Ω2(E0−E1). (2.4)
Substituindo a Equa¸c˜ao 2.4 na Equa¸c˜ao 2.3, temos:
P(E1) =
Ω(E0;E1)
PE0
E1=0Ω1(E1)Ω2(E0−E1)
. (2.5)
Como os dois subsistemas possuem energias diferentes, haver´a fluxo de energia do corpo
com maior energia para o de menor energia, at´e que o equil´ıbrio t´ermico se estabele¸ca.
Como consequˆencia, Ω(E) deve crescer com o aumento da energia E, pois devem aparecer mais estados microsc´opicos dispon´ıveis `a medida que a energia aumenta. Adotando que,
inicialmente, a energia do subsistema E1 ´e maior que a energia de E2, temos que Ω(E1)
cresce, enquanto Ω(E0−E1) decresce com a energiaE1, indicando que a fun¸c˜aoP(E1) deve
apresentar um m´aximo. Escrevendo o logaritmo natural da probabilidade:
lnP(E1) = lnc+ ln Ω1(E1) + ln Ω2(E0−E1). (2.6)
Na situa¸c˜ao de probabilidade m´axima, ondeE2 =E0−E1, temos:
∂lnP(E1)
∂E1
= ∂ln Ω1(E1)
∂E1 −
∂ln Ω2(E2)
∂(E2)
= 0. (2.7)
Em outras palavras, a condi¸c˜ao de equil´ıbrio ´e satisfeita quando a rela¸c˜ao 2.8 ´e obtida:
∂ln Ω1(E1)
∂E1
N1,V1
=
∂ln Ω2(E2)
∂(E2)
N2,V2
Figura 2.2: Sistema S em contato com o reservat´orio R (a temperatura T) [13].
Os passos anteriores permitem verificar que ao ser atingida a situa¸c˜ao de equil´ıbrio,
ln Ω(E1, E−E1) do sistema total encontra-se no m´aximo. Isto sugere que ln Ω(E1, E−E1),
que agora ser´a chamada de ln Ω, informa a entropia do sistema, concordando com a Segunda
Lei da Termodinˆamica: a entropia do sistema N, V e E ser´a m´axima quando ele estiver
em equil´ıbrio t´ermico. A rela¸c˜ao entre ln Ω e a entropia pode ser estabelecida atrav´es da
equa¸c˜ao: S(N, V, E) =kBln Ω(N, V, E), onde kB ´e a constante de Boltzman (1,38066.10
−23
J/K em unidades do S.I).
2.1.2
Ensemble Canˆ
onico
O Ensemble Canˆonico, tamb´em conhecido como NVT, ´e usado para descrever um sistema
(S) que que est´a em contato com um reservat´orio t´ermico (R). Esse sistema tem n´umero de part´ıculas (N) e volume (V) fixos, pois suas paredes s˜ao imperme´aveis e r´ıgidas. Al´em
disso, deve-se considerar as trocas de calor com o reservat´orio, tal que sua temperatura se
mantenha fixa e igual a do reservat´orio.
No ensemble canˆonico deseja-se saber qual a probabilidade de encontrar o sistema S em um
particular microestado j de energia Ej. Para responder esta quest˜ao, come¸ca-se a analisar o
sistema composto pelos dois subsistemas R e S na formula¸c˜ao microcanˆonica. Neste caso, a
energia total (E0) do sistema composto (S+R) ´e constante, ou seja,
E0 =Ej +E, (2.9)
dado por:
Ω(Ej, E) = ΩS(Ej)ΩR(E), (2.10)
onde ΩS(Ej) ´e o n´umero de estados microsc´opicos acess´ıveis ao reservat´orio S com energia
Ej e ΩR(E) ´e o n´umero de estados microsc´opicos acess´ıveis ao reservat´orio R com energia E.
