UNIVERSIDADE FEDERAL DO CEARÁ
FACULDADE DE MEDICINA
DEPARTAMENTO DE FISIOLOGIA E FARMACOLOGIA
CURSO DE PÓS-GRADUAÇÃO EM FARMACOLOGIA
AVALIAÇÃO FARMACOCINÉTICA DE DUAS
FORMULAÇÕES DE MINOCICLINA EM CONDIÇÕES DE
JEJUM E APÓS DIETA RICA EM CÁLCIO
ANA PAULA DE AZEVEDO LEITÃO
FORTALEZA – CE
L 548a Leitão, Ana Paula de Azevedo
Avaliação farmacocinética de duas formulações de minociclina em condições de jejum e após dieta rica em cálcio / Ana Paula de Azevedo Leitão – Fortaleza, 2001.
181 f.
Orientadora: Profa. Dra. Maria Elisabete Amaral de Moraes.
Dissertação (Mestrado). Universidade Federal do Ceará. Departamento de Fisiologia e Farmacologia.
1. Minociclina 2. Dieta rica em cálcio 3. Farmacocinética I. Título.
UNIVERSIDADE FEDERAL DO CEARÁ
FACULDADE DE MEDICINA
DEPARTAMENTO DE FISIOLOGIA E FARMACOLOGIA
CURSO DE PÓS-GRADUAÇÃO EM FARMACOLOGIA
AVALIAÇÃO FARMACOCINÉTICA DE DUAS
FORMULAÇÕES DE MINOCICLINA EM CONDIÇÕES DE
JEJUM E APÓS DIETA RICA EM CÁLCIO
ANA PAULA DE AZEVEDO LEITÃO
Dissertação submetida à Coordenação do Curso de Pós Graduação em Farmacologia do Departamento de Fisiologia e Farmacologia da Universidade Federal do Ceará, como requisito parcial para obtenção do título de Mestre em Farmacologia.
Orientadora: Profa. Dra. Maria Elisabete Amaral de Moraes
AVALIAÇÃO FARMACOCINÉTICA DE DUAS
FORMULAÇÕES DE MINOCICLINA EM CONDIÇÕES DE
JEJUM E APÓS DIETA RICA EM CÁLCIO
ANA PAULA DE AZEVEDO LEITÃO
Esta dissertação foi submetida como parte dos requisitos necessários para a obtenção do Grau de Mestre em Farmacologia, outorgado pela UFC e encontra-se a disposição dos interessados na Biblioteca do Centro de Ciências da Saúde da referida universidade.
A citação de qualquer trecho desta dissertação é permitida, desde que realizada de acordo com as normas da ética científica.
Dissertação aprovada em:22 / 08 / 2001
Profª Dra. Maria Elisabete Amaral de Moraes Orientadora
Profª Dra. Geanne Matos de Andrade Cunha
P
P
O
O
E
E
M
M
A
A
“Teu amor sem exigência me diminui,
tua exigência sem amor me desanima,
D
D
E
E
D
D
I
I
C
C
A
A
T
T
Ó
Ó
R
R
I
I
A
A
Dedico esta dissertação a todas as preciosidades que Deus me deu, meu pai,
Agradecimentos
A Deus, por se fazer sempre muito presente, dando-me forças para enfrentar todos os momentos dessa caminhada.
Minha Família, em especial ao Carmelo Filho, por me compreender e me ajudar sempre, participando de todos os momentos desse trabalho, em especial os mais difíceis, me apoiando em tudo que fosse necessário.
Dra. Mª Elisabete Amaral de Moraes, minha orientadora, a qual apoiou-me na construção do saber científico, através de seus ensinamentos, bem como, oferecendo a oportunidade de realização de cursos em outros estados brasileiros. Também pude observar o afinco, a dedicação e o respeito a pesquisa clínica, ao voluntário, o que a faz conquistar espaços ainda obscuros.
Dra. Geanne Matos de Andrade Cunha, por ter aceitado participar da banca examinadora da dissertação de mestrado. Por ser um exemplo de ser humano. Com sua sala sempre cheia de bolsistas e pós-graduandos de diversos laboratórios, sempre fui recebida com um sorriso largo e despedi-me com uma valiosa contribuição científica.
Dr. Manoel Odorico de Moraes, por colaborar sempre, seja com sua biblioteca, ou na confecção de slides, ou ainda com seus ensinamentos enriquecedores que muito contribuíram ao longo do Mestrado, bem como em sua participação no Exame de Qualificação.
Dr. Fernando Antônio Frota Bezerra, amigo e professor, por estar sempre disposto a nos ensinar a ciência com grande arte e sabedoria de um mestre.
Dra. Gisela Costa Camarão, por ter participado da banca do Exame de Qualificação contribuindo bastante para o meu engrandecimento acadêmico.
Dr. Jaime Ilha por estar sempre disposto à prestar seus valiosos esclarecimentos sobre bioestatística.
Professores do Curso de Pós-Graduação, por terem transmitido com muita sapiência seus conhecimentos.
Colegas de Pós-Graduação, por termos juntos vencidos tantas disciplinas, apresentações de artigos e provas com um saldo maravilhoso, que é nossa amizade.
À Maria Tereza e Flávia, da UNIFAC, pela presteza e competência com que colaboram, bem como pelos momentos agradáveis que tivemos.
Ao Ronaldo Soares, da UNIFAC, por sempre transmitir com boa vontade seu saber e sua experiência.
As secretárias do Departamento de Fisiologia e Farmacologia, Sílvia, Aura, Marta, Joana e Rejane que sempre estiveram dispostas a nos ajudar no que fosse preciso.
Aos técnicos, Silvana e Evanir pelo apoio na realização dos experimentos.
Laboratório Stiefel Ltda., Brasil, por ter patrocinado o estudo, oferecendo subsídios para que o mesmo fosse realizado.
Índice
Poema iv
Dedicatória v
Agradecimentos vi
Índice x
Lista de Abreviaturas xiv
Lista de Figuras xv
Lista de Tabelas xvi
Anexos xviii
Resumo xix
Abstract xx Prefácio xxi Definições xxiii Introdução 02
A Importância da Farmacologia Clínica 02
As Quatro Fases da Terapêutica 08
Fase Farmacêutica 08
Figura 1 - Fase Farmacêutica 09
Fase Farmacocinética 10
Absorção 10 Distribuição 12 Biotransformação 12
Eliminação dos Fármacos 14
Fase Farmacodinâmica 16
Fase Terapêutica 17
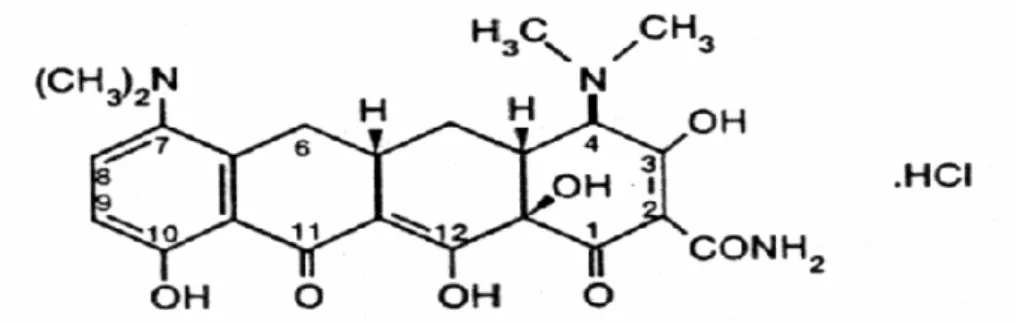
Minociclina 17 Figura 2 - Fórmula estrutural da minociclina 18
Figura 3 - Fórmula estrutural da tetraciclina 19
Farmacologia 19
Farmacologia Clínica 21
Farmacocinética 23 Biodisponibilidade 23
Distribuição 24 Metabolismo 24
Relação Dose-Efeito 25
Microbiologia 25
Espectro Antibacteriano 26
Bactérias Gram Negativas 26
Indicações Terapêuticas 27
Contra-indicações 28
Interações com Drogas 33
Exemplos de Interações Medicamentosas com Tetraciclinas
34
Interações com Alimento 35
Tetraciclinas versus Alimentos 37
Apresentação Farmacêutica 38
Objetivos 39
Material e Métodos 40
Protocolo Clínico 42
Delineamento do Estudo 42
Seleção de Voluntários 43
Exames Laboratoriais 43
Critérios de Inclusão 44
Critérios de Exclusão 45
Critérios para a Retirada do Estudo 46
Entrada do Voluntário no Estudo 47
Internamento 47 Restrições 48
Produtos Estudados 49
Esquema Experimental 49
Inventário das Amostras 50
Tabela 1 - Randomização da Minociclina 51
Coleta de Sangue 52
Efeitos Adversos 53
Definições de Efeitos Adversos quanto à Intensidade 53 Relacionamento suposto com a Droga Experimental 53
Etapa Analítica 54
Análise da droga 54
Preparo das Soluções Padrões Reagentes 55 Preparação das Amostras da Curva de Calibração e Controles de Qualidade
55
Pré-tratamento das Amostras de Plasma 56 Condições Cromatográficas e de Espectrometria de Massa
57
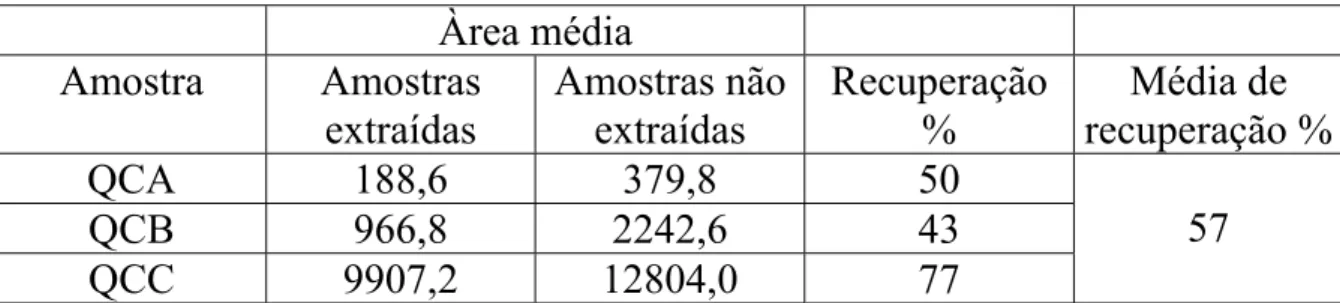
Recuperação 58
Precisão e Acurácia 58
