Universidade de Lisboa
Faculdade de Farmácia
Transition from the traditional typing methods to
the application of whole genome sequencing
(WGS) for surveillance of food- and waterborne
diseases
Marta Patrícia Ribeiro Ferreira
Dissertation to obtain the Master of Science Degree in
Pharmaceutical Engineering
Work supervised by:
Professor João Almeida Lopes, PhD (Supervisor)
Vítor Borges, PhD (Co-Supervisor)
ii
2016
Universidade de Lisboa
Faculdade de Farmácia
Transition from the traditional typing methods to
the application of whole genome sequencing
(WGS) for surveillance of food- and waterborne
diseases
Marta Patrícia Ribeiro Ferreira
Dissertation to obtain the Master of Science Degree in
Pharmaceutical Engineering
Work supervised by:
Professor João Almeida Lopes, PhD (Supervisor)
Vítor Borges, PhD (Co-Supervisor)
iii
2016
The experimental work was performed in the Bioinformatics Unit of
the Department of Infectious Diseases of the National Institute of
Health Dr. Ricardo Jorge (INSA). All the facilities, equipments,
materials and support were gently provided by INSA.
iv
Acknowledgments
Every successful individual knows that his or her achievement depends on a community of persons working together. Paul Ryan
Foram meses, semana, dias e horas de trabalho, alguma frustração e muita dedicação, apoio e orientação. Existiram pessoas que com palavras ou simplesmente sorrisos me ajudaram a completar esta fase da minha vida com sucesso. A todos eles deixo o meu mais sincero obrigado. No entanto, seria impossível não fazer referência a algumas em especial.
Ao meu orientador Professor João Pedro Martins de Almeida Lopes, obrigada por me ter apoiado nesta etapa e por ter ajudado a concretizar este trabalho no âmbito que sempre desejei.
Ao meu co-orientador Dr. Vítor Manuel Monteiro Borges, agradeço a excelente orientação e disponibilidade, garantindo que sem o seu apoio e confiança, este trabalho não seria possível. Ao mesmo tempo, a sua amizade tem sido, e certamente continuará a ser, um forte impulso para o meu desenvolvimento pessoal e profissional. Ao Dr. João Paulo Gomes, responsável pela Unidade de Investigação e pelo Núcleo de Bioinformática do Departamento de Doenças Infecciosas do Instituto Nacional de Saúde (INSA) Dr. Ricardo Jorge, por ter disponibilizado todos os meios necessários para desenvolver este trabalho.
À Dra. Mónica Oleastro, responsável pelo Laboratório Nacional de Referência de Infecções Gastrintestinais do INSA, e seus colaboradores, por todo o apoio e pelo trabalho prévio de caracterização do isolados bacterianos pelos métodos tradicionais de tipagem.
Ao Instituto Nacional de Saúde (INSA) Dr. Ricardo Jorge, em particular ao Departamento de Doenças Infecciosas, na pessoa do seu coordenador, Dr. Jorge Machado, por ter disponibilizado todas as condições para a melhor prossecução deste trabalho de Mestrado.
A todos os que me acompanharam neste percurso no INSA, a vossa amizade e disponibilidade foi importante para a construção deste trabalho, por isso sinceramente agradeço à Dra. Maria José Borrego e à Dra. Alexandra Nunes, ao Miguel Pinto, à Rita Caldeira e Aleksandra Azevedo.
A todas as pessoas que mesmo longe se faziam presentes e assim ajudaram a que isto se tornasse realidade. Um obrigado especial à Ana Correia, com o teu apoio teria sido muito mais difícil. O Hugo Silva, pela paciência, compreensão, amizade e amor ao longo dos anos.
Aos meus pais e irmã. Foi convosco e para vós. Não há palavras que descrevam a gratidão que tenho pelo vosso apoio, protecção e ajuda ao longo de toda a minha vida. Obrigada por me ajudarem e caminharem sempre ao meu lado para que pudesse
v sempre realizar os meus sonhos. Agradeço também à minha enorme e restante família pelo apoio e amizade que sempre demostraram. Um especial obrigada aos meus queridos avós maternos, desde pequena que apendi convosco e continuei a aprender, muito do que sou hoje a vocês o devo. Obrigada!
Por fim, deixar um agradecimento as meninas da residência. Todas as que se aqui cruzaram comigo de uma maneira ou de outra deixaram a sua marca. Um especial obrigada à Tayone, Rafaela, Catarina, Idalina, Diana, Victoria, Adriana por todos os bons momentos de convívio que me ajudaram a ultrapassar os momentos mais difíceis destes anos.
vi
Abstract
Infectious Diseases, in particular food- and waterborne diseases (FWD), are critical public health concerns nowadays, reinforcing that public health laboratories should establish accurate strategies that allow a proper epidemiological surveillance of the causative infectious agents. With the advances in the whole genome sequencing (WGS) technologies, and associated bioinformatics, genome-based typing tools are becoming the gold standard methodology for laboratory epidemiological monitoring of FWD, with recognized benefits for public health.
In the present study, we aimed at starting the implementation of WGS/bioinformatics techniques (in place of traditional typing methods) for routine epidemiological surveillance of FWD-associated pathogens, more specifically of four of the most clinically relevant bacterial pathogens: Listeria monocytogenes, Salmonella
enterica, verocytotoxin-producing Escherichia coli (VTEC) and Campylobacter jejuni.
The WGS of multiple bacterial isolates (n= 89) firstly allowed us to apply and test multiple bioinformatics tools for in silico capture of the typing data routinely provided by the traditional pheno- and genotyping techniques. In general, high levels of concordance were reached, opening good perspectives that backwards compatibility with “historical” typing data may be ensured during this technology transfer process. Subsequently, in order to test novel genome-based typing tools for routine surveillance of FWD, we implemented and assayed core genome multilocus sequence typing (cgMLST) schemes and core-genome SNP-based strategies, with unequivocal gains regarding the discriminatory power. Moreover, the results were particularly concordantly between the two strategies, which also stood when applying either “assembly-free” or “de novo assembly-based” core-genome SNP-based strategies. Finally, our insights on the potential of WGS/bioinformatics to predict bacterial virulence and antimicrobial susceptibility profiles provided very optimistic perspectives for the future routine application of WGS for such purposes.
In conclusion, the present study constitutes an unequivocally important step towards the application of genome-based typing tools as the future gold standard technique for routine epidemiological surveillance of FWD in Portugal.
Keywords: Food- and waterborne diseases, Epidemiological surveillance, Traditional typing methodologies, Whole-genome sequencing, Bioinformatics
vii
Resumo
As doenças infecciosas, em particular as doenças transmitidas por alimentos e água, são importantes problemas de Saúde Pública, apresentando taxas relevantes de morbilidade e mortalidade na população em geral. Neste contexto, e tendo também em conta o preocupante aumento do número de casos de resistência a agentes antimicrobianos, os laboratórios de Microbiologia clínica e Saúde Pública, nomeadamente os Laboratórios Nacionais de Referência, procuram continuamente implementar e validar estratégias que assegurem uma eficiente vigilância epidemiológica dos agentes infeciosos causadores dessas doenças. Em particular, é importante estabelecer metodologias que permitam, não só uma detecção inequívoca desses agentes patogénicos, mas também técnicas de “tipagem” que permitam a sua posterior discriminação e classificação. Com os tremendos avanços nas tecnologias de sequenciação total do genoma, e ferramentas bioinformáticas associadas, é esperado que técnicas de tipagem baseadas na análise dos genomas microbianos em larga escala se tornem as técnicas-padrão para vigilância laboratorial de doenças transmitidas por alimentos e água num futuro muito próximo. Espera-se que esta transição tecnológica traga benefícios inequívocos para a Saúde Pública, tais como: i) o aumento da capacidade de resolução na caracterização genotípica de agentes patogénicos, ampliando, por exemplo, a capacidade para detectar e investigar surtos e fontes de infecção; ii) uma melhor identificação e monitorização da emergência de agentes que possam constituir novas ameaças para a Saúde Pública (tais como estirpes multiresistentes) facilitando a resposta a situações de emergência; e, iii) uma maior capacidade para prever o potencial patogénico das estirpes, nomeadamente o seu perfil de resistências a agentes antimicrobianos ou outros fenótipos de virulência, podendo estes dados facilitar as tomadas de decisão clínica e terapêutica.