Quando o sistema composto (S+R) estiver isolado, com energia total E0, valem os
postu-lados fundamentais da mecˆanica estat´ıstica de equil´ıbrio. Nesse caso, a probabilidadePj de
encontrar o sistema S num estado microsc´opico particular j ser´a dada por:
Pj =
Ω(Ej, E) Ω0
. (2.11)
Em outras palavras,
Pj =
1 Ω0
ΩS(Ej)ΩR(E). (2.12)
Como o n´umero de microestados acess´ıveis ao sistema total (Ω0) ´e constante, a probabilidade
Pj de encontrar o sistema S em um dado estado de energia j pode ser escrita em termos da
constante (c):
Pj =cΩS(Ej)ΩR(E). (2.13)
Al´em disso, como o n´umero de microestados acess´ıveis ao sistema S ´e muito menor que o
n´umero de microestados acess´ıveis ao sistema R, temos que ΩS(Ej) = 1, ent˜ao:
ln ΩR(E) = ln ΩR(E0)+
∂ln ΩR(E)
∂E
E=E0
(−Ej)+
1 2
∂2ln Ω
R(E)
∂E2
E=E0
(−Ej)2+.... (2.15)
Usando a defini¸c˜ao de entropia proporcionada pelo segundo postulado da mecˆanica
es-tat´ıstica, onde T ´e a temperatura do reservat´orio, temos:
∂ln ΩR(E)
∂E =
1
kBT
, (2.16)
Como a energia do reservat´orio R ´e fixa, o termo quadr´atico da expans˜ao pode ser reescrito
como:
∂2ln ΩR(E)
∂E2 =
1 kB ∂ ∂E 1 T
→0, (2.17)
Diante destes argumentos, temos que a expans˜ao se reduz `a forma:
ln ΩR(E) = ln ΩR(E0)−
1
kBT
Ej, (2.18)
ou seja,
ΩR(E) = ΩR(E0) exp (−βEj), (2.19)
onde β = 1/(kBT).
Como ΩR(E0) ´e constante e independe do estado j, a probabilidade Pj pode ser reescrita
como:
c= P 1
jexp−βEj
, (2.21)
Ent˜ao, a probabilidade Pj pode ser escrita como:
Pj =
exp−βEj
Z , (2.22)
e
Z =X
j
exp(−βEj). (2.23)
A probabilidadePj´e conhecida como distribui¸c˜ao de probabilidade canˆonica, onde exp−βEj
´e o fator de Boltzman e Z ´e a fun¸c˜ao de parti¸c˜ao canˆonica do sistema.
Observa-se que a soma das energias ´e realizada sobre todos os estados macrosc´opicos.
Por-tanto, dado um certo valor de energia, pode haver v´arios termos iguais, correspondentes a
todos os estados microsc´opicos com este particular estado de energia. Levando em conta
esse fator de degenerescˆencia, pode-se escrever:
Z =X
j
exp(−βEj) =
X
E
Ω(E) exp(−βE), (2.24)
onde Ω(E) ´e o n´umero de estados microsc´opicos do sistema S com energia E. Substituindo a soma pelo seu termo m´aximo:
Z =X
E
exp[lnΩ(E)−βE] exp[−βminEE−T S(E)], (2.25)
onde foi utilizada a defini¸c˜ao de entropia S(E) = kBln Ω(E). Como a opera¸c˜ao de
Z →exp(−βF), (2.26)
onde F ´e a energia livre de Helmholtz. Como os n´ıveis de energia Ej devem ser fun¸c˜oes do
volume V e do n´umero de part´ıculas N, a fun¸c˜ao de parti¸c˜ao Z depende de T, V e N, ou
seja,Z =Z(T, V, N). Nesse caso, podemos escrever:
F =F(T, V, N)→ −1
β lnZ(T, V, N). (2.27)
A vantagem de se conhecer a fun¸c˜ao de parti¸c˜ao ´e que a partir dela ´e poss´ıvel determinar todas
as propriedades termodinˆamicas macrosc´opicas do sistema. Por exemplo, para o Ensemble
Canˆonico:
E =kBT2
∂lnZ
∂T
N,V
(2.28)
P =kBT
∂lnZ
∂V
N,T
(2.29)
S=kBT lnΛ +kB
∂lnZ
∂T
N,V
(2.30)
2.1.3
Ensemble das Press˜
oes
No Ensemble das Press˜oes, ou simplesmente ensemble NPT, o n´umero de part´ıculas (N), a