Especificidade 59 Parâmetros Farmacocinéticos Determinados 59
Análise Estatística 60
Comitê de Ética 32
Termo de Consentimento Livre e Esclarecido 62
Confidenciabilidade 64
Resultados e Discussão 65
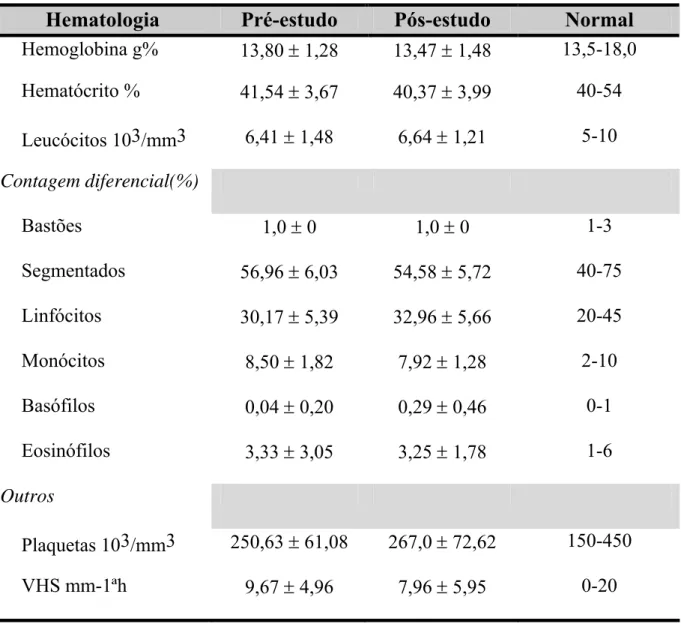
Resultados 66 Tabela 3 - Resultados Hematológicos das Determinações
Laboratoriais (média ± SD) dos 24 voluntários
68 Tabela 4 - Resultados Bioquímicos das Determinações Laboratoriais (média ± SD) dos 24 voluntários
69 Tabela 5 - Resultados das Determinações Laboratoriais do Exame de Sumário de Urina (média ± SD) dos 24 voluntários
70
Etapa Analítica 71
Figura 4 - Curva de Calibração - Linearidade 72 Figura 5 - Plasma em condições de jejum, hiperlipêmico e hemolizado.
74 Figura 6 - Área do pico padrão (analito e padrão interno) 76
Etapa Farmacocinética 77
Tabela 6 - Parâmetros farmacocinéticos após a administração de 100mg de minociclina (Minomax®) em 24 voluntários em condições de jejum
78
Tabela 7 - Parâmetros farmacocinéticos após a administração de 100mg de minociclina (Minomax®) + dieta padrão específica em 24 voluntários
79
Tabela 8 - Parâmetros farmacocinéticos após a administração de 100mg de minociclina (Minoderm) em 24 voluntários em condições de jejum
80
Tabela 9 - Parâmetros farmacocinéticos após a administração de 100mg de minociclina (Minoderm) + dieta padrão específica em 24 voluntários
81
Tabela 10 - Média dos parâmetros farmacocinéticos obtidos dos 24 voluntários após a administração de 100mg de minociclina em condições de jejum
82
Tabela 11 - Média dos parâmetros farmacocinéticos obtidos dos 24 voluntários após a administração de 100mg de minociclina + dieta padrão específica
83
Figura 7 - Média ± desvio padrão (SD) da concentração plasmática da minociclina (ng/mL) versus tempo (h) obtida após uma única administração oral de 100mg de minociclina, formulação referência (Minomax®), na ausência e na presença de dieta padrão específica em 24 voluntários sadios
84
plasmática da minociclina (ng/mL) versus tempo (h) obtida após uma única administração oral de 100mg de minociclina formulação teste (Minoderm), na ausência e na presença de dieta padrão específica em 24 voluntários sadios
Tabela 12 - Valores médios de concentração plasmática (ng/mL) de minociclina (Minomax®) sob condições de jejum e com dieta padrão específica durante o período médio de absorção de uma droga administrada por via oral
86
Tabela 13 - Valores médios de concentração plasmática (ng/mL) de minociclina (Minoderm) sob condições de jejum e com dieta padrão específica durante o período médio de absorção de uma droga administrada por via oral
87
Figura 9 - Resultados médios (n=24) obtidos das curvas de concentração plasmática (ng/mL) versus tempo (h) do medicamento referência (Minomax®) e do teste (Minoderm) sob condições de jejum
88
Figura 10 - Resultados médios (n=24) obtidos das curvas de concentração plasmática (ng/mL) versus tempo (h) do medicamento referência (Minomax®) e do teste (Minoderm) na presença de dieta padrão específica
89
Tabela 14 - Análise estatística das razões individuais de AUC(0-96h) e Cmax para as duas formulações de minociclina (Minoderm e Minomax®) na ausência e na presença de dieta padrão específica
90
Tabela 15 - Meia vida de eliminação-t1/2 (h) obtida das curvas de concentração plasmática (ng/mL) versus tempo (h) das duas formulações de minociclina (Minoderm e Minomax®) na ausência e na presença de dieta padrão específica
91
Discussão 92 Conclusão 97 Referências 100 Anexos 109
Anexo I - Ficha clínica do voluntário 01 SCC 110 Anexo II - Gráficos da concentração plasmática (ng/mL) versus tempo (h) dos 24 voluntários quando administrado minociclina (Minomax® e Minoderm) em condições de jejum e quando administrado minociclina (Minomax® e Minoderm) na presença de dieta padrão específica
130
Anexo III - Procedimento Operacional Padrão para Transporte das Amostras da UNIFAC para a Unidade Analítica Cartesius - ICB – USP
178
Lista de Abreviaturas
% porcentagem
± mais ou menos
≤ menor ou igual
µg/mL micrograma por mililitro
µL/min microlitro por minuto > maior art. artigo
cm centímetro
SD desvio padrão
eV elétron volt
h hora Kcal kilocaloria Kg kilograma
L litro
ln logaritmo neperiano
mg/dia miligrama por mililitro mg/Kg miligrama por kilograma mg/mL miligrama por mililitro mM milimolar
ng/mL nanograma por mililitro nº número
rpm rotação por minuto tto tratamento V volt
Lista de figuras
Figura 1. Fase farmacêutica
Figura 2. Fórmula estrutural da minociclina
Figura 3. Fórmula estrutural da tetraciclina
Figura 4. Curva de Calibração - Linearidade
Figura 5. Plasma em condições de jejum, hiperlipêmico e hemolizado
Figura 6. Área do pico padrão (analito e padrão interno)
Figura 7. Média ± desvio padrão (SD) da concentração plasmática da minociclina (ng/mL) versus tempo (h) obtida após uma única administração oral de 100mg de minociclina, formulação referência (Minomax®), na ausência e na presença de dieta padrão específica em 24 voluntários sadios
Figura 8. Média ± desvio padrão (SD) da concentração plasmática da minociclina (ng/mL) versus tempo (h) obtida após uma única administração oral de 100mg de minociclina formulação teste (Minoderm), na ausência e na presença de dieta padrão específica em 24 voluntários sadios
Figura 9. Resultados médios (n=24) obtidos das curvas de concentração plasmática (ng/mL) versus tempo (h) do medicamento referência (Minomax®) e do teste (Minoderm) sob condições de jejum
Lista de tabelas
Tabela 1. Randomização da Minociclina
Tabela 2. Parâmetros Farmacocinéticos
Tabela 3. Resultados Hematológicos das Determinações Laboratoriais (média ± SD) dos 24 voluntários
Tabela 4. Resultados Bioquímicos das Determinações Laboratoriais (média ± SD) dos 24 voluntários
Tabela 5. Resultados das Determinações Laboratoriais do Exame de Sumário de Urina (média ± SD) dos 24 voluntários
Tabela 6. Parâmetros farmacocinéticos após a administração de 100mg de monociclina (Minomax®) em 24 voluntários em condições de jejum
Tabela 7. Parâmetros farmacocinéticos após a administração de 100mg de minociclina (Minomax®) + dieta padrão específica em 24 voluntários
Tabela 8. Parâmetros farmacocinéticos após a administração de 100mg de monociclina (Minoderm) em 24 voluntários em condições de jejum
Tabela 9. Parâmetros farmacocinéticos após a administração de 100mg de minociclina (Minoderm) + dieta padrão específica em 24 voluntários
Tabela 10. Média dos parâmetros farmacocinéticos obtidos dos 24 voluntários após a administração de 100mg de minociclina em condições de jejum