Neste sentido, este estudo, desenvolvido no Instituto Nacional de Saúde Doutor Ricardo Jorge, teve como principal objectivo começar a implementar técnicas de tipagem baseadas na sequenciação total do genoma/bioinformática (em substituição das técnicas tradicionais de tipagem) para vigilância epidemiológica de agentes patogénicos associados a doenças transmitidas pelos alimentos e água, especificamente quatro agentes bacterianos com importante impacto na Saúde Pública: Listeria monocytogenes, Salmonella enterica, Escherichia coli produtora de verotoxinas (VTEC) e Campylobacter jejuni.
Através da sequenciação total do genoma de múltiplos isolados bacterianos (n=89), procurou-se em primeiro lugar testar ferramentas bioinformáticas para extracção/captura in silico de dados de tipagem fornecidos pelas técnicas tradicionais de “genotipagem“ (por exemplo, as “sequência-tipo” obtidas pela técnica de Multilocus
sequence typing – MLST) e “fenotipagem” (por exemplo, o serótipo). Em geral, os
resultados mostraram altos níveis de concordância, deixando, assim, boas perspectivas de que os dados históricos de tipagem poderão continuar a ser usados para melhor caracterização dos agentes patogénicos durante e após o processo de transição tecnológica. Esta valência conferida pela nova abordagem de tipagem baseada na análise dos genomas bacterianos é bastante relevante pois existe um
viii grande conhecimento científico sobre a distribuição geográfica de estirpes apresentando certos perfis genotípicos e fenotípicos, bem como sobre a sua associação com determinadas fontes de infeção e/ou manifestações clinicas.
De seguida, com o objectivo de testar novas estratégias de tipagem à escala do genoma bacteriano para vigilância laboratorial de doenças transmitidas por alimentos e água, foram implementadas e avaliadas, para os quatro agentes patogénicos do Homem em estudo, as seguintes abordagens bioinformáticas: core
genome multilocus sequence typing (cgMLST) e core-genome Single Nucleotide Polymorphisms (coreSNP). Os resultados obtidos pelas duas estratégias foram
particularmente concordantes, tendo claramente aumentado o poder de resolução em relação às técnicas tradicionalmente aplicadas para discriminação e classificação de estirpes daqueles agentes bacterianos. Além disso, foi também notória a concordância entre duas técnicas de coreSNP testadas: uma baseada no mapeamento bioinformático contra sequências conhecidas e outra baseada na montagem de novo da sequência do genoma dos isolados bacterianos. No caso desta última abordagem, o uso de diferentes software para montagem dos genomas de novo não teve impacto nas inferências filogenéticas construídas com base na diversidade genética do core-genoma.
Ainda no âmbito da implementação de novas estratégias de tipagem assentes na análise bioinformática dos genomas bacterianos, destaca-se o facto de o esquema de cgMLST aplicado para discriminação de isolados clínicos de L. monocytogenes ter grande potencial para vir a ser implementado e validado à escala mundial para vigilância epidemiológica deste importante agente patogénico humano. Neste estudo, os resultados gerados com esta nova abordagem (os quais foram corroborados pelas estratégias in house de coreSNP) permitiram já sinalizar clusters de isolados geneticamente muito próximos, os quais vão ser alvo de investigação epidemiológica retrospectiva. Destaca-se, ainda, o facto da flexibilidade conferida pelas estratégias de
coreSNP (i.e., a amplitude do genoma analisado é dependente do conjunto de estirpes
sob avaliação, com impacto no poder de discriminação) ter permitido, por exemplo, confirmar a identidade genética entre dois isolados clínicos de L. monocytogenes colhidos do mesmo paciente (de diferentes sítios anatómicos) e reforçar dados provenientes de métodos de tipagem tradicionais sugerindo que três estirpes de S.
enterica terão estado associadas a um surto.
Neste estudo foram também aplicadas ferramentas bioinformáticas para previsão da presencia/ausência de genes associados à virulência ou à resistência a antibióticos. Este primeiro passo no uso da técnica de sequenciação total do genoma/bioinformática para previsão rápida do arsenal de factores de virulência, bem como de perfis de resistência a agentes antimicrobianos deixa perspectivas muito optimistas para a futura aplicação por rotina destas novas abordagens com vista à previsão do potencial patogénico daqueles agentes infeciosos. De facto, foi possível determinar in silico o reportório de genes potencialmente mediadores de virulência apresentado por estirpes de E. coli, nomeadamente os genes que codificam a bem conhecida Shiga toxina. Por outro lado, foi já possível confirmar certas associações genótipo-fenótipo no que respeita aos perfis de resistência a antibióticos apresentados por isolados de S. enterica e C. jejuni. Adicionalmente, para melhor exposição dos resultados, tirou-se ainda partido de novas plataformas focadas em permitir uma melhor visualização e interpretação de dados de tipagem, as quais têm sofrido um forte desenvolvimento fruto do paralelo crescimento dos campos da genómica
ix microbiana e da bioinformática. A sua aplicação neste estudo permite perspetivar que estas ferramentas serão de grande utilidade para o futuro estabelecimento, de forma dinâmica e intuitiva, de correlações entre os perfis genéticos de certas estirpes e os dados clínicos/epidemiológicos a elas associadas. Por fim, é de ressalvar que a futura avaliação de todas as ferramentas bioinformáticas (e variáveis inerentes) aqui testadas a uma maior escala (envolvendo um grande número de isolados bacterianos) no decurso da processo de transição tecnológica irá certamente reforçar as nossas expectativas quanto à sua aplicabilidade e utilidade no contexto da vigilância de doenças infecciosas.
Em conclusão, o presente estudo constitui um passo inequivocamente importante para o estabelecimento de ferramentas de tipagem baseadas na sequenciação total do genoma (e análises bioinformáticas associadas) como técnicas-padrão a usar num futuro próximo para a vigilância epidemiológica de doenças infecciosas transmitidas por alimentos e água em Portugal. Por outro lado, os dados gerados com este trabalho permitiram já lançar importantes hipóteses científicas que estão actualmente a ser alvo de novos estudos, incluindo estudos focados em confirmar potenciais relações epidemiológicas aqui sugeridas, bem como estudos de investigação focados em estabelecer relações genótipo-fenótipo.
