Lopes Maria João Rhodes Silva Gaspar
Texto
Imagem

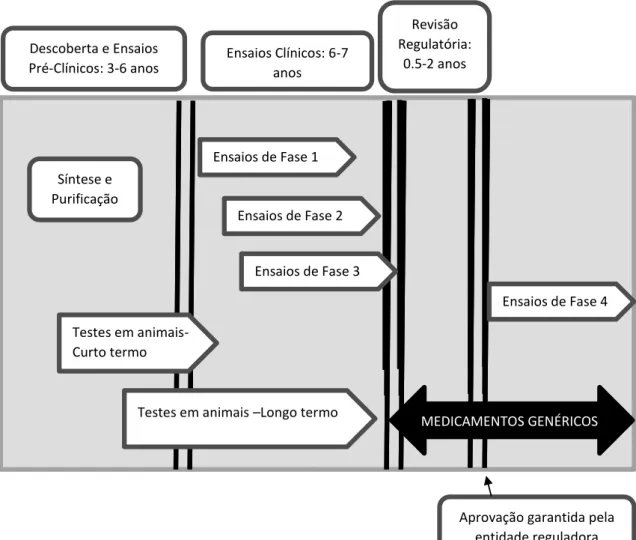








Outline
Documentos relacionados
No Estado, existem mais de duas mil empresas ata- cadistas e distribuidoras cadastradas, sendo 600 des- tas, inscritas no Contrato de Competitividade Comercial Atacadista
O presente regulamento é elaborado ao abrigo do disposto nos arti- gos 112.º e 241.º da Constituição da Republica Portuguesa, artigo 117.º e 118.º do Código do
Em suma, é no contexto do planejamento ambiental que se tem de dar conta do patrimônio cultural nacional, com todas as suas variantes regionais e locais, de modo a assegurar
Assim, como o folhetinista é definido por Machado de Assis como sendo um “colibri” a voar livremente sobre todos os fatos, o crítico de rodapé, como explica
34 — Cristiano Filipe Martins Conduto 35 — Rute Isabel Teixeira Lopes Monteiro 36 —joão Manuel Figueiredo Duarte 37 — António José Moinheiro dos Reis 38 — Maria de Fátima
**Fará jus também, conforme tulação apresentada, aos seguintes valores (pagos pelo tulo de maior grau, de forma não cumula va, sendo necessária a apresentação do diploma
Em relação à comenda em estudo, cujo inventário não está datado, encontra-se descrita entre os fólios 17v. Vicente, incluindo Vila Chã, São Martinho, Vila Nova e Lageosa. Um
[...] necessidade do diálogo no relacionamento com os demais, compreender a natureza dos conflitos que aumentam nos dias de hoje, e acima de tudo perder o