Tabela 12. Valores médios de concentração plasmática (ng/mL) de minociclina (Minomax®) sob condições de jejum e com dieta padrão específica durante o período médio de absorção de uma droga administrada por via oral
Tabela 13. Valores médios de concentração plasmática (ng/mL) de minociclina (Minoderm) sob condições de jejum e com dieta padrão específica durante o período médio de absorção de uma droga administrada por via oral
Tabela 14. Análise estatística das razões individuais de AUC(0-96h) e Cmax para as duas formulações de minociclina (Minoderm e Minomax®) na ausência e na presença de dieta padrão específica
Anexos
Anexo I. Ficha clínica do voluntário 01 SCC
Anexo II. Gráficos da concentração plasmática (ng/mL) versus tempo (h) dos 24 voluntários quando administrado minociclina (Minomax® e Minoderm) em
condições de jejum e quando administrado minociclina (Minomax® e Minoderm) na presença de dieta padrão específica
Anexo III. Procedimento Operacional Padrão para Transporte das Amostras da UNIFAC para a Unidade Analítica Cartesius - ICB – USP
Resumo
Abstract
Prefácio
A importância de um estudo clínico que avalie a interferência da alimentação na biodisponibilidade de fármacos é de extrema importância sob os aspectos farmacocinéticos e principalmente dentro do contexto atual da Política Nacional de Medicamentos adotada em nosso país.
Primeiramente, através da realização desse estudo, desmistificamos o fato de que as tetraciclinas, especificamente a minociclina, apresenta alterações significativas na sua biodisponibilidade quando ingerida com alimentos, principalmente, aqueles ricos em metais como cálcio.
Outro aspecto relevante é o estudo da bioequivalência em si, termo até bem pouco tempo desconhecido, uma vez que nosso país aprovou apenas recentemente a Lei de Medicamentos Genéricos (Lei 9787 de 1999) e a Resolução que regulamenta esta lei (Res. 391/97 e RDC nº 10, de 2 de janeiro de 2001-ANVISA). Estas dizem exatamente que para um medicamento ser bioequivalente, ter-se-á que ser intercambiável com o medicamento de marca ou inovador.
permitindo que a classe menos favorecida tenha acesso, possibilitando o tratamento farmacológico completo, minimizando os riscos de não adesão ao tratamento ou interrupção do mesmo.
Não podemos esquecer que para alguns pacientes o preço de uma prescrição é o principal fator, portanto, sugiro aos profissionais médicos que tenham conhecimento de quanto custa o tratamento, pois o compromisso do paciente com o mesmo depende também da possibilidade em adquirir a medicação.
D
Definições
Biodisponibilidade Absoluta (F): a fração da droga sistemicamente absorvida e a disponibilidade da droga da forma farmacêutica quando comparada com a biodisponibilidade da droga após administração intravenosa. É usualmente calculada como a razão da área sobre a curva (AUC) da concentração plasmática versus tempo, para a forma farmacêutica administrada oralmente em relação a AUC obtida após administração intravenosa da droga. Um valor F de 0,80 ou 80% indica que apenas 80% da droga ficou disponível sistemicamente da forma farmacêutica.
Biodisponibilidade Oral: indica a velocidade e a extensão de absorção de um princípio ativo em uma forma de dosagem, a partir de sua curva concentração plasmática versus tempo na circulação sistêmica ou a partir de sua excreção urinária. É a fração da dose de um medicamento, oralmente administrado, que foi absorvida e atinge a circulação como princípio ativo.
biodisponibilidade apartir da forma farmacêutica é a mesma, mas não indica a completa absorção sistêmica da droga. A determinação da biodisponibilidade é muito importante em estudos de medicamentos.
Bioequivalência: dois medicamentos são ditos bioequivalentes quando são administrados na mesma dose molar, nas mesmas condições experimentais e não apresentam diferenças estatisticamente significativas em relação à biodisponibilidade, segundo a Resolução RDC nº 10, de 2 de janeiro de 2001-ANVISA.
Bioequivalência: existe quando a natureza e a extensão dos efeitos tóxicos e terapêuticos são iguais seguindo a administração de dose iguais. É usada para comparar duas formulações ou rotas diferentes de administração de uma droga.
Food and Drug Administration (USA).
Curva de Calibração: é preparada usando plasma humano controle com soluções padrões contendo o analito a ser quantificado.
Denominação Comum Internacional: denominação do fármaco ou princípio farmacologicamente ativo, e recomendada pela Organização Mundial de Saúde.
Droga: é a matéria prima mineral, vegetal ou animal da qual se podem extrair um ou mais princípios ativos. De acordo com esta acepção, os agentes terapêuticos de origem sintética não são drogas.
Droga Referência: medicamento inovador registrado no órgão federal responsável pela vigilância sanitária e comercializado no país cuja eficácia, segurança e qualidade foram comprovadas cientificamente junto ao órgão federal competente, por ocasião do registro. É o primeiro medicamento que foi registrado no país. Seu nome é determinado pela Denominação Comum Brasileira (DCB) e na ausência desta, seguir-se-á a Denominação Comum Internacional (DCI).
Droga Teste: medicamento que se propõe a ser intercambiável com o medicamento referência.
Equivalentes Farmacêuticos: são medicamentos que contém o mesmo princípio ativo, ou seja, mesmo sal ou éster da mesma molécula terapeuticamente ativa, na mesma quantidade e forma farmacêutica, podendo ou não conter excipientes idênticos. Devem cumprir as mesmas especificações atualizadas da Farmacopéia Brasileira; no caso desta não abordar, deve-se seguir à outros códigos autorizados pela legislação vigente ou ainda com outros padrões aplicáveis de qualidade, relacionados à identidade, dosagem, pureza, potência, uniformidade de conteúdo, tempo de desintegração e velocidade de dissolução, quando for o caso.
Estudo Aberto: tanto o pesquisador responsável, como o sujeito da pesquisa, é informado e têm conhecimento sobre a medicação que será administrada.
Estudo Cruzado: em uma das fases da pesquisa administra-se ao sujeito da pesquisa (voluntário) o medicamento referência e na outra o medicamento teste, ou vice–versa. O mesmo voluntário deve ser submetido às duas formulações.
Índice de Massa Corpórea (IMC): é a razão do peso (Kg) pela altura ao quadrado (m). É o melhor indicador de obesidade, pois enfatiza a adiposidade relativa dos indivíduos e minimiza os efeitos da altura. O valor ótimo é de 20 - 27 segundo Ministry of National Health and Welfare, Canadá, entretanto, a National Academy of Sciences enfatiza que o IMC aumenta com a idade, de 19 – 24 para adultos jovens e de 24 – 29 para acima de 65 anos.
