Exploring the use of cytochrome oxidase c subunit 1 (COI) for DNA barcoding of free-living marine nematodes.
Texto
Imagem

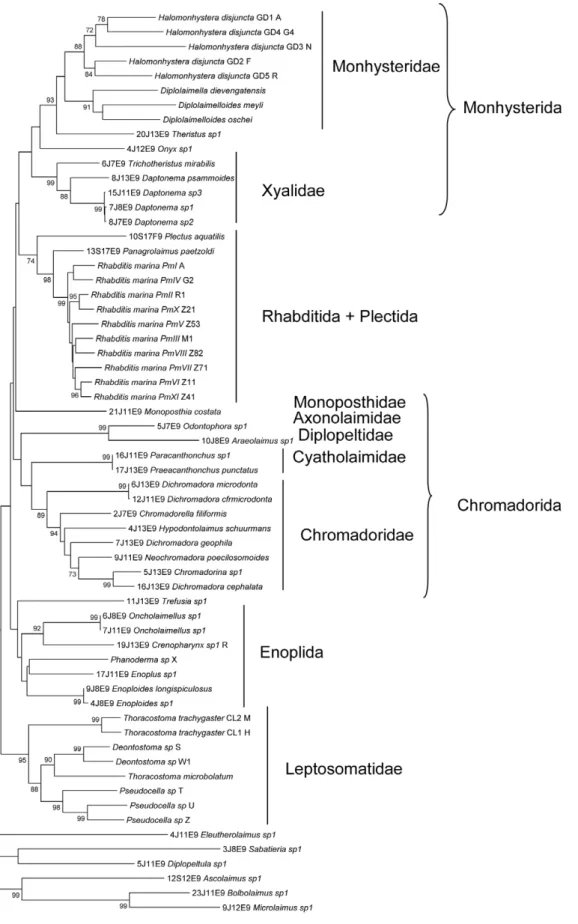
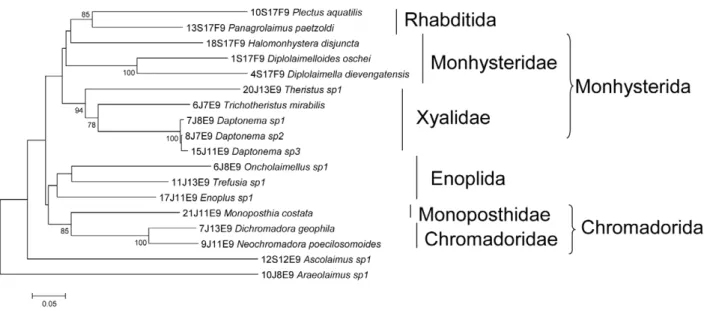
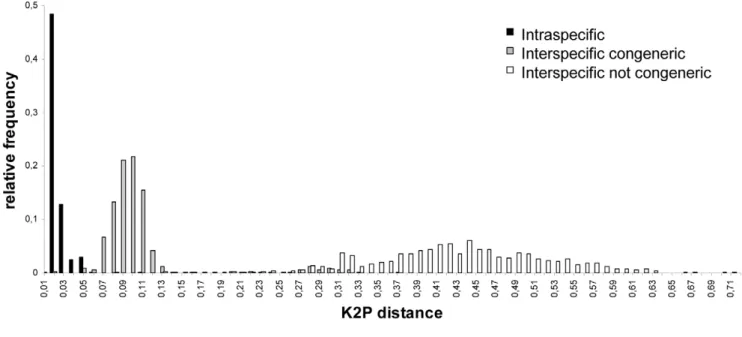
Documentos relacionados
Recentemente, o Ministério da Defesa Nacional conseguiu disponibilizar ainda algumas dotações extraordinárias, na ordem de 1,5 milhões de euros, para inter- venções imediatas
3.3.3 Perseverança e esperança na ciência e na divulgação da doença Os familiares têm esperança na ciência, acreditando que a divulgação da doença pode ajudar no campo
A distribuição espacial deste índice orienta a sequência de cálculo de balanço hídrico e a propagação dos escoamentos de cada célula; (ii) a determinação das
The population of the plant- parasitic nematodes slightly increased from the initial level of 1286/250 g soil to 2288 in untreated beds, whereas the treatments with
Root-knot nematodes are considered among the top 5 major plant pathogens and the first among the 10 most important genera of plant parasitic nematodes in the world (Mukhtar et
Pela importância de que se reveste este livro na Obra literária da Escritora, pela novidade temática e pelo caráter ambivalente do texto em termos de receção, exploramos
Portanto, é pertinente para esta pesquisa olhar para esse quadro e pensar quais tipos de representação estão contidas no documentário, as diferentes funções
Com isso, um país pode ser ineficiente ainda que tenha uma gestão adequada de seus recursos públicos (AFONSO; AUBYN, 2006). Entretanto, quais variáveis de fato interferem