press˜ao (P) e a temperatura (T) s˜ao fixos. Uma das particularidades deste ensemble ´e que
reservat´orio R com temperatura e press˜ao fixas. O sistema composto est´a isolado, com
energia total E0 e volume total V0. A parede ideal que separa os subsistemas ´e diat´ermica e
m´ovel, mas imperme´avel `a passagem de part´ıculas. Portanto, a probabilidade do sistema S
ser encontrado num particular estado microsc´opico j, com energiaEj e volume V0, pode ser
escrita como:
Pj =cΩR(E, V). (2.31)
onde c ´e uma constante e ΩR(E, V) ´e o n´umero de estados microsc´opicos acess´ıveis ao
reservat´orio R, com energia E e volume V. Podemos, ent˜ao, escrever a expans˜ao:
lnPj =constante+
∂ln ΩR
∂E
E0,V0
(−Ej) +
∂ln ΩR
∂V
E0,V0
(−Vj) +.... (2.32)
Usando a defini¸c˜ao de entropia dada pelo segundo postulado da mecˆanica estat´ıstica, onde
T e p s˜ao a temperatura e a press˜ao do reservat´orio, temos:
∂ln ΩR(E)
∂E =
1
kBT
e∂ΩR
∂V =pkBT (2.33)
No limite de um reservat´orio t´ermico suficientemente grande, a expans˜ao pode ser escrita
como:
Pj =c−
Ej
kBT −
pVj
kBT
. (2.34)
ou seja,
Pj =
1
Y =
j
exp(−βEj−βpVj). (2.36)
Neste ensemble, o volume, entalpia e entropia do sistema s˜ao dados por
V =−kBT
−∂lnY ∂P
N,T
(2.37)
H =−kBT
−∂lnY ∂T
N,P
(2.38)
S=kBT lnY +kBT
−∂lnY ∂T
N,P
(2.39)
2.2
Dinˆ
amica Molecular
Na Dinˆamica Molecular (DM), as equa¸c˜oes do movimento s˜ao resolvidas levando em
con-sidera¸c˜ao todas part´ıculas que interagem no sistema. As equa¸c˜oes b´asicas do movimento
s˜ao:
~
FA(t) = m~aA(t), (2.40)
~aA(t) =
d~vA(t)
dt , (2.41)
~vA(t) =
d~rA(t)
era¸c˜ao e for¸ca resultante sobre uma part´ıcula A, em um instante t.
A for¸ca ´e obtida a partir do potencial de intera¸c˜ao para as part´ıculas do sistema. No caso
deste trabalho, os potencias empregados para o estudo do espectro vibracional dos
nanotu-bos de carbono foram o Adaptive Intermolecular Reactive Empirical Bond Order Potential
(AIREBO) [3] e oReactive Force Field(ReaxF F) [6]. Nas pr´oximas se¸c˜oes estes potenciais ser˜ao descritos com mais detalhes.
Ap´os a obten¸c˜ao das for¸cas de intera¸c˜ao entre as part´ıculas, ´e poss´ıvel determinar a
acel-era¸c˜ao, a velocidade e a posi¸c˜ao de cada uma delas.
O c´alculo das posi¸c˜oes, velocidades e acelera¸c˜oes das part´ıculas ´e realizado atrav´es da
inte-gra¸c˜ao das equa¸c˜oes de Newton do movimento. Um dos algoritmos que foram concebidos
para esta tarefa ´e o Algoritmo de Verlet [7].
2.2.1
Algoritmo de Verlet
O Algoritmo de Verlet ´e um dos mais simples para integra¸c˜ao num´erica das equa¸c˜oes de
movimento. Neste m´etodo a expans˜ao em s´eries de Taylor da posi¸c˜ao (~rA) de uma part´ıcula
em torno do tempo (t), fornece uma equa¸c˜ao que descreve sua trajet´oria:
~rA(t+ ∆t) =~rA(t) +~vA(t)∆t+
1
2~aA(t)∆t
2+ ∆t3
3! ...
rA+ Θ∆t4, (2.43)
onde t ´e o instante de tempo presente, ∆t ´e o intervalo de tempo,~vA e~aA s˜ao,
respectiva-mente, os vetores velocidade e acelera¸c˜ao da part´ıcula A.