Medicamento: produto farmacêutico, tecnicamente obtido ou elaborado, com finalidades profiláticas, curativas, paliativas ou para fins de diagnóstico. É uma forma farmacêutica terminada que contém o fármaco, geralmente em associação com adjuvantes farmacotécnicos (Resolução 391, de 9 de agosto de 1999). É o mesmo que fármaco, mas especialmente quando se encontra na sua forma farmacêutica (Andrejus Korolkovas, Joseph H. Burckhalty. Química Farmacêutica. Editora Guanabara Koogan S.A., 1988, Rio de Janeiro – RJ).
Padrão Interno: substância que apresenta semelhanças com a droga em questão, fórmula estrutural e tempo de retenção diferente na determinação analítica. Deve apresentar concentração conhecida, pois funcionará como um controle de qualidade do funcionamento do aparelho que irá quantificar o princípio ativo e/ou seus metabólitos.
Parâmetros Farmacocinéticos
Área sob a Curva da Concentração Plasmática versus Tempo, de
Zero ao Infinito – AUC(0-∞): relaciona-se com a quantidade ou a extensão de absorção da droga. A quantidade da absorção sistêmica da droga é diretamente relacionada com AUC que é geralmente calculado pelo método trapezoidal e expresso em unidades de concentração vezes tempo. A extrapolação dessa área para o infinito (AUC(0-∞)) é feita adicionando-se o valor Cx/K ao calculado (AUC(0-xh)) [onde Cx = concentração plasmática calculada da equação da regressão log-linear obtida através de K xh após a dose]. X é o último horário de coleta de sangue.
Constante de Eliminação K: representa a soma de todas as taxas constantes para remoção da droga do organismo, incluindo a taxa constante da excreção renal e metabolismo (biotransformação) como descrito através da equação: K = Ke + Km, onde Ke = constante de excreção renal e Km = constante de metabolismo. Determina a taxa de eliminação da droga dos vasos sangüíneos. Serve para determinar a concentração da droga em um tempo t. É inversamente relacionada à meia vida do fármaco: K= 0,693/t1/2.
Meia Vida de Eliminação da Droga t1/2: representa o tempo em que a concentração da droga no plasma é reduzida a metade. Calcula-se através do logaritmo neperiano (ln) de 2 (ln2= 0,693) dividido pela constante de eliminação
t
1/2 = ln2/KTempo correspondente à Concentração Máxima Atingida no Plasma -
I
INTRODUÇÃO
A IMPORTÂNCIA DA FARMACOLOGIA CLÍNICA
Até meados do século XIII a terapêutica era realizada sem qualquer embasamento científico, portanto, era na maioria dos casos ineficiente. Nessa época ainda não havia conhecimentos sobre farmacologia, fisiologia, bioquímica e fisiopatologia, o que levou o escritor inglês Richard Gordon a afirmar: “A história da medicina foi, em grande parte, até os fins do século passado, a substituição da ignorância por mentiras”.
Pode-se tentar definir a Farmacologia Clínica como sendo uma ciência que define métodos científicos, a fim de desmistificar a terapêutica medicamentosa, promovendo a cura ou aliviando o estado patológico.
Atualmente, a Farmacologia Clínica desenvolve principalmente duas atividades, ou seja, estudos farmacocinéticos e elaboração, execução e análise de ensaios clínicos para verificar a segurança, qualidade e eficácia dos medicamentos em seres humanos.
Os testes de bioequivalência são de extrema importância, pois através deles assegura-se que o medicamento pesquisado apresentará eficazmente o efeito esperado, após ter passado com sucesso pelas etapas que antecedem a etapa terapêutica, as quais sejam farmacêutica, farmacocinética e farmacodinâmica.
Após 12 anos dos primeiros estudos de bioequivalência pode-se observar um grande avanço nessa área, pois no princípio trabalhava-se com estrutura precária e com enorme ceticismo da comunidade científica. Atualmente o Brasil conta com tecnologia de ponta, estrutura física adequada e corpo científico qualificado, o que contribui para a credibilidade com que se faz jus desta nova área de pesquisa, a Farmacologia Clínica. Diante de tantas mudanças pode-se afirmar que o objetivo foi mantido, verificar se duas ou mais formulações farmacêuticas podem ser intercambiáveis, termo muito popular na atualidade frente a política de medicamentos genéricos.
Uma parcela da indústria farmacêutica sempre demonstrou interesse em avaliar alguns de seus produtos, antes mesmo que houvesse uma regulamentação própria pelo Sistema de Vigilância Sanitária do Brasil. Até a década de 90, foi realizado o primeiro Teste Fase I no Brasil (Frota Bezerra, 1999), estudo que corresponde à primeira administração de uma nova entidade química em humanos, além de vários estudos de interação de drogas em voluntários sadios, assim como avaliação de eficácia terapêutica em suas diversas Fases, em voluntários pacientes.
O tempo para realização de um estudo de bioequivalência deve-se à instância de tramitação com o Conselho de Ética em Pesquisa (CEP) credenciado pela Comissão Nacional de Ética em Pesquisa (CONEP-CNS) e às etapas de seleção e internação dos voluntários, terminando com a quantificação do medicamento no líquido biológico e análise estatística.
Para realização dos estudos clínicos de bioequivalência, voluntários de ambos os sexos são selecionados através de rigorosa avaliação clínica e laboratorial, só sendo internado depois de comprovado estado de higidez.
momento em que realiza a avaliação clínica e laboratorial, o número de formulações a serem testadas, o número de internamentos e o tipo de desenho do estudo, se dose única ou múltipla. A complexidade da etapa analítica, como se há ou não a necessidade de quantificar metabólitos, não é preditivo para o ressarcimento do voluntário, mas é um dos fatores que contribui substancialmente com o orçamento do estudo.
As normas que regem a vigilância sanitária no país permitem assegurar ao medicamento genérico sua intercambialidade com o medicamento inovador. Ou seja, através de estudos in vitro (equivalência farmacêutica) e estudos in vivo em voluntários sadios (bioequivalência) verifica-se a qualidade, eficácia e segurança do medicamento teste em relação à referência.
Na realidade, pequenas diferenças, mesmo que significativas, são aceitas, desde que contidas dentro de uma faixa validada cientificamente e estabelecida por lei. Este processo de absorção é quantificado através dos parâmetros AUC, Cmax e em alguns casos o Tmax é também levado em consideração.
O cronograma de coletas das amostras deve contemplar um tempo igual ou superior a 3 a 5 (três a cinco) vezes a meia-vida de eliminação (t1/2) do fármaco ou do metabólito, caso este seja quantificável e ativo.
O intervalo entre os dois períodos de coleta (estudo cruzado) deve ser de, no mínimo, 7 (sete) vezes a meia-vida de eliminação (t1/2) do fármaco ou do metabólito ativo, o que assegura a depuração completa do fármaco, evitando um efeito de carreamento (Resolução – RDC nº 10/2001-ANVISA).
AS QUATRO FASES DA TERAPÊUTICA
Para que um medicamento possa atuar em seus sítios receptores, deve concluir com êxito uma série de etapas, as quais são: farmacêutica e farmacocinética. Após essas duas etapas ele passará a fase farmacodinâmica, a qual nos referimos anteriormente e a terapêutica, objetivo final de toda administração de fármaco.
Fase Farmacêutica
A fase farmacêutica inicia-se, a depender da forma farmacêutica, mesmo antes da administração, pois em relação aos medicamentos de uso parenteral, o manuseio ou o preparo para administração é fundamental para o êxito da terapêutica. Tratando-se de medicamentos de uso enteral, como sob a forma sólida, a fase farmacêutica ocorre após a administração do medicamento e antes que ele seja absorvido. Essa fase agrega as seguintes etapas: desintegração
liberação da droga do produto farmacêutico, formando grânulos ou agregados;
Figura 1: Fase Farmacêutica. Adaptado do gráfico de Leon Shargel. Comprehensive Pharmacy Review. Second edition. Harwal Publishing, 1994, Unites States of America.
As soluções, misturas homogêneas de um ou mais solutos dispersos molecularmente em um solvente, são ditas apresentarem a maior biodisponibilidade, pois já estando dissolvidas, não há necessidade da dissolução antes da absorção. Portanto, em relação as formulações orais sólidas são tidas como referência. Quando o solvente utilizado é hidroalcoólico (elixir), a droga forma um precipitado fino no trato gastrointestinal, de tal modo que serão redissolvidas antes da absorção. O mesmo ocorre com as suspensões, onde as partículas encontram-se finamente dispersas no líquido.