Palavras-chave: Doenças infecciosas transmitidas por alimentos e água, Vigilância epidemiológica, Métodos tradicionais de tipagem, Sequenciação total do genoma, Bioinformátic
x
Contents
Acknowledgments ... iv Abstract ... vi Resumo ... vii Contents ... xList of figures ... xii
List of tables ... xiv
Notes of the author: thesis organization, format and outline ... 1
1. Introduction ... 2
1.1. Molecular epidemiology for the surveillance of infectious diseases: Whole-genome sequencing (WGS) as the future gold standard typing method ... 2
1.2. FWD pathogens: a brief overview on diversity, molecular epidemiology and impact on human health ... 4
1.2.1. Listeria monocytogenes ... 4
1.2.2. Salmonella enterica. ... 5
1.2.3. Escherichia coli ... 7
1.2.4. Campylobacter jejuni... 8
1.3. General overview on the current development of WGS-based strategies for epidemiological surveillance of Food and Waterborne Diseases. ... 9
2. Material and Methods ... 12
2.1. Bacterial FWD pathogens ... 12
2.2. Data from traditional geno- and phenotyping methods... 12
2.3. Whole-genome sequencing ... 12
2.4. Quality assurance / Quality control (QA/QC) meausres on the raw sequencing data . 13 2.5. de novo assembly ... 14
2.6. de novo assembly-based genome alignments ... 14
2.7. Reference-based mapping... 15
2.8. Core-genome SNP-based phylogeny and global analysis of genetic diversity ... 15
2.9. Other bioinformatics platforms and species-specific analysis ... 15
3. Results and Discussion ... 17
3.1. Overall results on quality statistics of sequencing runs and de novo genome assembly measures for all bacterial FWD pathogens1 ... 17
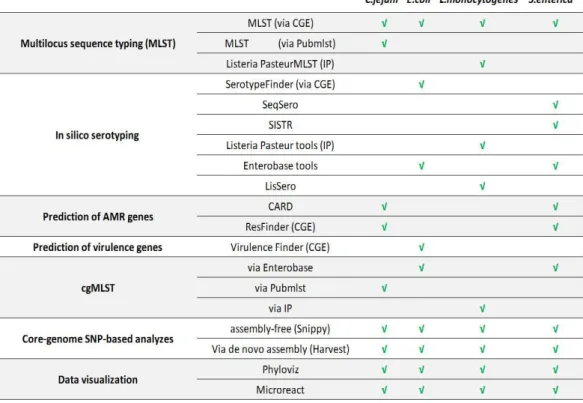
3.2. Overview on the bioinformatics analyses carried for each FWD pathogen ... 19
3.3. Listeria monocytogenes ... 21
3.4. Salmonella enterica ... 33
3.5. Escherichia coli ... 45
xi 4. General Conclusion... 63 References ... 67 Anexo A ... 73 Anexo B ... 78 Anexo C ... 81 Anexo D ... 82
xii
List of figures
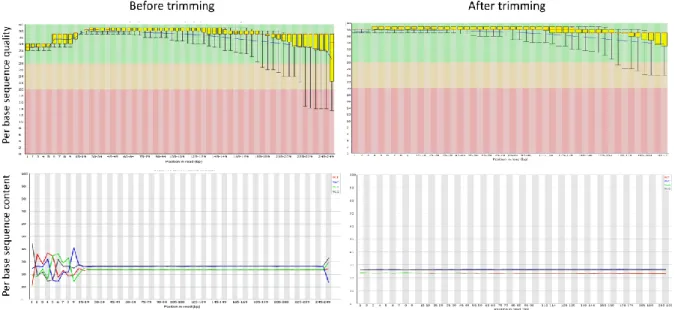
Figure 3.1. Example of the quality parameters of a representative read file before and after trimming measures in FastQC software.
17 Figure 3.2. Prediction of L. monocytogenes serotype/serogroup by using distinct approaches: traditional PCR and two different bioinformatics tools.
22 Figure 3.3. Impact of applying distinct de novo assemblers on the topology of the core-genome-based phylogeny of L. monocytogenes human isolates.
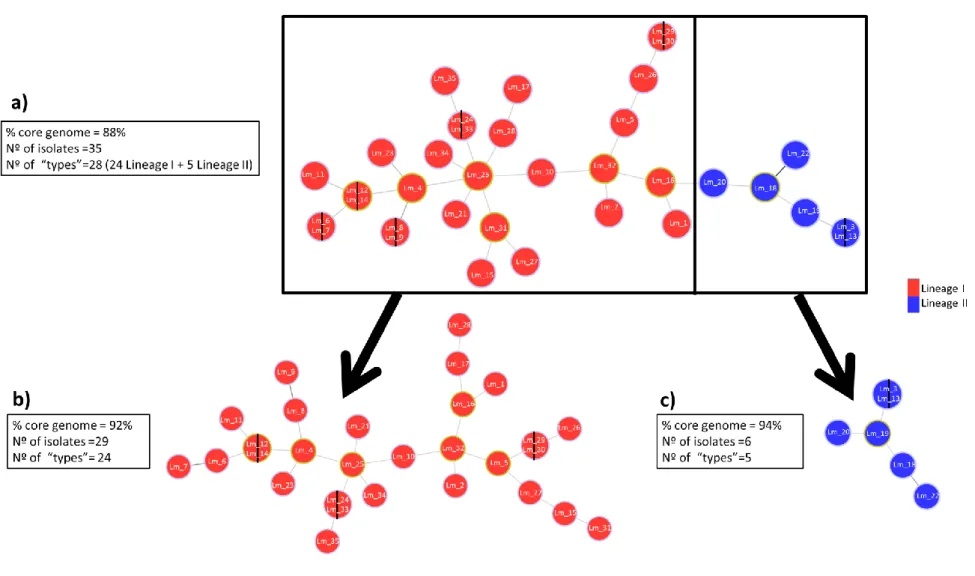
23 Figure 3.4. cgMLST-based analysis of Listeria monocytogenes human isolates.
25 Figure 3.5. Schematic representation of the correlation between L.
monocytogenes lineages and serogroups.
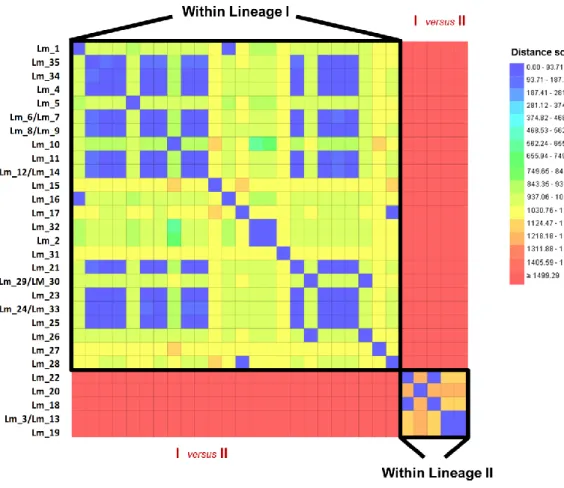
26 Figure 3.6. A distance matrix cgMLST-based analysis of 35 Listeria
monocytogenes human isolates.
27 Figure 3.7 Clusters of L. monocytogenes human isolates detected by cgMLST analyses.
28 Figure 3.8. Impact of applying distinct core-genome SNP-based strategies on inferring the phylogeny of Listeria monocytogenes human isolates.
29 Figure 3.9. Core-genome SNP-based analyze per Lineage. 31 Figure 3.10. Genetic relationships of 35 L. monocytogenes human isolates using different genome-based analyses
32 Figure 3.11. In silico serovar prediction by different bioinformatics platforms, and comparison with the traditional phenotyping method.
35 Figure 3.12. Impact of applying distinct de novo assemblers on the topology of the core-genome-based phylogeny of Salmonella enterica isolates.
36 Figure 3.13. cgMLST-based analysis of Salmonella enterica isolates. 37 Figure 3.14. Impact of applying distinct core-genome SNP-based strategies on inferring the phylogeny of Salmonella enterica isolates.
38 Figure 3.15. Genetic relationships of 16 isolates of Salmonella enterica isolates inferred by using different genome-based approaches.
39 Figure 3.16. Overview on the in silico predicted profiles of “presence/absence” of genes potentially associated with antibiotic resistance for 16 S. enterica isolates as predicted by two open-accessible bioinformatics tools.
41
Figure 3.17. Correlation between phenotypic results from antibiotic susceptibility tests and the in silico prediction of the presence of AMR genes for S. enterica
42
Figure 3.18. Impact of applying different criteria and inputs on the SeroTypeFinder bioinformatics tool on the number of isolates with predicted serotype.
47
Figure 3.19. Impact of applying distinct de novo assemblers on the topology of the core-genome-based phylogeny of Escherichia coli isolates.
48 Figure 3.20. Impact of applying distinct core-genome-based strategies on inferring the phylogeny of Escherichia Coli isolates.
49 Figure 3.21. Relationships between phylogenetic relatedness, serotype, ST and arsenal of virulence genes for the studied E. coli isolates.
50 Figure 3.22. Repertoire of E. coli virulence genes detected per isolate using VirulenceFinder with different input files.
51 Figura 3.23. Genetic relationships of 25 isolates of Escherichia coli isolates inferred by using different analytic approaches.