Para a trajet´oria da part´ıcula no instantet−∆t, pode-se expandir~rAem torno det, obtendo
a equa¸c˜ao:
~rA(t−∆t) =~rA(t)−~vA(t)∆t+
1
2~aA(t)∆t
2
−∆t
3
3! ...
rA+ Θ∆t4, (2.44)
em outras palavras:
~rA(t+ ∆t)≈2~rA(t)(∆t)−~rA(t−∆t) +~aA(t)∆t2 (2.46)
A estimativa da nova posi¸c˜ao possui um erro de ordem ∆t4, onde ∆t ´e o passo de tempo
na DM. Pode-se notar que o algoritmo de Verlet n˜ao usa a velocidade para calcular a nova
posi¸c˜ao. Por´em, ela pode ser obtida a partir da trajet´oria da part´ıcula fazendo:
~rA(t+ ∆t)−~rA(t−∆t) = 2~vA(t)(∆t) + Θ∆t3, (2.47)
onde a Equa¸c˜ao 2.47 pode ser escrita na forma da Equa¸c˜ao 2.48 tornando poss´ıvel a
deter-mina¸c˜ao da velocidade~vA da part´ıcula:
~vA(t) =
~rA(t+ ∆t)−~rA(t−∆t)
2∆t + Θ∆t
3. (2.48)
O algoritmo de Verlet simula um sistema isolado, ou seja, um ensemble NVE. Neste tipo de
sistema, o n´umero de part´ıculas (N), o volume da caixa de simula¸c˜ao (V) e a energia (E)
permanecem fixos, por´em, a temperatura e a press˜ao variam.
2.3
O Potencial de Intera¸c˜
ao
Na Dinˆamica Molecular (DM), as mol´eculas s˜ao tratadas como um conjunto de ´atomos
que podem ser descritos por for¸cas newtonianas. O conjunto completo dos potenciais de
intera¸c˜ao entre as part´ıculas ´e chamado de campo de for¸ca. Um campo de for¸ca emp´ırico,
descrito como a soma dos v´arios termos de energia, incluindo os termos para os ´atomos
ligados (comprimentos e ˆangulos de liga¸c˜ao, ˆangulos diedros) e os termos para ´atomos
n˜ao-ligados (intera¸c˜oes de van der Waals e de Coulomb).
Um potencial t´ıpico ´e representado pela Equa¸c˜ao 2.49:
V(r) =XVl+
X Vθ+
X
VvdW +
X
Velec, (2.49)
onde, Vl ´e a energia de estiramento da liga¸c˜ao em rela¸c˜ao a seu valor de equil´ıbrio, Vθ ´e
a energia de deforma¸c˜ao do ˆangulo de liga¸c˜ao em rela¸c˜ao a seu valor de equil´ıbrio, VvdW
representa a energia das intera¸c˜oes de van der Waals eVelec representa as energias de atra¸c˜ao
ou repuls˜ao eletrost´atica entre duas cargas.
Os diversos potenciais existentes foram desenvolvidos de maneira independente, envolvendo
conjuntos de parˆametros espec´ıficos. Alguns incluem termos para descrever especificamente
as liga¸c˜oes de hidrogˆenio ou para acoplar oscila¸c˜oes entre ˆangulos e comprimentos de liga¸c˜ao,
com o objetivo de se obter uma melhor concordˆancia com espectros vibracionais. A
con-fiabilidade dos resultados ´e baseada na elabora¸c˜ao de um potencial com parˆametros bem
definidos. A escolha do potencial depende, em grande parte, do sistema a ser estudado e
das propriedades que ser˜ao investigadas. No caso dos sistemas estudados neste trabalho,os
potenciais utilizados foram: o Adaptive Intermolecular Reactive Empirical Bond Order
Po-tential (AIREBO) e o Reactive Force Field (ReaxFF).
