Escola de Engenharia
Pedro Miguel Azevedo Veloso
Improving derived
Listeria
phage endolysins
properties at low temperatures
Escola de Engenharia
Pedro Miguel Azevedo Veloso
Improving derived
Listeria
phage endolysins
properties at low temperatures
Tese de Mestrado
Mestrado em Bioengenharia
Trabalho realizado sob a orientação de
Doutor Leon D. Kluskens
É autorizada a reprodução parcial desta tese apenas para efeitos de investigação, mediante declaração escrita do interessado, que a tal se compromete,
Universidade do Minho, __ /__ /____
Aknowledgements
Agradecimentos
x
Agradecimentos
A vida é uma longa caminhada e, apesar de este ser apenas mais um capítulo que agora se encerra, dela fazem parte várias pessoas. Pessoas essas que de forma direta ou indireta me ajudaram a conseguir alcançar objetivos, metas. É a essas pessoas que expresso, de seguida, os meus sinceros agradecimentos por me terem apoiado ao longo deste projeto.
Em primeiro lugar quero agradecer ao meu orientador, Doutor Leon Kluskens, por me ter dado a oportunidade de estar presente num projeto científico ambicioso, por todo o conhecimento e conselhos transmitidos, por ter permitido aumentar o meu gosto pela área da biologia molecular assim como pela disponibilidade que sempre demonstrou ao longo de todo este projeto.
Agradecer à Graça, que desde o primeiro dia me ajudou de forma incansável, demonstrando sempre enorme disponibilidade, espírito de entreajuda, apoio e compreensão mesmo nos momentos mais difíceis. Obrigado pelo conhecimento e experiência transmitidos.
Agradecer ao Hugo, que apesar de ter entrado um pouco mais tarde no projeto, me ajudou com todo o seu conhecimento, pragmatismo e criatividade ajudando a contornar obstáculos que, por diversas vezes, se colocaram no caminho.
Aos meus colegas de laboratório: Luís, pelos conselhos prestados, boas conversas e momentos de bom humor; à Catarina pela sua grande simpatia; ao José pelas boas conversas tecnológicas que tivemos. À Marta e Patrícia, colegas de mestrado, pela sua prontidão e disponibilidade em ajudar. Aos meus amigos de sempre, Pedro, Rui, João e Maria João pelo apoio prestado e pela boa disposição em todos os momentos.
À Ana, meu ombro amigo, meu porto de abrigo, minha conselheira. Obrigado teu apoio incondicional e preciosos conselhos transmitidos durante este capítulo da minha vida.
E por fim, o mais importante, à minha família, mãe, pai, irmã e avós. Obrigado por todo o esforço que fizeram para que conseguisse alcançar os meus objetivos pessoais e académicos. Obrigado por todo o apoio nos bons e maus momentos. Obrigado por me terem incutido desde sempre bons valores humanos. Espero um dia conseguir retribuir-vos tudo aquilo que me deram. Ser-vos-ei eternamente grato!
Abstract
xiv
Abstract
Listeria monocytogenes is a Gram-positive opportunistic pathogen that can grow in a wide variety of conditions and is responsible for listeriosis, a potential fatal disease, associated to the ingestion of contaminated food. The concerns about the upsurge of widespread reported cases, combined with emerging antibiotic-resistance amongst pathogenic bacteria, such as L. monocytogenes, demand for the development of novel preservation techniques that ensure the safety of food products.
Endolysins, which originate from virulent bacteriophages, are responsible for the hydrolysis of the covalent bonds in peptidoglycan layer of the host cell. These enzyme properties represents a good alternatively approach against Gram-positive foodborne pathogens without altering the organoleptic properties of food products.
However, in most of the cases, the activity and stability of naturally occurring enzymes is significantly lower than the biotechnological industry needs. Besides, there is a lack of research advances in lytic activity improvements of endolysins in food storage conditions.