Partículas finas
Comprimido ou cápsula
Droga em solução Grânulos ou
agregados Dissolução
Desagregação
Desintegração
Dissolução
Fase Farmacocinética
A fase farmacocinética, a qual teve por objetivo essa dissertação,
compreende os processos de absorção, distribuição, metabolização e eliminação. Uma das definições clássicas de farmacocinética é exatamente o que o
organismo faz com a droga.
Absorção
Primeiramente, a droga é absorvida do sítio de administração, atinge circulação sistêmica e se distribui pelo organismo; a velocidade e o padrão de distribuição podem variar largamente de uma droga para outra. Finalmente, a droga é removida do plasma e eliminada do organismo através de biotransformação metabólica e/ou excreção da droga inalterada.
As drogas são absorvidas a partir dos muitos sítios de administração, mas cada rota leva a um diferente acesso da droga a circulação sistêmica. A escolha da via de administração é feita, entre outras, pela via oral, sublingual, subcutânea, intravenosa, intra-arterial, intramuscular, tópica, pulmonar ou retal.
de alimentos no trato gastrointestinal, como é o caso da interação que ocorre com alguns antibióticos tetraciclínicos com o íon cálcio principalmente; veículo de administração; o pH gástrico; o tamanho e a formulação das partículas; a velocidade de dissolução para formas sólidas que é o fator limitante da absorção, e 2) fatores fisiológicos, como as condições intrínsecas do sítio de administração, como a circulação para o local de absorção; adiposidade do tecido; área da superfície absorvente; motilidade gastrointestinal e o fluxo sangüíneo esplâncnico o qual é reduzido em estados hipovolêmicos diminuindo a absorção.
A taxa de absorção reflete o tempo para obter a concentração máxima da droga no plasma, inferindo dois parâmetros muito importantes analisados, os quais são: tempo para atingir a concentração máxima no plasma (Tmax) e a própria concentração plasmática máxima (Cmax).
Distribuição
Após a absorção da droga, atingindo a circulação sistêmica, inicia-se o processo de distribuição para os tecidos do organismo.
Os fatores principais que podem interferir nessa fase são: 1) o fluxo sangüíneo, levando o fármaco para os órgãos de melhor perfusão como coração, cérebro, rins e fígado; 2) a lipossolubilidade devido a natureza extremamente permeável das membranas endoteliais capilares; 3) a ligação às proteínas plasmáticas; 4) o elevado peso molecular levando ao confinamento no espaço intravascular, prolongando a ação do fármaco no mesmo tecido ou em local distante atingido através da circulação. Muitas drogas são suficientemente pequenas e lipossolúveis fazendo com grande parte da dose administrada que foi absorvida, seja distribuída para o espaço extravascular. É importante lembrar que o término da ação do fármaco é dependente do processo de biotransformação e de excreção.
Biotransformação
exemplo, onde elas têm que se tornar mais hidrofílicas para posterior eliminação.
O metabolismo de fármacos depende de vários fatores, sejam internos como sexo, idade, peso, estado nutricional, gestação, fatores de administração dos fármacos, via, local e velocidade de administração, volume administrado, veículo e fatores ambientais externos como temperatura, umidade, luz e hora do dia.
As reações de biotransformação podem ser de oxidação, redução, hidrólise, hidroxilação ou dealquilação dos sítios ativos e ocorrem nas frações microssomais hepáticas das células do fígado. Essas reações são mediadas por uma classe de enzimas conhecidas por sistema do citocromo P-450. Através dessa classe de enzimas os metabólitos das drogas sofrem essas reações, ditas reações de Fase I, tornando-as mais hidrofílicas que seus progenitores. Em geral as reações de Fase I alteram muito pouco a estrutura estérica da droga, formando com freqüência substâncias farmacológicas ativas.
de drogas que as inibam ou por patologias hepáticas como é o caso de cirrose e hepatite.
O processo de biotransformação também pode ser acompanhado de reações de Fase II, consumindo energia, através das quais ocorre a adição de grupos químicos à droga, também conhecida por reações de conjugação. As enzimas que promovem esse tipo de reação são chamadas transferases ou de conjugação, facilitando a substituição de grupos covalentes por ácido glicurônico, glicina, sulfato, acetil ou glutationa para a droga. Em geral os metabólitos de Fase II são inativos.
Predominantemente as drogas são metabolizadas através de biotransformação hepática numa série de reações de Fase I terminando na Fase II, com a conjugação do ácido glicurônico a glicuronídeo, o qual é inativo, hidrofílico e excretado pelos rins.
Eliminação de fármacos
Os fármacos podem ser excretados do organismo sob sua forma inalterada, ou seja, sem sofrer metabolização, e sob a forma de metabólitos. Com exceção dos pulmões, os órgãos excretores eliminam mais facilmente os compostos polares em comparação às substâncias lipossolúveis, as quais devem sofrer biotransformação até tornar-se mais polares.
O órgão que mais se destaca na eliminação de drogas e/ou metabólitos é o rim. A via fecal restringe-se às substâncias administradas por via oral que não sofrem absorção, ou os metabólitos excretados na bile que não são reabsorvidos pelo trato gastrointestinal.
A excreção no leite materno é importante pelo fato de, mesmo em baixas concentrações, serem fontes potenciais de efeitos farmacológicos indesejáveis ao lactante. A importância da excreção pulmonar deve-se à eliminação de vapores e gases anestésicos.
Fase Farmacodinâmica
A farmacodinâmica é a fase que culmina com a conclusão das etapas da fase farmacocinética. É nesta fase que ocorre a interação fármaco-receptor no tecido alvo. Pode ser definida como o estudo dos efeitos bioquímicos e fisiológicos das drogas e os seus mecanismos de ação, ou seja, é a avaliação quantitativa dos efeitos biológicos e terapêuticos das drogas. Através dela, demonstra-se o efeito terapêutico esperado da droga e, quando possível, o seu mecanismo de ação. Além deste, a farmacodinâmica também pesquisa os efeitos nos principais sistemas do organismo, os efeitos terapêuticos e os efeitos tóxicos.
No estudo farmacodinâmico, o mecanismo de ação é investigado de modo íntimo e específico em todos os níveis possíveis, do tecidual até o bioquímico e o molecular.
Fase Terapêutica
Nessa fase observa-se se o efeito farmacológico (farmacodinâmica), está sendo transformado no efeito terapêutico adequado. Sendo o paciente
beneficiado com a terapêutica medicamentosa, o efeito farmacológico deve ser transformado em benefício clínico, podendo ser verificado através de sinais e sintomas, ou indiretamente através de exames complementares.
MINOCICLINA
A minociclina foi o centro de estudo dessa pesquisa por ser um derivado da tetraciclina, a literatura ainda é muito controversa, no que diz respeito da interferência de alimentos ricos em metais como o cálcio, na sua biodisponibilidade.
A minociclina foi desenvolvida na década de 1960 através de fermentação seguida de transformação química. É um antibiótico de amplo espectro indicado para o tratamento de infecções causadas por organismos sensíveis à tetraciclina bem como é eficaz no tratamento de organismos tetraciclina-resistente, como é o caso de cepas de estafilococos (Brogden et al 1975). Com relação ao uso no tratamento de estafilococos meticilina-resistente há relatos de que sua atividade é limitada (Smilack, 1999) e de que apresenta boa eficácia sem causar resistência (Qadri et al, 1994). É considerado o tratamento de escolha para infecção por clamídia, micoplasma e brucelose (Wrightson et al, 1998). É uma tetraciclina lipídica, solúvel freqüentemente usada em casos de acne resistente à tetraciclina (Leyden, 1985).
A minociclina por ser um derivado semi-sintético da tetraciclina apresenta quatro anéis benzeno em sua fórmula estrutural. Seu nome químico é [4S-(4α, 4aα, 5aα, 12aα)]-4, 7-bis (dimetilamino) - 1, 4, 4a, 5, 5a, 6, 11, 12a-octahidro- 3, 10, 12, 12a, tetrahidroxi-1, 11 dioxo-2- naftacenecarboxamido monohidrocloridrato.
Figura 3: Fórmula estrutural da tetraciclina
Apresenta peso molecular de 493,9, mas sob a forma de base livre o peso molecular é de 457,5. A presença de vários grupos ionizáveis na molécula confere diferentes graus de dissociação (pKa: 2.8, 5.0, 7.8, 9.3) em solução. Possui solubilidade em água maior que no álcool, sendo de aproximadamente 1:30 e 1:100 respectivamente. Apresenta sabor amargo, é ligeiramente higroscópica e sensível à luz. O composto clinicamente útil é a forma levógira. A minociclina não se encontra disponível combinada a outros produtos.