52 Figure 3.24. MLST-based representation of the worldwide genetic diversity among C. jejuni isolates.
55 Figure 3.25. MLST-based representation of the genetic diversity among C.
jejuni human isolates.
55 Figure 3.26. MLST-based representation of the genetic diversity among C.
jejuni human isolates according to the clonal complex.
xiii Figure 3.27. Impact of applying distinct de novo assemblers on the topology of
the core-genome-based phylogeny of Campylobacter jejuni human isolates.
57 Figure 3.28. Correlation between phenotypic results from antibiotic susceptibility tests and the in silico prediction of the presence of AMR genes or mutation associated with resistances for C. jejun.
58
Figure 3.29. cgMLST-based analysis of blood-invasing Campylobacter jejuni human isolates
59 Figure 3.30. Impact of applying distinct core-genome-based strategies on inferring the phylogeny of Campylobacter jejuni isolates.
xiv
List of tables
Table 2.1. Overview on the data available for the isolates under evaluation. 12 Table 2.2. Overview on the bioinformatics tools applied. 16 Table 3.1. Overview on the bioinformatics tools and strategies applied for each FWD pathogen.
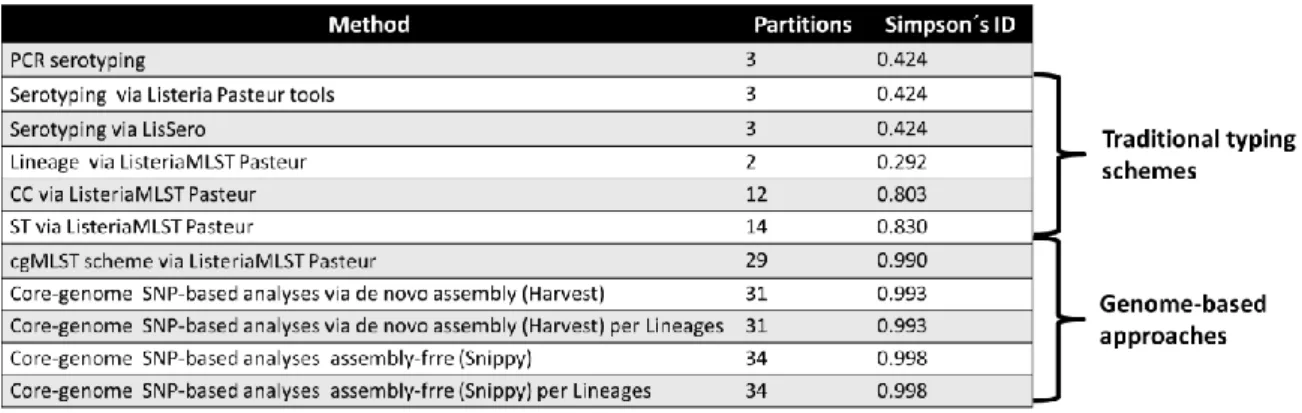
20 Table 3.2. Insight on the discriminatory power provided by distinct typing methodologies L.monocytogenes using the Simpson’s Index of diversity.
30 Table 3.3. Insight on the discriminatory power provided by distinct typing methodologies for S. enterica using the Simpson’s Index of diversity.
43 Table 3.4. In silico ST prediction using the CGE MLST tool with different sequence inputs and EnteroBase.
45 Table 3.5. Insight on the discriminatory power provided by distinct typing methodologies for E. coli using the Simpson’s Index of diversity
50 Table 3.6. Comparison between in silico extracted STs and one previously obtained by the traditional method.
54 Table A1. Overview on the some measures extracted by analyses in FASTQC 71
Table B1. List of AMR genes as predicted by CARD. 76
Table C1. List of AMR genes as predicted by ResFinder. 79 Table D1. List of antibiotics for which data regarding the susceptibility testing was available.
1
Notes of the author: thesis organization, format and outline
This MSc thesis describes the work developed on behalf of the Master´s Degree in Pharmaceutical Engineering coordinated by the Faculty of Pharmacy of the University of Lisbon. All work was carried out in the Bioinformatics Unit of the
Department of Infectious Diseases of the National Institute of Health (NIH) Dr. Ricardo Jorge.The document is organized in four main sections as follows:
Introduction. This section consists of a general introduction aiming at describing:
i) an overview on the relevance of molecular epidemiology studies for the surveillance of infectious diseases and on the context behind the current need to transit from the traditional typing methods to the application of whole genome sequencing (WGS) for surveillance of food- and waterborne diseases (FWD);
ii) a brief overview on diversity, molecular epidemiology and the impact on human health of each one of the FWD pathogens under study:
Listeria monocytogenes, Salmonella enterica, verocytotoxin-producing Escherichia coli (VTEC) and Campylobacter jejuni; and,
iii) the current development of WGS-based strategies for epidemiological surveillance of FWD.
Material and Methods. This section describes all procedures followed during this work, in particular all bioinformatics tools and strategies implemented and/or tested for WGS data processing and analysis.
Results and Discussion. This key chapter is subdivided in six sections. In the two first sections, it is provided technical data regarding the main quality statistics of sequencing runs and genome assembly obtained for the multiple bacterial isolates subjected to WGS, and also a brief overview on the bioinformatics analyses carried out for each FWD pathogen. Each one of the following four sections is focused on describing and discussing the results obtained for each FWD pathogen on behalf of this work that constitutes the first important step towards the implementation of genome-based typing tools for the surveillance of FWD in the Portuguese NIH. New research lines raised with this work that can be addressed in the future follow-up studies are also presented.
General conclusion. This section provides a global overview of the subjects addressed throughout the previous section, highlighting the main results and conclusions achieved in this MSc dissertation and pointing future steps to be carried out on the application of WGS in surveillance of infection diseases.
All section have been formatted in a unique style, with all references being cited by sequential numbers (in superscript) and listed in the "References" section according to the order in which they appear in the text. Finally, annexes are also compiled in a last section of this thesis.
2
1.
Introduction
1.1. Molecular epidemiology for the surveillance of infectious diseases: Whole-genome sequencing (WGS) as the future gold standard typing method
Bacteria have been present on Earth for around 3,5 billion years, are continuously evolving and, consequently, the worldwide bacterial population is very large and has very high levels of diversity (1). Furthermore, due to the never-ending contact with the environment, bacteria have developed multiple mechanisms to change their phenotype towards adaptation and survival, such as the fixation of adaptive genetic mutations or the acquisition of virulence genes and other defense mechanisms (2-4). Not surprisingly, during this long-term evolutionary pathway, multiple bacterial organisms have acquired the ability to colonize and further become pathogenic to humans, being now considered as major threats to the human health. While this scenario have turned out some Infectious Diseases as critical public health concerns nowadays, it also reinforces that health authorities should develop and implement strategies that allow a proper epidemiological surveillance of the bacterial causative agents. Among other goals, these efforts should aim not only at establishing methodologies that allow an unambiguously detection of the infectious agents, but also at setting up techniques that allow their further discrimination and classification. In this regard, the specific application of geno- and pheno-typing methodologies have historically proven to be pivotal for properly monitoring the emergence and spread of human pathogens and, consequently, for implementing strategies focused on the prevention and control of Infectious Diseases. In particular, molecular typing approaches have been fundamental to increase our understanding of human pathogens by means of increasing our knowledge on their genetic diversity, population structure and worldwide geographical distribution (2). In fact, most of these techniques have not only the important advantages of being faster, sensitive, highly specific and effective, but also the great benefit of being able to be experimentally adapted for surveillance of multiple organisms. As a consequence, the establishment of accurate geno- and phenotyping strategies for routine surveillance of infectious diseases provides tremendous practical benefits (2, 3, 5), such as:
1. to obtain baseline information about the evolutionary patterns of the infectious agents and further react on changing patterns;
2. to identify common sources of outbreaks;
3. to identify factors that contribute to the persistence and spread of endemic clones; 4. to allow identifying and monitoring critical points behind the cross contamination in
food-processing environments;
5. to complement diagnosis by identifying, for instance, virulent subtypes; and
6. to guide the treatment not only by providing information about the antibiotic resistance profile of the infecting bacteria, but also by helping in the evaluating the reasons behind treatment failure.