The experimental work developed in the scope of this thesis aimed at directing endolysin activity towards refrigeration temperatures against L. monocytogenes through the use of directed evolution strategies – error-prone PCR and cryodrilling.
Different attempts were done for the isolation of listerial phages from livestock industries effluents and consequently identification and improvement of lytic activity of its derived endolysins.
An in silico analysis of two different lysins – Ply500 and Ply511 – were performed to provide contextualization about their structure and domains. Although both proteins possess modular structure, Ply511 has a central catalytic domain and a not well characterized binding domain which contrasts to Ply500 domain organization. Protein expression in large and micro-scales of wild-type proteins was successfully done and confirmed by performing antibacterial tests against L. monocytogenes 5725.
To enhance the activity of endolysins against L. monocytogenes cells, modified endolysins were constructed by amplifying their sequences using error-prone PCR technique and cloning into pQE-30 vector. The cloned vectors were transformed in E. coli JM109 competent cells, however no colonies were obtained.
At the same time, using PlyP100 endolysin, a second approach based on the biotic interaction between phage-host at successively temperature was done to promote phage adaptation and consequently enzymatic evolution.
xv
Resumo
Listeria monocytogenes é um agente patogénico oportunista Gram-positivo, responsável por provocar listeriose, doença potencialmente mortal associada ao consumo de alimentos contaminados. A preocupação inerente à sua capacidade de sobreviver numa grande variedade de condições, o crescente número de surtos da doença e o aumento da resistência a antibióticos obrigam a que novas estratégias de conservação e preservação dos alimentos sejam desenvolvidas.
Endolisinas derivadas de fagos são enzimas responsáveis pela lise das células do hospedeiro. Uma vez que não alteram as propriedades organoléticas dos alimentos, o uso destas enzimas representa uma boa alternativa na eliminação de agentes patogénicos Gram-positivos.
Contudo, a baixa atividade e estabilidade das enzimas no seu estado natural torna-se incompatível com as necessidades industriais.
Este trabalho experimental visou o melhoramento das propriedades líticas das endolisinas a temperaturas de refrigeração recorrendo a técnicas de evolução direta – error-prone PCR e cryodrilling.
Numa primeira abordagem foram efectuadas tentativas para o isolamento de fagos de Listeria a partir de efluentes de indústria pecuária com posterior identificação e melhoramento das propriedades líticas das respetivas endolisinas. No entanto, as tentativas não foram bem-sucedidas. Foi efetuada a análise bioinformática das duas diferentes endolisinas – Ply500 and Ply511 – para se obter informações precisas sobre a sua estrutura. Apesar das duas proteínas possuírem uma estrutura modular, Ply511 apresenta um domínio catalítico central com função desconhecida e por conseguinte, pouco caracterizado, relativamente à organização modular de Ply500. A expressão das respetivas endolisinas wild-type em larga e micro escalas foi efetuada com sucesso e confirmada através de testes antibacterianos contra L. monocytogenes 5725.
Para melhorar a atividade lítica das respetivas endolisinas, as sequências foram amplificadas por error-prone PCR, clonadas no vetor pQE-30 e transformados em células competentes E. coli JM109. No entanto, não foram obtidas quaisquer colónias.
Ao mesmo tempo, usando a endolisina PlyP100, uma nova abordagem baseada no princípio de interação biótica entre fago-hospedeiro, foi efetuada a temperaturas sucessivamente mais baixas de forma a promover a evolução/adaptação do fago e consequentemente da endolisina.