2.3.1
Potencial AIREBO
O potencial Adaptive Intermolecular Reactive Empirical Bond Order Potential (AIREBO)
foi desenvolvido por Stuart, Tutein e Harrison [18], a partir do Reactive Empirical Bond
Order Potential (REBO), proposto por Don Brenner [3]. Estes dois potenciais s˜ao baseados
em um modelo desenvolvido por Tersoff. O potencial REBO foi originalmente elaborado
para modelar deposi¸c˜ao qu´ımica a vapor (CVD) de hidrogˆenio em filmes de diamante, e
posteriormente foi usado para v´arios sistemas de hidrogˆenio [3].
sim-como um potencial interessante para simula¸c˜ao de materiais com estas caracter´ısticas, sim-como
´e o caso do grafeno e dos nanotubos de carbono.
A natureza dinˆamica do potencial AIREBO permite descrever com precis˜ao intera¸c˜oes
intermoleculares n˜ao ligadas e liga¸c˜oes de car´ater covalente. Al´em disso, o AIREBO ´e
parametrizado para os sistemas que contˆem ´atomos de carbono e de hidrogˆenio.
O potencial AIREBO ´e representado por uma soma sobre as intera¸c˜oes entre pares de ´atomos,
incluindo intera¸c˜oes entre liga¸c˜oes covalentes, Lenard Jones e intera¸c˜oes de tor¸c˜ao [18]:
E = 1 2
X
i
X
j$1
[EijREBO +E LJ
ij +
X
k$i,j
X
l$i,j,k
Ekijltors], (2.50)
onde cada par de ´atomos ligados covalentemente interage conforme a equa¸c˜ao:
EREBO
ij =V
R
ij +bijVijA (2.51)
Em potenciais como REBO e AIREBO [18, 20], a intera¸c˜ao covalente, Vij, entre os ´atomos
i e j ´e composta por contribui¸c˜oes de um potencial repulsivo, VR
ij , e um potencial atrativo,
VA
ij . O valor do termo EijREBO depende da posi¸c˜ao e da identidade qu´ımica de ´atomos
atrav´es do termo bij, conhecido como “ordem de liga¸c˜ao”. Este termo ´e respons´avel pela
modifica¸c˜ao das condi¸c˜oes de equil´ıbrio do sistema, tais como o limite de atra¸c˜ao entre as
part´ıculas e distˆancia de equil´ıbrio da liga¸c˜ao com base nas mudan¸cas no ambiente onde
ela ser´a analisada. Em outras palavras, constitui um meio de modifica¸c˜ao da for¸ca de uma
liga¸c˜ao devido a altera¸c˜oes no ambiente local. Este modelo tem sido amplamente usado para
an´alise de sistemas a base de carbono e hidrocarbonetos, pois considera os efeitos de radicais
e conjuga¸c˜oes.
O termo repulsivoVR
VR
ij =ωijrij
1 + Qij
rij
Aije
−αijrij
, (2.52)
onde os parˆametrosQij, Aij e αij dependem dos tipos de ´atomos i e j. Valores para esses e
todos os outros parˆametros poss´ıveis est˜ao na Tabela 2.1.
Tabela 2.1: Parˆametros para o potencial AIREBO, obtidos a partir de Stuart et. all [18].
Parˆametro CC CH HH
Qij (˚A) 0.313460 0.340776 0.370471
αij (˚A
−1
) 4.7465391 4.1025498 3.5362986
Aij(eV) 10953.544 149.94099 32.817356
Bij(1)(eV) 12388.792 32.355187 29.632593
Bij(2)(eV) 17.567065 ... ...
Bij(3)(eV) 30.714932 ... ...
βij(1) (˚A
−1
) 4.7204523 1.4344581 1.7158922
βij(2) (˚A
−1
) 1.4332132 ... ...
βij(3) ˚A−1
) 1.3826913 ... ...
ρij ˚A
−1
) .. 1.09 0.7415887
ǫij (eV) 0.00284
√
ǫccǫHH 0.00150
σij ˚A
−1
) 3.40 1
2(σcc+σHH) 2.65
ǫiccj(eV) 0.3079 0.1787 0.1250
Na equa¸c˜ao 2.52, o termo ωij desliga as intera¸c˜oes covalentes quando o par de ´atomos
ultra-passa distˆancias de liga¸c˜ao t´ıpicas.