The advances in the microbial typing and detection techniques have been remarkable in the last few decades. However, one of the most significant changes is now occurring due to transition from the traditional typing methods to the application of
3 whole genome sequencing (WGS), which is expected to take place in a near future for the surveillance of almost all human bacterial pathogens. In parallel with this transition, huge progression has been naturally done to develop and implement proper bioinformatics tools for analysis and interpretation of WGS data. Not surprisingly, there is currently some uncertainty on how this transition should be carried out and, importantly, how WGS data should be handled and interpreted in order to provide useful information for public health (2). In fact, while the diversity of tools for WGS data is continuously increasing, which naturally implicates that efforts are needed to test, validate and ultimately standardize them for routine application, acquisition of knowledge and training on this the novel genome-based typing tool is also needed for all personnel staff involved in continuous surveillance of Infectious Diseases, from laboratory technicians, microbiologists and bioinformaticians to epidemiologists or clinicians (2).
The field of the Infectious Diseases that has been in the frontline in the implementation of WGS and bioinformatics tools for routine surveillance is the food and waterborne diseases (FWD). In fact, increased movements of people, expansion of international trade in foodstuffs and biological products, together with social and environmental changes linked to urbanization and deforestation highlight the need to concert efforts at cross-sectorial level (enrolling food, veterinary and public health sectors, industry, etc) to rapidly set up internationally agreed and harmonized WGS and bioinformatic tools for surveillance of FWD (2, 4). In fact, as referred above, this never-ending changing nature of the world directly implicates a never-ending adaptation of microorganisms, in particular of FWD pathogens, with consequent impact on public health such as, the return of old diseases and the emergence of new ones or the increase of antimicrobial resistance rates (4). Overall, the implementation of genome-based typing tools for FWD diseases, together with the evaluation the backward compatibility with the "historical" typing traditional methods is of major relevance. In Portugal, the “Instituto Nacional de Saúde (INSA) Doutor Ricardo Jorge” (the Portuguese NIH) is simultaneously the State Laboratory in the Health Sector, the National Reference Laboratory and the National Health Observatory. As such, one of its specific responsibilities relies on performing epidemiological surveillance of infectious diseases, namely FWD, implicating that INSA develops a continuous work on setting up accurate tools that allow an efficient health observation. In this context, this thesis, whose work was carried at INSA, is focused on starting the implementation and validation of WGS/bioinformatics techniques (in place of traditional methods) for typing of FWD pathogens, more specifically of four of the most important FWD pathogens:
Listeria monocytogenes, Salmonella enterica, verocytotoxin-producing Escherichia coli
(VTEC) and Campylobacter jejuni.
The following four sections will be focused on providing a brief overview on diversity, molecular epidemiology and the impact on human health of each one of the FWD pathogens under study. Subsequently, in the last section of the Introduction, it is summarized the main genomics and evolutionary traits of each of the four bacterial pathogens and also the current state-of-development of WGS-based strategies for epidemiological surveillance of FWD.
4 1.2. FWD pathogens: a brief overview on diversity, molecular epidemiology
and impact on human health
1.2.1. Listeria monocytogenes
Listeria spp. is a group of Gram-positive bacteria encompassing the following
species: L. monocytogenes, L. innocua, L. welshimeri, L. seeligeri and L. ivanovi. The one that is most associated with human diseases is L. monocytogenes. Listerioses, i.e., the disease caused by L. monocytogenes, can occur in healthy people, but has been of particular concern in immunocompromised people, such as patients with HIV, elderly, children and pregnant women (6, 7). Moreover, as L. monocytogenes can cause severe disease, such as septicemia or meningitis, it is normally associated with high hospitalization rate (>90%) and the case-fatality rate is the highest (>30%) among the food and waterborne bacterial pathogens(8-11).
In Europe, cases of human listerioses have been increasing since 2000 (7, 11). A recent European Union (EU) report, carried out by the European Food Safety Authority (EFSA) and the European Centre for Disease Prevention and Control (ECDC) indicated a notification rate of 0.52 cases per 100000 population, represents a 30 % increase from 2013 to 2014 (11). In Portugal, the number of cases of listeriosis is also thought to be increasing. For instance, a mean of 0.14 cases per 100 000 inhabitants have been estimated in 2003, but a total of 0.23 cases per 100,000 inhabitants have been described four years later (12, 13).
L. monocytogenes is present in the environment (water, soil and vegetation)
and can survive in food products and also in food processing environments. Moreover, besides this remarkable capacity to grow in multiple environments (including wide rage temperatures), it has the ability to form biofilms (3, 14). As a consequence, these traits are believed to be key factors contributing for its well-known persistence in food or food-processing environments for a long time (3) thus potentiating the risk of contamination of food products and ultimately the risk of human infection(15)´.
L. monocytogenes has been historically classified by lineages and serogroups/serotypes (7). At the present, it can be divided into four evolutionary lineages(7, 16), serotypes and four PCR serogroups(17, 18). Still, the four different lineages are normally associated with different groups of serogroups/serotypes. Lineage I includes serotypes 1/2b, 3b, 3c, 4b, 4d, 4e and it is the lineage most associated not only with outbreaks and sporadic cases, but also with cases of invasive and febrile gastroenteritis. Lineage II is constituted by the serotypes 1/2a, 1/2c, where serotype 1/2a is more associated with human disease. Lineage III is essentially composed by serotype 4a and 4c strains, but also some strains of serotype 4b. This lineage has been mainly associated with cases of animal infection. Finally, Lineage IV, which resulted from the recent reclassification of Lineage IIIB, is more associated with ruminants(7, 19, 20). Overall, L. monocytogenes strains from serotypes 1/2a, 1/2b and 4b are the most commonly associated with human infection (98% cases)(3). Moreover, besides the current existence of an important knowledge on the association between specific L. monocytogenes “sub-types” and their impact on the human disease, it is also known that the establishment of methods to detect L. monocytogenes and further sub-typing this pathogen is critical for rapid outbreak detection, sources identification and tracking of transmission routes (14, 21, 22). In particular, epidemiological
5 investigations supported by geno- and phenotyping data have been important not only for solving outbreaks, but also because they provide critical information about food sources of listeriosis and contribute to unveil food safety gaps (22). At this particular, the pulsed-field gel electrophoresis (PFGE) technique, which has been the gold standard method for L. monocytogenes typing, has historically proved to be very useful in detecting and controlling listeriosis clusters(21-23). PFGE, which is based on total bacterial DNA restriction patterns (24), has in general been displaying better discriminatory power and/or inter-laboratory reproducibility than other typing methods that have been tested, such as multilocus sequence typing (MLST), multiple-locus variable number tandem-repeats analysis (MLVA) (15) or PCR-based serotyping (17, 25). For instance, the seven loci-based MLST schemes tried for L. monocytogenes demonstrated consistent results with the PFGE typing, but provides insufficient discriminatory power for outbreak investigations (9), whereas MLVA, which relies on determining the number of tandem repeat sequences at different loci in the bacterial genome, has higher discriminatory power compared to MLST, but is known not to be 100% reproducible between laboratories. Finally, the multiplex PCR molecular serotyping scheme that is currently under application for L. monocytogenes (9) has a limited use for outbreak investigation due to its poor discriminatory power(22, 25), but is considered important since, as referred above, some serotypes are highly associated with cases human infection. Still, PFGE is labor-intensive, technically demanding and time-consuming. Moreover, strains known to be highly genetically distant may display an indistinguishable PFGE profile, which often hampers the differentiation between outbreak and sporadic cases of listeriosis (22). This is even more problematic considering that human listeriosis outbreaks are often difficult to detect, since cases may be geographically dispersed and may occur over long periods of time, again partially as a consequence of the persistent nature of L. monocytogenes in the food and food-processing environments (22, 23). Overall, the lack of powerful discriminatory power of the traditional methods and the need of international surveillance of L.
monocytogenes has rapidly driven researchers to implement methods not only
providing high resolution and reproducibility, but that can be harmonized and universally applied. All this requirements can be fulfill through the application of the powerful epidemiological surveillance tool, the whole-genome sequencing (WGS) (21). On this behalf, due to the high impact on public health of L. monocytogenes and its population and genetic traits, the change for the application of WGS-based methods at worldwide scale is expected to be fast.