xvi
xviii
Index
Agradecimentos ... x
Abstract ... xiv
Sumário ... x
List of abbreviations ... xxiv
List of figures ... xxviii
List of tables ... xxiv
CHAPTER 1 – Introduction and Background ... 1
1. Listeria monocytogenes and listeriosis ... 3
1.1. Adaptation mechanisms ... 4
1.2. Antimicrobial resistance ... 4
2. Bacteriophages as potential means to battle listeriosis ... 5
2.1. Listeria phages ... 7
3. Endolysins ... 9
3.1. Different types of endolysis ... 9
3.2. Endolysin structure ... 10
4. Strategies for endolysins improvement ... 11
4.1. Phage adaptation “cryodrilling” ... 12
4.2. Directed evolution: site-directed mutagenesis Vs. random mutagenesis ... 12
4.3. Site-directed mutagenesis ... 13
4.3.1. Domain swapping ... 14
4.4. Random mutagenesis for in vitro directed enzyme evolution ... 15
4.4.1. Chemical mutagenesis... 15
4.4.2. Mutator strains ... 16
4.4.3. Site-saturanting mutagenesis ... 16
4.4.4. Error-prone PCR ... 17
5. Advances in molecular engineering of endolysins ... 18
5.1. Advances using site-directed mutagenesis ... 19
5.1.1. Advances using domain-swapping ... 19
5.2. Advances using random-mutagenesis ... 20
xix
CHAPTER 2 – Materials and Methods ... 25
1. Bacterial strains, endolysins and plasmids ... 25
2. Listeria phage isolation ... 26
2.1. Non-lysogenic strains selection ... 26
2.2. Effluent samples and phage detection ... 27
2.3. Phage isolation and propagation ... 27
3. Protein production ... 28
3.1. Large-scale production ... 28
3.1.1. Culture and induction ... 28
3.1.2. Cell lysis... 28
3.1.3. Purification ... 28
3.1.4. Polyacrylamide gel electrophoresis (SDS-PAGE) ... 29
3.1.5. Protein quantification ... 29
3.2. Micro-scale protein production experiments ... 30
3.3. Host preparation and antibacterial assays ... 30
4. Bioinformatic tools ... 31
4.1. In silico analysis of bacteriophage endolysins ... 31
4.2. Primers design ... 32
5. Cloning ... 32
5.1. Polymerase chain reaction (PCR) techniques ... 33
5.1.1. Error-prone PCR ... 33
5.1.2. Colony-PCR ... 33
5.2. Plasmid extraction and digestion ... 34
5.3. Ligation ... 35
5.4. Agarose gel electrophoresis ... 35
6. Transformation ... 35
6.1. Competent cells ... 36
6.1.1. Chemio-competent cells ... 35
6.1.2. Electrocompetent cells ... 36
6.2. Transformation and plasmid replication ... 36
6.2.1. Heat-shock transformation ... 36
xx
7. Directed evolution screening assay ... 37
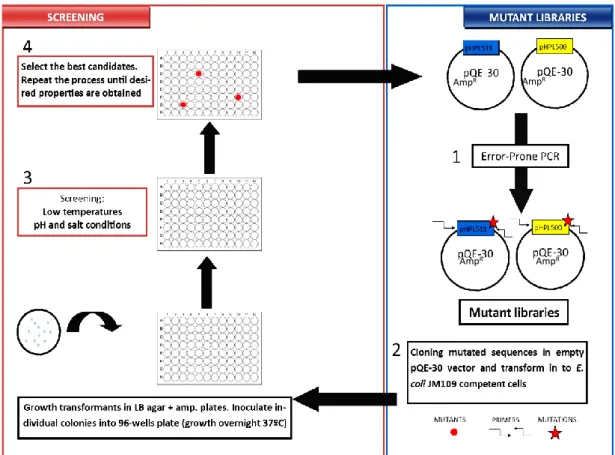
7.1. Mutant libraries ... 37
7.2. Screening tests ... 37
7.2.1. Low temperature tests ... 38
7.2.2. High salt conditions ... 38
8. Cryodrilling ... 39
CHAPTER 3 – Results and Discussion ... 41
1. Background ... 45
2. Listeria phage isolation... 45
3. In silico analysis of bacteriophage endolysins... 46
4. Protein expression preliminary tests ... 48
4.1. Large-sclae protein production ... 48
4.2. Micro-scale protein expression and preliminary tests ... 48