FARMACOLOGIA
alumínio no trato gastrointestinal (Dollery, 1993), o que mais uma vez nos impulsionou ao estudo realizado, frente a essas contradições existentes na literatura sobre esse tipo de interação alimentar.
A minociclina é mais lipofílica que as outras tetraciclinas e passa diretamente pela bicamada lipídica da parede celular bacteriana, além de apresentar um transporte ativo por um sistema de bombas através da membrana citoplasmática. Seu mecanismo de ação antibacteriano, assim como a tetraciclina, ocorre através da inibição da síntese protéica na porção 30S ribossômica. A droga parece inibir o acesso do RNAt ao sítio receptor do complexo RNAm-ribossomo, o que impossibilita a adição de aminoácidos para o desenvolvimento da cadeia peptídica. Em altas concentrações, a minociclina inibe a síntese protéica em células de mamíferos (Dollery, 1993).
De acordo com estudos recentes a minociclina causa aumento na expressão da ciclooxigenase do tipo 2 (COX2) elevando os níveis de prostaglandinas do tipo E2 (PGE2) (Patel et al, 1999; Attur et al, 1999).
teratogênicos foram demonstrados em ratos e coelhos (mas não em cães ou macacos) (Jackson, 1975).
FARMACOLOGIA CLÍNICA
A minociclina é rapidamente absorvida pelo trato gastrointestinal após administração por via oral. Dose única de dois comprimidos de 100mg de minociclina, administrada em 18 voluntários adultos sadios, atingiu a concentração máxima sangüínea de 2,1 e 5,1µg/mL (média de 3,5µg/mL) entre 1 e 4 horas (média de 2,1 horas). A meia-vida em voluntários sadios variou entre 11,1 e 22,1 horas (média de 15,5 horas) (PDR®, 1998).
Quando a minociclina é administrada simultaneamente com refeições, que incluem alimentos de consumo diário, a extensão de sua absorção não é consideravelmente influenciada. O pico de concentração plasmática é reduzido de forma insignificante (11,2%) e retardado por uma hora quando administrada com refeições comparado quando administrada fora das refeições.
entre 0,82 e 2,64µg/mL (média de 1,38µg/mL). Em um grupo de 5 voluntários sadios do sexo masculino, níveis séricos de 1,4 a 1,8µg/mL foram mantidos entre 12 a 24 horas quando a minociclina foi administrada por 3 dias na dose de 100mg a cada 12 horas. Quando administrado 200mg a cada 24 horas por 3 dias, os níveis séricos caíram para aproximadamente 1µg/mL em 24 horas. A meia-vida plasmática de doses intravenosas de 100mg de 12/12h ou 200mg a cada 24 horas não diferiu significativamente e variou de 15 a 23 horas (PDR®, 1998).
A administração oral de 100mg em 10 voluntários sadios apresentou níveis plasmáticos variando entre 0,74 e 4,45µg/mL em 1 hora (média de
2,24µg/mL), que após 12 horas alcançou uma faixa de 0,34 a 2,36µg/mL (média de 1,25µg/mL) (PDR®, 1998).
A quantidade de minociclina recuperada nas fezes e na urina quando administrada por via oral em 12 voluntários sadios é metade a um terço da quantidade de outras tetraciclinas (PDR®, 1998).
17 horas em 7 pacientes com disfunção hepática, e de 18 a 69 horas em 5 pacientes com insuficiência renal (Welling et al, 1975).
Farmacocinética
O método analítico de escolha é a cromatografia líquida de alta pressão (HPLC) com capacidade de detecção de até 0,5µg/mL, com especificidade de 93,6% (Leenheer et al, 1979). Já o método de cromatografia líquida acoplada a espectrometria de massa (LC-MS-MS) oferece uma significante vantagem para a confirmação das tetraciclinas residuais (Oka et al, 1998).
Biodisponibilidade
função renal ou hepática normal (Carney et al, 1974). Em pacientes com função renal comprometida, a meia-vida aumenta para 32 horas (Devulder et al, 1974; Kunin et al, 1967).
Distribuição
A minociclina é amplamente distribuída nos líquidos e tecidos corporais. Ela tem uma penetração tecidual maior quando comparada às oxitetraciclinas, e na maioria das vezes atinge níveis teciduais que excedem os níveis plasmáticos em tecidos como bile, tireóide, pulmão e fígado, mas também tem boa penetração na mama, pele e seios paranasais (Kunin, 1967). A minociclina atinge o líquido cefalorraquidiano melhor que as outras tetraciclinas, porém em concentrações ainda mais baixas. Possui boa penetração na saliva e lágrimas, além de ser eliminada no leite materno e atravessar a placenta.
A minociclina se liga mais fortemente as proteínas que as oxitetraciclinas (76% a 30%) (Kunin, 1967). Com meia-vida de 12-16 horas, apresenta um volume de distribuição de 78,6 ± 10,8L.
Metabolismo
sob pesquisa, estudos em animais demonstraram a conversão em 4-epiminociclina. A excreção da minociclina administrada por via intravenosa parece ser similar a doses orais. A minociclina é mais lentamente excretada na urina que as oxitetraciclinas, somente 5% nas primeiras 24 horas. A minociclina é extremamente solúvel no tecido adiposo. Cerca de 32% é eliminada como droga ativa, sendo 12% na urina e 20% nas fezes. Altas concentrações são encontradas na bile.
RELAÇÃO DOSE-EFEITO
A faixa terapêutica da minociclina dependerá da concentração inibitória mínima (CIM) do antibiótico para o microrganismo em questão, ou seja, da menor concentração do antibiótico capaz de inibir a replicação bacteriana.
A sensibilidade completa ocorre quando o CIM é ≤ 4µg/mL, e a suscetibilidade intermediária quando CIM estiver entre 4,0 e 12,5µg/mL e resistente se > 12,5µg/mL.
MICROBIOLOGIA
incluindo a minociclina, têm similar espectro antibacteriano contra Gram-negativos e Gram-positivos. A resistência cruzada desses microorganismos é comum (Wrightson et al, 1997).
Espectro antibacteriano
∗ Bactérias Gram negativas
Bartonella barciliformis, Brucella species, Campylobacter fetus,
Francinella tularensis, Haemopphillus ducrey, Haemophillos influenzae,
Listeria monocytogens, N. gonorrhoeaae, V. cholereae e Yersini2a pestis. Algumas bactérias Gram-negativas têm demostrado ser resistentes às tetraciclinas, deste modo, recomenda-se teste de sensibilidade e cultura para os microorganismos: Acinetobacter, Bacteroides, Enterobacter aerogenes, E. coli, Klebsiella species,e Shigella.
∗ Bactérias Gram positivas
trachomatis, Chlamidia psittaci, Entamoeba, Clostridium, Mycoplasma
pneumoniae, Rickettsiae, Treponema pallidum, Treponnema pertenue,
Ureoplasma urrealyticum.
INDICAÇÕES TERAPÊUTICAS
A minociclina está indicada no tratamento das seguintes patologias: acne vulgaris, cólera, gonorréia, granuloma e cancro inguinais, infecções estafilocócicas, infecções por clamídia, infecções respiratórias, nocardiose, quimioprofilaxia de infecções meningocócicas e sífilis (Baytch, 1974).
A suspensão oral e os comprimidos de minociclina são indicados no tratamento das seguintes patologias: febre das montanhas rochosas, febre tifóide, febre Q, infecção respiratória por micoplasma, linfogranuloma venéreo, tracoma, uretrites não gonocócica e infecções retal e endocervical porureoplasma, cancróide, tularemia, peste, brucelose, granuloma inguinal, infecções por Vibrio species e outras infecções por germes sensíveis (Smilack, 1999).
tetraciclinas podem ser usadas como coadjuvante amebicida. A minociclina também pode ser usada no tratamento da acne vulgar (Freeman, 1989). Minociclina oral é contra indicada para o tratamento de infecção meningocócica (PDR®, 1998).
A minociclina também pode ser usada em associação no tratamento de infecções anaeróbias, fibrose cística, gengivite, malária e lepra.
CONTRA-INDICAÇÕES
A Hipersensibilidade a tetraciclinas embora rara, pode acarretar
dentição permanente, hipoplasia do esmalte, e deprime o crescimento ósseo,
pacientes com Lupus Eritematoso Sistêmico podem apresentar exacerbação
dessa condição (Doniz, 1969).