1.2.2. Salmonella enterica.
Salmonella is a genus of Gram-negative bacteria that is composed by two
species: S. bongori and S. enterica. While S. bongori is not associated with human infection, S. enterica is a common cause of both animal and human diseases(3, 26). The latter can be subdivided in six subspecies, namely S. enterica subsp. enterica (I),
S. enterica subsp. salamae (II), S. enterica subsp. arizoanae (IIIa), S. enterica subsp. diarizonae (IIIb), S. enterica subsp. houtenae (IV) and S. enterica subsp. indica (VI),
where S. enterica subsp. enterica is the one responsible for the highest number of cases of human disease(27-29). S. enterica is normally acquired through food, namely poultry, meat or eggs (11) and represents a significant global public health concern due
6 to its associated morbidity and mortality rates (30). In fact, this important human foodborne pathogen caused 88,715 confirmed cases of human disease in EU in 2014, with a case fatality rate of 0,15 (11). In Portugal, in the same year, 261 confirmed cases were reported. In comparison to other EU countries, Portugal had the lowest incidence of Salmonellosis, but the rate of hospitalization was high. Of note, although people with diarrhea due to a S. enterica infection usually recover completely, infections with this human foodborne pathogen can progress to severe clinical onsets, which are more often developed in young children, older adults, and people with weakened immune systems (31-33).
On behalf of routine epidemiological surveillance S. enterica infections, public health laboratories around the world have historically employed serotyping. The reference method for serotyping of S. enterica uses the White-Kauffmann-Le Minor scheme (recently reviewed by Grimont and Weill (2007); https://www.pasteur.fr/sites/www.pasteur.fr/files/wklm_en.pdf), which relies on surface antigen identification using polyclonal antiserum to determine both lipopolysaccharide (LPS) (O) and flagellar (H) antigens. This method, which has been pivotal not only for the understanding the global epidemiology of S. enterica for decades, but also for tracking common sources of infection during outbreak investigation (e.g., contaminated food or an infected animal), can currently divide this human foodborne pathogen in more 2300 serotypes/serovars (34). Importantly, for multiple serotypes, there is nowadays an important knowledge on their distribution and association with human disease. In fact, while some serotypes are essentially found in a scarce number of animals or geographical locations, others are known to be able to infect many different hosts or to be widespread around the world (35, 36). On the other hand, while the degree of the severity of the S. enterica-related human diseases can somehow be associated with specific serotypes, one also know that some serotypes, such as the
Salmonella serotype Typhimurium and Salmonella serotype Enteritidis, are historically
linked to highest number of cases of human infection (5). In Portugal, the National Reference Laboratory (NLR) for Gastrointestinal Infections (GI) of the Department of Infectious Disease at INSA received more 6000 S. enterica isolates between 2000 and 2013. Serotyping classification identified S. Enteritidis and S. Typhimurium as the more common serotypes, which is concordant to what has been observed in most Europe countries (11, 37).
Nevertheless, this serotyping scheme is expensive and intensive, and it can take several days to complete (38). Moreover, it is known that serotyping does not provide per si an accurate perspective over the evolutionary trends of S. enterica, as antigenic profiles might not reflect the genetic relationships among the different serovars (29), which is concordant with the high genomic diversity (marked by the frequent occurrence of horizontal gene transfers) found among S. enterica isolates (3). In this context, and also as a means to increase the throughput and discriminatory power associated with serotyping, public health laboratories normally employ other typing methods, in particular DNA-based methods such as PFGE and MLVA, and less often MLST (3, 39). While these molecular techniques have been unequivocally useful for providing additional information to understand common serotypes and, importantly, for identifying, investigating, and tracing S. enterica outbreaks (40, 41), it is at the
7 present well accepted that WGS-based methods are needed to properly perform epidemiological surveillance of this important pathogen (3, 29, 42).
1.2.3. Escherichia coli
Escherichia coli is a Gram-negative bacterium that is a normal inhabitant of the
human gastrointestinal tract, more specifically, the colon mucosa. Although most strains of E. coli are harmless, some of them have acquired virulence factors that allow them to infect healthy people, and further cause disease (3, 43). When E. coli is pathogenic, it has been normally associated not only with diarrhea, often hemorrhagic, but also with cases of illness outside of the intestinal tract. Cases of severe disease, including severe diarrhea, haemorrhagic colitis (HC) and the life-threatening hemolytic uremic syndrome (HUS), can progress to more complicated clinical frames and eventually be fatal (43). This entero-pathogenic E. coli can be transmitted after consuming contaminated food or water, or through contacting with infected animals or people (44).
E. coli is a highly diverse and versatile pathogen marked by a huge capability to
exchange genetic information as a means to continuously adapt to environmental changes. As such, a plenty of E. coli diarrheagenic “pathotypes” have been described, which can in general be distinguished by the diversity of virulence traits, namely the ability to produce specific toxins and/or proteins associated to the colonization and invasion of eukaryotic cells. Diarrheagenic E. coli have been historically categorized in the following pathotypes: enteropathogenic E. coli (EPEC), enterotoxigenic E. coli (ETEC), enterroaggrative E. coli (EAEC), enteroinvasive E. coli (EIEC), diffusely adherent E. coli (DAEC) and enterohemorrhagic E. coli (EHEC) (3, 45, 46). The latter, which may also be generally referred to as Verocytotoxin-producing E. coli (VTEC) or Shiga toxin-producing E. coli (STEC), is able to produce a toxin called Shiga toxin and is the pathotype most commonly associated with foodborne outbreaks of HC and HUS (3, 46).
In Europe, 5955 cases of VTEC human infection have been reported in 2014, although the number seems to be decreasing since 2011 (11). In Portugal, the NRL-GI at INSA receives E. coli strains from primary laboratories spanned throughout the country, which are routinely tested for the presence of specific virulence factors (including Shiga toxins). For instance, from the 1038 strains of E. coli received between 2002 and 2012, 497 were positive for the presence of one or more pathogenic factors. In particular, these pathogenic E. coli could be classified in ETEC (36.6%), EAEC (26,7%) and VTEC (25,3%), with a frequency lower are EPEC (8,5%) and the cases when the pathogenicity of patotypes ETEC/VTEC were detected in simultaneous are representative of 2,9% (47).
While the discrimination between commensal and pathogenic E. coli cannot be straightforwardly performed by either plate isolation or biochemical assays(3), the further determination of the molecular traits behind the phenotypic differences displayed by distinct diarrheagenic E. coli pathotypes may also not be a trivial task because strains may present a huge variation in their arsenal of the virulence factors. In fact, the genome of E. coli is very plastic, so genetic traits underlying pathogenicity
8 may be encoded in genetic elements as diverse as transposons, phages, plasmids or the so called pathogenicity islands. Because of this, it is critical to set up geno- and phenotyping methods that allow not only a reliable routine epidemiological surveillance of E. coli infections, but also a proper characterization of the repertoire of virulence traits carried by phenotypically different strains, in particular by clinical-associated strains (e.g, genes associated with antibiotic resistance; specific toxin-encoding genes, etc) (48). In order to achieve this, microbiological laboratories have traditionally been applying PCR-based assays (usually through multiplex schemes) for detecting specific virulence factors whose presence is known to correlate with specific diseases or clinical outcomes. For instance, the Shiga toxin type 2 (stx2) carried by some STEC has been strongly associated with confirmed cases of human diseases and with severe illness (3). On the other hand, most laboratories have also implemented methods for serotype determination, again taking advantage of the well-known association between specific serotypes and disease severity. A remarkable example stands for the O157:H7 serotype, which has been the most associated with outbreaks (3, 49, 50).