4.3. Lytic activity – Low temperatures vs. Room temperatures ... 51
5. Cloning ... 52
5.1. Gene amplification ... 52
5.2. Plasmid linearization and ligation ... 54
5.3. Transformation ... 54
6. Cryodrilling ... 56
CHAPTER 4 – Conclusions and Future Perspectives ... 59
1. Conclusions and future perspectives ... 63
Bibliography ... 66
Annexes ... 76
xxiv
List of abbreviations
BCA - Bicinchoninic acid assay CaCl2 – Calcium chloride
CBD – Cell-Binding Domain
CEB – Centre of Biological Engineering DLA – Double-layer agar
DNA – Desoxyribonucleic Acid ds – Double-stranded
ECD – Enzymatic Catalytic Domain
ECDC – European Centre Disease Prevention and Control EFSA – European Food Safety Authority
ep – Error-prone
FDA – U.S. Food and Drug Administration GAD – Glutamate Decarboxylase System GlcNAc – N-acetylglucosamine acid GRAS – Generally Recognized as Safe
ICTV – International Committee on the Taxonomy of Viruses IPTG – Isopropyl β-D-1-thiogalactopyranoside
LB – Lysogeny Broth
mDAP – meso-2,6-diaminopimelic acid MurNA – N-acetylmuramic acid PBS – Phosphate Buffered Saline PCR – Polymerase Chain Reaction PEG – Polyethyleneglycol
PG – Peptidoglycan
PPI – Protein-protein interactions RCA – Rolling-circle
rT – Room temperature RM – Random Mutagenesis RNA – Ribonucleic Acid
xxv
SDM – Site-Directed Mutagenesis
SDS-PAGE - Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis ss – Single-stranded
SSM – Site-Saturating Mutagenesis Tm – Melting temperature
Tris – Tris(hydroxymethyl)aminomethane TAE – Tris-Acetate-EDTA
xxvi
List of figures
Chapter 1
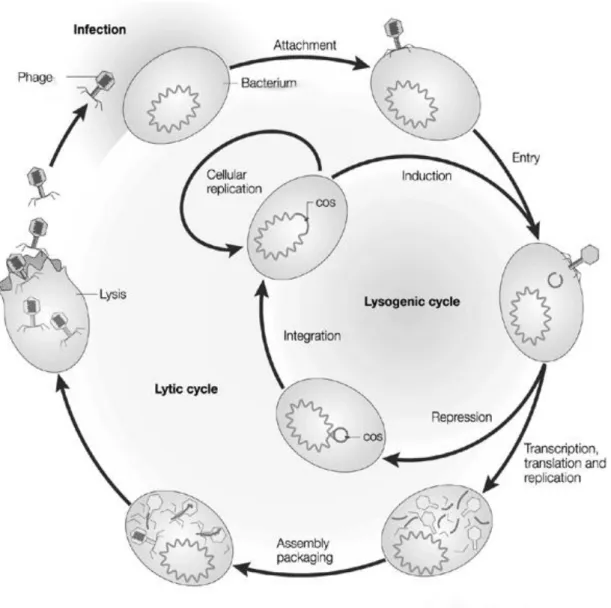
Figure 1 – Phage life cycle: lytic and lysogenic. Phage attaches to host and injects genetic material. In lytic cycle the genomic material is transcripted and replicated by replication and protein synthesis mechanisms of the host cell used to assemble new phage particles and subsequent release by cellular lysis. In the lysogenic cycle the phage genome integrates chromosomal DNA of the host without cell death Adapted from Campbell (2003). ... 6
Figure 2 - Representation of peptidoglycan differences between positive (left) and Gram-negative (right) cells and different type of endolysins mode of action. 1) Glycosidases; 2) Muramidases; 3) lytic transglycosidases; 4) Amidases; 5), 6), 7) e 8) Endopeptidases. Withdrawn from Oliveira et al., (2012) ... 10
Figure 3 – Overview of the QuickChange Lightning site-directed mutagenesis method. ... 14 Figure 4 – Diagram scheme comparing the most conventionally random mutagenesis procedures to ep-RCA evidencing the complexity and time-consuming of each method. ... 18
Chapter 2
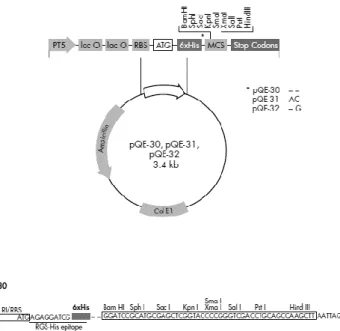
Figure 1 - Representation scheme of pQE30 cloning vector used to clone the endolysins sequences of ply500 and ply511 ... 25