Recomenda-se que todos os pacientes recebendo minociclina, principalmente por mais de 1 ano, necessitam de “screening” para desenvolvimento de pigmentação (Eisen & Hakin, 1998). Até o momento não há relatos de efeitos letais.
Todas as tetraciclinas formam um complexo estável com o cálcio no tecido ósseo em formação. Uma redução da taxa de crescimento da fíbula tem sido observada em crianças prematuras que receberam doses de 25mg/kg a cada 6 horas. Essa reação foi mostrada ser reversível quando a droga é descontinuada (PDR®, 1998).
Na presença de insuficiência renal e, principalmente, na gravidez, a terapia diária intravenosa com tetraciclina em doses superiores a 2gm leva a morte por falência hepática. Quando é necessário um tratamento intensivo deve-se considerar deve-seu potencial prejudicial, associado principalmente com a gravidez e a presença de insuficiência renal ou hepática, e investigar, portanto, a função renal e hepática antes e durante a terapia, bem como as concentrações séricas de tetraciclina.
A fotosensibilidade é observada também como uma reação adversa em indivíduos que, em uso de tetraciclinas, têm história de exposição exagerada ao sol. No caso desses pacientes recomenda-se o uso de protetores solares, bem como a orientação de não se expor a radiação ultravioleta. O tratamento deve ser interrompido ao primeiro sinal de eritema na pele. A fotosensibilidade tem sido relatada raramente com o uso de minociclina (PDR®, 1998).
Alguns dos efeitos que estão associados ao sistema nervoso central são: cefaléia e vertigem. Pacientes que trabalham no controle de máquinas ou conduzindo veículos devem tomar minociclina com cautela. Esses sintomas desaparecem com a interrupção da droga (PDR®, 1998).
Algumas suspensões orais contêm sulfito de sódio, substância que pode causar reação alérgica incluindo sintomas anafiláticos ou efeitos menos severos em indivíduos suscetíveis. Pacientes asmáticos têm maior freqüência de reatividade ao sulfito que indivíduos normais. É desconhecida a prevalência de sensibilidade ao sulfito de sódio na população geral, mas estima-se que seja baixa (PDR®, 1998).
caso de superinfecção a droga deve ser descontinuada e instituída uma nova terapia apropriada (PDR®, 1998).
Em adultos, um pseudotumor cerebral (hipertensão intracraniana) tem sido associado com o uso de tetraciclina. As manifestações usuais são cefaléia e visão turva. A minociclina pode causar tontura, ataxia e náusea por disfunção vestibular, o que parece ser mais freqüente em mulheres e reversível com a descontinuação da terapia.
INTERAÇÕES COM DROGAS
O número de interações com drogas é enorme, principalmente em pacientes hospitalizados os quais, na grande maioria, necessitam de seis ou mais medicamentos. Embora muitas das interações não sejam observadas clinicamente ou os casos reportados sejam inconclusivos, observa-se que vem aumentando a cada ano.
Um aspecto que não pode ser esquecido é que o fato de uma interação maléfica ou benéfica já ter sido relatada, não necessariamente ocorrerá em todos os pacientes e que o oposto também não garante que não venha a aparecer.
As interações classificam-se de acordo com o mecanismo da interação e podem ser divididas em: 1) físico-química na qual há uma incompatibilidade física ou química de uma droga com a outra e ocorre antes da administração das drogas, ainda fora do organismo; são muito comuns em misturas destinadas a administração intravenosa; 2) farmacocinética onde há interação em um ou mais de um dos quatro processos que compõem a etapa farmacocinética, seja na absorção, distribuição, metabolismo ou excreção e existe também a interação 3)
Exemplos de interações medicamentosas com tetraciclinas
As tetraciclinas têm sido envolvidas na redução da atividade das protrombinas plasmáticas, pacientes em terapia anticoagulante precisam de ajuste da sua posologia.
Drogas bacteriostáticas como as tetraciclinas, interferem com a ação bactericida das penicilinas, por isso é aconselhável evitar a administração concomitante de drogas das classes das tetraciclinas com penicilinas.
A absorção da tetraciclina é prejudicada por antiácidos contendo alumínio, cálcio ou magnésio, e preparações contendo ferro.
O uso concomitante de tetraciclina e metoxiflurano tem sido relacionado com a ocorrência de toxicidade renal fatal (Kuzneu, 1970).
As tetraciclinas não devem ser usadas concomitantes com contraceptivos orais, por reduzirem sua eficácia e aumento de sangramento intermenstrual.
interação esta que foi estudada nesta dissertação, portanto será abordada com mais ênfase.
INTERAÇÕES COM ALIMENTO
As interações de drogas com alimento devem ser consideradas como uma interação da formulação com o alimento, devido as evidências de que a droga pode ser afetada diferentemente pelo alimento quando administrada em formulações diferentes. Podem ser classificadas em 5 categorias: as que causam redução, atraso, aumento ou aceleram a absorção e ainda as que a alimentação não exerce efeito sobre a droga.
Os efeitos da alimentação na biodisponibilidade de drogas ou bioequivalência entre elas depende de propriedades da droga, sejam físico-químicas como solubilidade ou farmacocinéticas como sítio de ação, taxa e extensão de absorção e metabolismo de 1ª passagem, bem como da liberação do princípio ativo do produto farmacêutico. Os efeitos da co-administração de alimentos com drogas é máximo quando o produto é administrado imediatamente após a alimentação. O conteúdo da alimentação, o volume de fluido, a temperatura e o conteúdo calórico dos alimentos influenciam a magnitude da alteração fisiológica que pode afetar a absorção da droga. Alimentos ricos em calorias, gorduras e de alta densidade são os mais prováveis de levar a alterações na biodisponibilidade de drogas (Guidance for Industry. Food-Effect Bioavailability and Bioequivalence Studies. U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER) - October 1997 BP X). Welling e colaboradores (1977) relataram que uma dieta rica em gordura, carboidratos e proteínas produz 50% de inibição na absorção de tetraciclina e 20% de inibição na absorção de doxiciclina.
circulante devido à presença de alimento e o possível impacto clínico (Welling, 1996).
Tetraciclinas versus alimento
Yu e colaboradores (1990), Baggot (1992) e Sutter e colaboradores (1993); mostraram que a presença de alimento no trato gastrointestinal pode afetar a absorção de drogas administradas por via oral, e que o grau da interação depende da droga em questão bem como da espécie animal.
Segundo Palmer e colaboradores (1983) e Kniffen e colaboradores (1989), a administração de tetraciclinas com alimento pode resultar em interações que influenciam na biodisponibilidade da droga, alterando a possibilidade de obter concentrações terapeuticamente ativas no plasma e nos tecidos.
biodisponibilidade é grandemente reduzida quando a tetraciclina é misturada ao alimento.
Apresentação farmacêutica
O laboratório inovador nos Estados Unidos e Reino Unido é o Lederle Laboratories que comercializou a minociclina com o nome de Minocin. No Brasil a minociclina de referência é o Minomax® do Laboratório Wyeth.
Comprimidos de 50 e 100mg de minociclina estão disponíveis tanto nos Estados Unidos como no Reino Unido. O período de validade é de 3 anos, desde que armazenados em ambiente de temperatura variando de 10 a 30ºC, na embalagem original ou em recipientes protegidos contra a umidade.
O
OBJETIVOS
Determinar a farmacocinética da minociclina administrada em condições de jejum e após uma dieta padrão específica.
Comparar a biodisponibilidade de duas formulações de minociclina, fabricadas por dois laboratórios, um teste e outro de referência, em condições de jejum eapós dieta padrão específica.
M
MATERIAL E MÉTODOS
PROTOCOLO CLÍNICO
Delineamento do Estudo
A pesquisa consistiu de um estudo aberto, com replicação, de 24 voluntários sadios, adultos (doze homens e doze mulheres, não grávidas). Depois da seleção e observado um período de pelo menos 4 semanas sem fazer uso de qualquer medicação, os voluntários qualificados para participar do estudo foram internados por quatro períodos de aproximadamente 36 horas, com duas semanas de intervalo entre os internamentos. Em cada internamento, os voluntários receberam a formulação teste ou referência acompanhada ou não de uma dieta padrão específica a qual foi elaborada objetivando-se um alto teor de cálcio.
Em dois dos quatro internamentos foi administrado minociclina com uma dieta padrão específica, sendo uma vez com a minociclina referência (Minomax®) e em outra a minociclina teste (Minoderm). Quinze minutos após a ingestão da dieta padrão específica, administrou-se a minociclina conforme uma tabela de randomização para quatro períodos.