Noteworthy, public health laboratories have however historically employed other techniques for subtyping of diarrheagenic E. coli, namely restriction fragment length polymorphism (RLFP), PFGE, MLVA, single locus sequence typing (SLST) and MLST. Still, even though the latter methodologies have provided the basis of our current knowledge on the worldwide molecular epidemiology of STEC (in particular MLST, which has been routinely performed throughout the world), none of them has the same potential as WGS to fulfill the requirements (high discriminatory power, multiple traits prediction, etc) needed for a proper surveillance of this important human pathogen.
1.2.4. Campylobacter jejuni
Infections by Gram-negative Campylobacter spp. are considered the most prevalent cause of human bacterial gastroenteritis worldwide, with the Campylobacter
ejuni species estimated to be responsible by ~90% of the cases (3, 51, 52). C. jejuni is
mostly associated with sporadic cases of human infection, but rare outbreaks have been reported. While the latter have been associated with consumption of contaminated water or raw milk (53), most of C. jejuni human infections are normally due to manipulation and consumption of infected poultry (3, 52, 53). In general, C.
jejuni causes mild and self-limiting gastroenteritis that can be associated with fever,
vomiting, headaches and/or abdominal pain with watery or bloody (52, 54). Still, several cases have been described where C. jejuni infections can lead to septicemia, extra-intestinal diseases and neurological sequelae (52). In this regard, blood-invading
C. jejuni strains are of particular concern, being targets of several studies focused on
understanding the mechanisms underlying their potential enhanced virulence (55). Remarkably, it is estimated that approximately 1% of European population develops Campylobacteriosis every year (52). For instance, 236 851 confirmed cases of human Campylobacteriosis have been confirmed in 2014 in the European Union (EU) (11), and this incidence is likely increasing (11, 53). But, C. jejuni epidemiology is only incompletely understood as efforts to shed new lights on this area are normally difficult to carry out because the large majority of infection cases are sporadic and
9 remain unreported (56). In particular, concerns have been raised since Campylobacter spp. strains are often resistant to one or multiple antibiotics of human use, a trend that is believed to be emerging (54). In Portugal, the NRL-GI at INSA receives
Campylobacter spp strains from laboratories spanned throughout the country, including
17 primary laboratories that routinely send all isolates together with clinical and demographic data, thus constituting a network for surveillance of campylobacteriosis. These strains (e.g., more than 200 were received in 2014) are routinely genotyped (by MLST) and tested regarding their antibiotic susceptibility profile, thus allowing getting insight on the Campylobacteriosis trends in Portugal, as well as on the evolution of the rates of antimicrobial resistance.
Public health laboratories have traditionally performed several geno- and phenotyping methods (such as, MLST, PFGE, antibiotic susceptibility resistance testing and methods to specifically detect virulence genes) to reinforce the surveillance of
Campylobacter spp. infections (54, 56). These methods have been shown to be
important working tools for the understanding the epidemiology of C. jejuni infections, and have provided relevant contributes to the current knowledge on the nature of C.
jejuni infections in human patients and their potential sources. Nevertheless, these
methods, in particular the worldwide applied MLST scheme (57), show in general low discriminatory power, which have been hampering to discriminate genetically related isolates (56). In this context, it is necessary to develop and implement methods that allow not only an efficient epidemiological surveillance efficient, but also an accurate comparison of molecular data between different areas, which is relevant considering the worldwide scale of Campylobacteriosis (56). The high discrimination power and the expected easier padronization of WGS place this technique in the frontline to become the gold standard typing method for surveillance of C. jejuni infections (53, 56).
1.3. General overview on the current development of WGS-based strategies for epidemiological surveillance of Food and Waterborne Diseases. The four bacterial pathogens under study presents some differences on their biological, genomics and evolutionary traits that should be briefly highlighted as they naturally impact the development, implementation and the upcoming routine application of genome-based typing tools for surveillance of the associated FWD. First of all, these bacteria have considerable differences in their genome size. In fact, taking into account the hundreds of genomes available at the NCBI database, while C. jejuni presents a mean genome length of 1.7 Mbp (~1650 genes) and L. monocytogenes displays a ~3.0 Mbp genome (~2900 genes), E. coli and S. enterica present largest genomes of around 5.2 Mbp (~5000 genes) and 4.8 Mbp (~4500 genes), respectively. Not surprisingly, this important difference has a direct influence on the cost, which is a relevant decisive factor when switching to novel typing techniques. In fact, the number of samples that can be processed in a single WGS run is inversely proportional to the genome size (2). Depending on the amount of samples to be process, the genome size also directly impacts the data storage, sharing and analysis procedures, thus influencing the computing capacity requirements needed to handle each pathogen-derived WGS data (2). Secondly, the relative weight of the distinct evolutionary forces (i.e., horizontal gene transfer, single point mutations, homologous recombination events, etc) that may drive the evolutionary adaptation of these bacteria, and, consequently, shape their genome content and organization, is also expectedly
10 different. This has a natural impact on the global genetic diversity of the organisms, and as a consequence, on the laboratory strategies to be carried out while routinely monitoring the epidemiological trends of these worldwide distributed human pathogens. For instance, epidemiological and comparative genomic studies have been pointing that much of the diversity among C. jejuni isolates is likely generated by horizontal genetic exchange, with recombination presumably having a considerable high role on diversification than mutation (58-60). Also, the highly diverse S. enterica and E. coli species, which are believed to be even more profoundly marked by abundant homologous recombination events and by the exchange of large proportions of horizontally acquired genomic regions (the so called “genomic islands” that are often enriched by antibiotic resistance and virulence-associated genes), present genomes with an extensive mosaic nature (61, 62). On the other hand, L. monocytogenes, which is also a highly heterogeneous species, it presents a clonal structure (as revealed by the existence of four evolutionary lineages) that seems to impose less constraints to the development of WGS-based typing methodologies (18, 21). Taking all together, all these distinct evolutionary and genomic traits also naturally implicate that these bacteria present rather different dynamics on their pan-genome (i.e., the set of genetic loci observed in one species) mostly to what refers to the size and content stability. This is highly relevant because genome-based typing methods are currently being designed in order to target one or both parts of the pan-genome, i.e., the core-genome (i.e., the subset of loci that is theoretically common to all members of a given species) and the accessory genome (i.e., the subset of loci that is not common to all strains of one species) (11). In fact, since the exact size of the accessory- and core- genome is dependent on the amount of genomes under analysis and this amount is constantly increasing due to huge progress in the WGS technologies, researchers have been struggling to understand: i) whether some species have more “open” pan-genome structure than others (63, 64) and ii) which typing strategies (e.g. core-genome- or pan-genome-based strategies) confers enough discriminatory power for epidemiological surveillance (namely outbreak detection) of each FWD pathogen. As such, huge research advances have been done towards the development of core genome multilocus sequence typing (cgMLST) or whole genome multilocus sequence typing (wgMLST) schemes, which refers to strategies scaling-up (in terms of the amount loci analysed) the rationale behind the traditional seven-loci MLST (2, 21, 22, 38, 59, 65, 66). Alternatively, methods have been tried to rapidly infer phylogenies based on single nucleotide polymorphisms (SNPs), which are expected to be become particularly useful for discriminating closely related isolates that might be linked to the same public health event (2). Still, although WGS-based approaches tested for these four human pathogens unequivocally have the potential to offer an unprecedented discriminatory power for routine surveillance and outbreak investigation of FWD (2, 21, 22, 38, 59, 65, 66).no scheme have yet become universally applicable and standardized, thus implicating that public health laboratories should make efforts to test, validate and ultimately standardize novel WGS-based typing approaches that are continuously being made available. Moreover, as stated above, during the gradual switch from traditional typing to WGS typing-based surveillance, it is also necessary to check the consistency between data retrieved by the both schemes, thus avoid losing historical typing data generated by traditional pheno-genotyping (5).