O termo atrativo da equa¸c˜ao 2.51 ´e:
VijA=−ωij(rij)
3
X
n=1
Bij(n)e
β(ijn)rij, (2.53)
em que os parˆametros Bij(n) eβ
(n)
ij s˜ao dados na Tabela 2.1.
A principal diferen¸ca entre os potencias AIREBO e REBO refere-se `a inclus˜ao das
Eij =VijREBO +V tors
ij +V
vdW
ij (2.54)
Intera¸c˜oes de van der Waalls s˜ao muito importantes nas simula¸c˜oes, principalmente nas
situa¸c˜oes em que ´atomos que n˜ao est˜ao ligados covalentemente, como nas situa¸c˜oes em que
mol´eculas de hidrogˆenio inclu´ıdas nas proximidades do nanotubo de carbono.
As intera¸c˜oes entre ´atomos n˜ao ligados s˜ao modeladas utilizando o potencial de
Lennard-Jones (LJ), modificado por um conjunto de fun¸c˜oes de comuta¸c˜ao S(t):
ELJ
ij =S(tr(rij))S(tb(b
∗
ij))CijVijLJ(rij) + [1−S(tr(rij))]CijVijLJ(rij) (2.55)
Que inclui o termo tradicional LJ:
VLJ ij = 4ǫij
" σij rij 12 − σij rij 6# (2.56)
O termoCij, baseado na intera¸c˜ao entre os ´atomos i e j, ´e usado para examinar as intera¸c˜oes
entre os ´atomos associados ao potencial LJ e vizinhos pr´oximos, devido `as intera¸c˜oes
molecu-lares via potencial REBO entre o primeiro e segundo vizinhos e o termo de tor¸c˜ao descrevendo
terceiro vizinhos de um ´atomo.
A fun¸c˜ao de comuta¸c˜ao S(tr(rij)) ´e usada para modificar a for¸ca de intera¸c˜ao entre os
´atomos com base no potencial LJ. A fun¸c˜ao de comuta¸c˜aoSS(tb(b
∗
ij)) ´e usada para filtrar as
intera¸c˜oes repulsivas LJ entre os ´atomos que s˜ao encontrados em distˆancias mais longas do
que as distˆancias de liga¸c˜ao covalente, mas possuem uma certa intera¸c˜ao devido ao n´umero
de ´atomos vizinhos. Os parˆametros σij e εij s˜ao apresentados na Tabela 2.1.
A contribui¸c˜ao dos ˆangulos diedros (por exemplo, o termo energia de tor¸c˜ao) para o total de
Etors
kijl =ωki(rki)ωijr(ij)ωjlr(jl)Vtors(ωkijl), (2.57)
onde:
Vtors(ω kijl) =
256
405ǫkijlcos
10(ω
kijl/2)−
1
10ǫkijl, (2.58)
O parˆametro ǫkijl encontra-se na Tabela 2.1. Al´em disso, o uso de pesos, tais como ωij(rij)
asseguram que a energia de tor¸c˜ao associada a um dado ˆangulo de diedro ωij possa ser
removida sem problemas, quando qualquer um dos constituintes das liga¸c˜oes s˜ao quebrados.
A vantagem desta formula¸c˜ao para a energia de tor¸c˜ao ´e que a simetria do potencial de
ˆangulo entre dois ´atomos de carbono ´e totalmente ditada pela simetria da mol´ecula, e pode
mudar `a medida que ocorrem as rea¸c˜oes.
2.3.2
Potencial Reax
O Reactive Force Field (ReaxFF) ´e um potencial originalmente escrito por Adri van Duin
[6]. Trata-se de um potencial dinˆamico que considera a ocorrˆencia de rea¸c˜oes durante a
simula¸c˜ao. Este tipo de potencial emp´ırico ´e chamado de reativo e da mesma forma que os
potenciais n˜ao-reativos, o Reax divide o sistema em v´arias contribui¸c˜oes parciais de energia,
conforme est´a demonstrado na Equa¸c˜ao 2.59.