Nos outros dois internamentos, a administração constou apenas da formulação de minociclina referência ou a minociclina teste.
Seleção do Voluntário
Os voluntários foram selecionados através de uma avaliação clínica completa, conduzida por um profissional médico, realização de exames laboratoriais para avaliar as funções renal, hepática e hematológica, bem como avaliação da função cardíaca através de um eletrocardiograma (ECG).
Exames Laboratoriais:
1) Análise Hematológica: Hemoglobina, Hematócrito, Contagem total e diferencial de leucócitos, Contagem de plaquetas e Velocidade de Hemossedimentação (VHS).
2) Análise Bioquímica: Uréia, Creatinina, Bilirrubina Total, Proteína Total, Albumina, Glicose em jejum, Fosfatase Alcalina, Aspartato – amino - transferase (AST), Alanina - amino - transferase (ALT), Colesterol Total, Triglicérides, Ácido Úrico e Gama GT.
3) Sumário de Urina
4) Exames realizados somente no período pré-estudo: Parasitológico de Fezes; Análise Sorológica para Hepatite B, Hepatite C, HIV (Síndrome da Imunodeficiência Adquirida-AIDS) e Teste sorológico para gravidez (Beta – HCG) no caso de tratar-se de voluntários do sexo feminino.
Critérios de Inclusão
Os seguintes critérios foram satisfeitos para que o voluntário participasse do estudo:
2. Peso corporal entre 55 e 95Kg dentro de uma variação de 15%, de acordo com seu índice de massa corpórea (IMC);
3. Ser submetido a uma história clínica e exame físico, e ser considerado saudável;
4. Concordar livremente e assinar o termo de consentimento, após todos os elementos essenciais do protocolo serem esclarecidos, antes de qualquer procedimento.
Critérios de Exclusão
Qualquer um dos seguintes critérios excluía o voluntário do estudo:
1. Os resultados dos exames laboratoriais estavam fora dos valores considerados normais (± 10%), a menos que fossem considerados clinicamente irrelevantes;
2. O voluntário participou de qualquer estudo experimental ou ingeriu qualquer droga experimental nos últimos três meses que antecederam o início do estudo;
3. Fez uso de medicação regular nas últimas 4 semanas que antecederam o inicio do estudo, ou fez uso de qualquer medicação uma semana antes do início do estudo;
5. Apresentava história de abuso de álcool ou drogas, ou tinha ingerido bebidas alcoólicas nas 48 horas que antecederam o período de internação para início do estudo;
6. Possuía história de doença hepática, renal, pulmonar, gastrointestinal, epiléptica, hematológica ou psiquiátrica;
7. Apresentava hipotensão ou hipertensão de qualquer etiologia que necessitasse de tratamento farmacológico;
8. Apresentava história ou tinha tido infarto do miocárdio, angina e/ou insuficiência cardíaca;
9. O voluntário tinha doado ou perdido 450mL ou mais de sangue nos últimos três meses que antecederam ao estudo;
10. Mulher com teste imunológico positivo para gravidez;
11. O voluntário tinha qualquer condição que o impedia de participar do estudo pelo julgamento do investigador.
Critérios para Retirada do Estudo:
As seguintes condições foram consideradas como critérios de retirada do estudo:
intolerância aos procedimentos do estudo. Efeitos adversos da droga, testes laboratoriais anormais julgados de relevância clínica, doença intercorrente requerendo medicação.
Entrada do Voluntário no Estudo:
Os voluntários foram aceitos no estudo somente se fossem considerados saudáveis, como determinado pela história médica, exame físico e os exames laboratoriais que antecederam o início do estudo.
Uma vez avaliada a higidez, os voluntários foram submetidos a uma entrevista para avaliação das condições emocionais para participar da investigação. Após todas as dúvidas, por parte dos voluntários, terem sido esclarecidas, e os mesmos tendo concordado com o protocolo clínico, assinaram o termo de consentimento (Anexo IV) para participação no estudo.
Internamento:
anterior à administração do medicamento, e que permanecesse na mesma até 24 horas após a administração da medicação.
Restrições:
Todos os voluntários chegaram à Unidade de Farmacologia Clínica tendo feito uma refeição normal noturna (jantar). A partir das 23:00 horas da noite do internamento, os voluntários ficaram em jejum até a manhã seguinte e durante o período de 2 horas após a administração da medicação quando um desjejum foi servido. O almoço foi servido entre 5 e 6 horas após administração e o jantar após 12 horas. Não foram permitidos outros alimentos no período de internação. Em dois dos quatro períodos, os voluntários tomaram um café da manhã padronizado 15 minutos antes da administração da medicação e as demais refeições seguiram o mesmo protocolo. Líquidos foram permitidos ad libitum após as refeições, mas bebidas contendo xantinas (incluindo chá, café e cola) foram evitadas.
Produtos Estudados
Formulação Teste Formulação Referência
Nome Minoderm Minomax®
Ingrediente Ativo Minociclina Minociclina
Forma Comprimidos Comprimidos
Potência 100mg 100mg
Lote CT 010/99 CT 011/99
Fabricante Laboratórios Stiefel Laboratórios Wyeth
Esquema Experimental
Os voluntários receberam em cada uma das fases (período) de internamento, de acordo com a randomização (tabela 1), os seguintes produtos:
Minomax®, comprimido contendo 100mg de cloridrato de minociclina (formulação referência), da Wyeth, em jejum.
Minoderm, comprimido contendo 100mg de cloridrato de minociclina (formulação teste), da Stiefel, em jejum.
Minoderm, comprimido contendo 100mg de cloridrato de minociclina (formulação teste), da Stiefel com dieta padrão específica.
Inventário das amostras
Minoderm 100mg Stiefel Minomax® 100mg Wyeth
Tabela 1: Randomização da Minociclina
Vol. Fase I Fase II Fase III Fase IV
1. Minomax® Minoderm Minomax® + dieta Minoderm + dieta
2. Minomax® + dieta Minoderm Minoderm + dieta Minomax®
3. Minoderm + dieta Minomax® Minoderm Minomax® + dieta
4. Minoderm Minomax® + dieta Minoderm + dieta Minomax®
5. Minomax® Minomax® + dieta Minoderm Minoderm + dieta
6. Minoderm + dieta Minomax® Minoderm Minomax® + dieta
7. Minomax® + dieta Minoderm + dieta Minomax® Minoderm
8. Minoderm Minoderm + dieta Minomax® Minomax® + dieta
9. Minoderm Minoderm + dieta Minomax® + dieta Minomax®
10. Minomax® + dieta Minoderm Minomax® Minoderm + dieta
11. Minomax® Minomax® + dieta Minoderm + dieta Minoderm
12. Minoderm + dieta Minomax® Minomax® + dieta Minoderm
13. Minomax® + dieta Minoderm + dieta Minoderm Minomax®
14. Minoderm + dieta Minomax® Minoderm Minomax® + dieta
15. Minoderm Minomax® + dieta Minomax® Minoderm + dieta
16. Minoderm + dieta Minomax® Minomax® + dieta Minoderm
17. Minoderm Minoderm + dieta Minomax® + dieta Minomax®
18. Minoderm Minomax® + dieta Minomax® Minoderm + dieta
19. Minomax® Minoderm Minoderm + dieta Minomax® + dieta
20. Minomax® Minomax® + dieta Minoderm + dieta Minoderm
21. Minomax® + dieta Minomax® Minoderm + dieta Minoderm
22. Minomax® Minoderm + dieta Minoderm Minomax® + dieta
23. Minoderm + dieta Minoderm Minomax® + dieta Minomax®
Coleta de Sangue
As amostras de sangue para determinação da concentração plasmática de minociclina foram obtidas, através de escalpe heparinizado introduzido em veia superficial do antebraço do voluntário, imediatamente antes da administração de uma das preparações de minociclina (tempo ZERO), e aos seguintes intervalos a partir da administração: 0:30, 1:00, 1:30, 2:00, 3:00, 4:00, 6:00, 8:00, 10:00, 12:00, 16:00, 24:00, 48:00, 72:00 e 96:00 horas.
A cada intervalo de tempo foram coletados 8mL de sangue e colocados em tubos vacutainer de vidro contendo 30µL de heparina. Depois de centrifugadas a 3.000 rpm, na temperatura de 8°C, durante 12 minutos, o plasma foi separado, colocado em dois tubos e em seguida os tubos devidamente identificados foram mantidos em freezer, numa temperatura de –20ºC, até sua análise.