11 Taking all together, on behalf of the ongoing/upcoming worldwide transition from the traditional typing methods to the application of WGS for surveillance of Infectious Diseases, this study, aimed at starting transition for the surveillance of four FWD pathogens in the Portuguese NIH, has the following specific objectives:
i) to apply and test multiple bioinformatics tools for in silico prediction/capture of the typing data routinely provided by the traditionally applied pheno- and genotyping techniques, as a means both to give the first step towards their replacement and to ensure backwards compatibility with “historical” typing data;
ii) to implement novel genome-based typing strategies, as a means to start testing their applicability for the future WGS-based routine surveillance of FWD diseases.
iii) to get insight on the potential offered by WGS to predict bacterial virulence and antimicrobial susceptibility profiles.
12
2. Material and Methods
2.1. Bacterial FWD pathogens
This work is focused on the implementation of genome-based typing tools (in place of traditional geno- and phenotyping methods) for surveillance of FWD. To achieve this, we took advantage of the vast strain collection of the National Reference Laboratory for Gastrointestinal Infections (NRL-GI) of the National Institute of Health (NIH) Dr. Ricardo Jorge to select isolates of four important FWD bacterial pathogens: L. monocytogenes (n = 35), C. jejuni (n= 13), S. enterica (n = 16) and E. coli (n = 25). The rationale behind the strain selection criteria is presented throughout the thesis, but varied with the bacterial species under evaluation, and it was in general dependent on: i) the study design outlined for each pathogen; ii) on the data available for each isolate (e.g., geno- and phenotyping data, epidemiological data and clinical data); and iii) the overall cost of the study.
2.2. Data from traditional geno- and phenotyping methods
The NRL-GI at INSA routinely performed multiple techniques for the characterization of FWD pathogens (37, 47, 54, 67, 68). In order to compare the results of the traditional typing methods with the results generated with novel genome-based typing tools, geno- and/or phenotyping data (as well as clinical data or other epidemiological data, when available) regarding the bacterial isolates evaluated in the present study was preliminarily collected and will be exposed throughout the text when the need arises. Still, a brief overview on the available data is provided in the Table 2.1.
Table 2.1. Overview on the data available for the isolates under evaluation.
2.3. Whole-genome sequencing
Genomic DNA has been previously extracted from pure cultures of each isolate under study using a silica-based automatic DNA extractor (NucleiSENS® easyMAG®, bioMeriux). Exceptionally for L. monocytogenes, due to its Gram positive nature, an extra lysis step with lysozyme (10mg/ml) at 56ºC for 30 minutes was carried out before DNA extraction. The yield and quality of all purified DNA samples was subsequently assessed by using the Qubit assay (Quanti-it dsDNA Assay Kit, Broad Range; Lifetechnologies, MA, USA) and agarose gel (0.7%) electrophoresis, respectively. High-quality DNA samples were then used to prepare Nextera XT dual-indexed Illumina libraries that were further subjected to paired-end (2 x 250bp) sequencing on a
13 Next-generation Sequencing (NGS) MiSeq equipment (Illumina Inc., San Diego, CA, USA) available at the Portuguese NIH. All NGS-related procedures were carried out by a sequencing-specialized team (Innovation and Technology Unit), who have previously optimized the procedures according to the manufacturer’s instructions. Of note, L.
monocytogenes isolates were exceptionally subjected to Illumina WGS outside the
Portuguese NIH, as they are also being studied on behalf of an international project run by the European Centre for Disease Prevention and Control (ECDC).
2.4. Quality assurance / Quality control (QA/QC) meausres on the raw sequencing data
Raw sequence data generated in each Illumina run was firstly analyzed regarding the overall mean quality obtained across samples (as provided in the Miseq report). The quality of each base within a given read generated by automate DNA sequencing is normally expressed through the “PHRED quality score” as
where refers to the base calling error probability. For instance, a PHRED score above 30 (which is normally considered as “high quality”) means that the chance that a given base is called incorrectly is less than 1 in 1000, while a PHRED score of 20 (the typical minimum quality cut-off) indicates that there is a chance of 1 in 100 that the base has been wrongly called i.e., sequenced. Although the overall mean quality provides a good indication of the run performance, raw sequence data (i.e., sequence reads in fastq format files) generated for each bacterial isolate was
subsequently inspected in detail by using FastQC
(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). This software provides a set of well-established statistics on reads quality in very comprehensive way, and thus, it was applied to check reads quality before (raw reads) and after the implementation of quality measures. For each of one of the parameters scrutinized in each fastq file (e.g., “Per Base Sequence Quality”, “Per Base Sequence Content”, “Per Sequence GC Content”), FastQC provides a quick report highlighting which statistics should be improved, thus guiding about the optimal downstream read processing measures to implement for each file. In this study, bioinformatics measures to improve the quality of raw sequence data have been carried by using the command-line-based FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Finally, two additional features have been checked regarding the data quality: the guanine-cytosine content (GC content) and “Depth of coverage”. The mean value % GC, which is expectedly of similar range for same-species bacterial strains, was inspected as it could be indicative of contamination. On the other hand, the depth of coverage, i.e., the average number of times each base in the genome is contained in individual raw reads (ECDC), is a critical aspect to take into account when evaluating the confidence of NGS data. As such, we inferred the theoretical mean “depth of coverage”, through the formula:
14 Calculations were performed before (raw reads) and after the implementation of quality measures (processed reads), as a mean to evaluate not only the impact of reads processing on the data confidence, but also whether the expected depth of overage fits the desired values. Of note, while an optimal minimum coverage cut-off is difficult to establish as it may depend, for instance, on the species under evaluation and the ultimate goal of the WGS, values above 50x-70x are normally pointed to provide enough quality for routine surveillance.
2.5. de novo assembly
After improving the read’s quality, we proceeded to the assembly of draft genomes. In this work, we selected the two mostly worldwide applied de novo assembly software, SPades (version 3.8.1) (http://spades.bioinf.spbau.ru/) (69) and Velvet (version 1.2.10) (70). Velvet runs were optimized by taking advantage of the VelvetOptimiser script version 2.2.5 (http://bioinformatics.net.au/software.velvetoptimiser.shtml). For Spades runs, K-mer length were also automatically set in order to optimize the contigs length and the settings “—careful” and “only-assembler” were chosen. Run statistics comparisons were carried out by checking the performance of all assembly outputs, which mostly relied on the evaluation of number of contigs, the N50 value and length of the largest contig/scaffold.
2.6. de novo assembly-based genome alignments
After draft genome assemblies, the genetic relationships between isolates of each FWD pathogen were studied by performing multiple genome alignments followed by phylogenetic inferences. In this study, we took advantage of two widely used genome alignment software: Harvest suite 1.1.2 (https://harvest.readthedocs.io/en/latest/) (71) and Mauve software (version 2.3.1) (http://darlinglab.org/mauve/mauve.html) (72, 73). Basically, Harvest tools were applied to perform core-genome SNP-based alignments from draft complete genome sequences followed by both rapid visualization and extraction of SNP profiles (here referred as a “de novo assembly-based core-genome SNP strategy”). In order to potentiate the use of high-confident SNPs, only variant calls outside regions more prone to technical artefacts (namely, regions enrolling Local collinear Blocks smaller than 200bp, and regions where < 50% ID or > 20 indels are observed within the 100 bp window where the SNP falls) were considered for downstream analysis. Of note, given its rapid performance, Harvest tools were also used to perform a draft insight on the impact of using distinct de novo assemblers on the core-genome phylogenetic tree topology. On the other hand, MAUVE was strictly used for visualizing and confirming the homology of particular genomic regions and also for performing pairwise alignments of the draft genome sequences of isolates presenting high genomic similarity (e.g., isolates collected from the same patient). For the latter, both whole-genome and core-genome alignments were constructed by using the progressiveMauve and stripSubsetLCBs algorithms of Mauve software, respectively. Core-genome regions where extracted when sequences aligned over at least 500 bp and further concatenated to build core-genome alignments.