No ReaxFF, as ordens de liga¸c˜ao s˜ao obtidas a partir de distˆancias interatˆomicas e s˜ao
continuamente atualizados em cada itera¸c˜ao, permitindo mudan¸cas de conectividade. Al´em
disso, o ReaxFF descreve intera¸c˜oes n˜ao-ligadas entre todos os ´atomos, independentemente
da conectividade, podendo ser intera¸c˜oes de van der Waals ou de Coulomb. A seguir ser˜ao
apresentadas as principais caracter´ısticas deste potencial baseando-se na referˆencia [6].
Etotal=Ebond+Eover+Eunder+Eval+Epen+Efors+Econj+EvdW+ECoul (2.59)
Cada uma das contribui¸c˜oes energia parciais ser˜ao descritas a seguir. As Tabelas 2.2 - 2.7
Parˆametro Valor Descri¸c˜ao Equa¸c˜ao
γ1 50,0 corre¸c˜ao de ordem de liga¸c˜ao sobrecoordenada 3c
γ2 15,61 corre¸c˜ao de ordem de liga¸c˜ao sobrecoordenada 3d
γ3 5,02 1−3 corre¸c˜ao de ordem de liga¸c˜ao 3e,f
γ4 18,32 1−3 corre¸c˜ao de ordem de liga¸c˜ao 3e,f
γ5 8,32 1−3 corre¸c˜ao de ordem de liga¸c˜ao 3e,f
γ6 −8,90 energia de sobre-coordena¸c˜ao 6
γ7 1,94 energia de sub-coordena¸c˜ao 7a
γ8 −3,47 energia de sub-coordena¸c˜ao 7a
γ9 5,79 energia de sub-coordena¸c˜ao 7b
γ10 12,38 energia de sub-coordena¸c˜ao 7b
γ11 1,49 energia de valˆencia 8b
γ12 1,28 energia de valˆencia 8b
γ13 6,30 energia de valˆencia 8c
γ14 2,72 energia de valˆencia 8c
γ15 33,87 energia de valˆencia 8c
γ16 6,70 energia de valˆencia 8d
γ17 1,06 energia de valˆencia 8d
γ18 2,04 energia de valˆencia 8d
γ19 36,0 energia de penalidade 9a
γ20 7,98 energia de penalidade 9a
γ21 0,40 energia de penalidade 9b
γ22 4,00 energia de penalidade 9b
γ23 3,17 energia de tor¸c˜ao 10b
γ24 10,00 energia de tor¸c˜ao 10c
γ25 0,90 energia de tor¸c˜ao 10c
γ26 −1,14 energia de conjuga¸c˜ao 11a
γ27 2,17 energia de conjuga¸c˜ao 11b
![Figura 1.2: NTCMs obtidos por Iijima. a) 5 folhas de grafeno com 6,7 nm de diˆametro; b) 2 folhas de grafeno com diˆametro externo de 5,5 nm; c) 7 folhas de grafeno com diˆametro externo de 6,5 nm e diˆametro interno de 2,2 nm[14].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15747252.637354/16.892.271.620.148.544/figura-obtidos-iijima-grafeno-diˆametro-diˆametro-diˆametro-diˆametro.webp)
![Figura 1.5: NTCs produzidos pela t´ecnica de Ablas˜ao por Laser [14].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15747252.637354/19.892.186.725.139.555/figura-ntcs-produzidos-pela-ecnica-ablas-por-laser.webp)
![Figura 1.6: Os NTCs formam um emaranhado de filamentos de 10 a 20 nm de diˆametro e 100µm de comprimento [14].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15747252.637354/20.892.300.611.143.457/figura-os-ntcs-formam-emaranhado-filamentos-diˆametro-comprimento.webp)
![Figura 1.7: Vista transversal de um dispositivo de descarga a arco para produ¸c˜ao de NTCs [14].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15747252.637354/21.892.178.735.181.575/figura-vista-transversal-dispositivo-descarga-arco-produ-ntcs.webp)
![Figura 2.1: Dois fluidos separados por uma parede adiab´atica fixa e imperme´avel [13].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15747252.637354/35.892.271.629.149.323/figura-dois-fluidos-separados-parede-adiab-atica-imperme.webp)