Figure 2 - 96-wells plate scheme of antibacterial assay against L. monocytogenes 5725 using Ply500 expressed in micro-scale conditions. Protein expression was tested for 24 and 48 h. Endolysin 68 was used as positive control against outer membrane permeabilzed of P. aeruginosa cells. Negative controls are present in dashed lines wells. Blue wells represents protein expression in 4mL tube; yellow wells the protein expression in micro-scale; green wells protein expression of 200 µL collected from 4 mL expression tube and pink wells represent the protein production in 1 mL collected from four wells. ... 31
Figure 3 - 96-wells plate scheme of antibacterial assay against L. monocytogenes 5725 using Ply500 expressed in micro-scale conditions. Protein expression was tested for 24 and 48 h. Endolysin 68 was used as positive control against outer membrane permeabilzed of P. aeruginosa cells. Negative controls are present in dashed lines wells. Blue wells represents protein expression in 4mL tube; yellow wells the protein expression in micro-scale; green wells protein expression of 200 µL collected
xxvii
from 4 mL expression tube and pink wells represent the protein production in 1 mL collected from four wells. ... 38
Chapter
3
Figure 1 – SDS- PAGE electrophoresis gel 12%, stained with Coomassie Blue, for Ply511 (A) and Ply500 (B) proteins. M, Protein Ladder (10-250 kDa) from NEB; F1, 2, 3, and 4, fractions 1, 2, 3 and 4 of eluted proteins; W, wash; FT, flow through. The proteins were eluted with elution buffer containing 250 mM imidazole concentration. ... 48
Figure 2 - Antibacterial assays of Ply500 endolysin against normal and permeabilized L. monocytogenes 5725 cells. The endolysins were expressed in 4 mL broth and cell lysis was done by sonication (A). Protein expression was done in 5 wells (200 µL each well) then joined (1 mL) and sonicated (B). Endolysins were expressed in 200 µL of a 96-wells plate (C). Also protein expression was induced in 4 mL broth (D). Next 200 µL were transferred to 96-wells. Cell lysis was done by chloroform vapors. In all the cases the expression conditions were 16°C during 24 h or 48 h with 1.5 mM IPTG concentration. E. coli JM109 wild-type was used as negative control. ... 49
Figure 3– Kinetics of antibacterial assays of Listeria phage endolysins Ply500 and Ply511 at refrigeration temperatures (A) and room temperatures (B). Negative control was done using 180 µL bacterial suspension mixed with 20 µL of PBS. ... 51
Figure 4. – PCR of amplified protein genes using temperature gradient between 55-65°C. Gradient temperatures from left to right: 55°C, 57°C, 60°C, 62°C, 65°C. Agarose gel 1% concentration, stained with Sybr Safe and run at 90 V. Legend: M – DNA 1 kb ladder. ... 52
Figure 5 – Amplified endolysins sequences by ep-PCR. The generated products possess 1020 bp (plyP500) and 1176 bp (plyP511) and were then double digested by using selected restriction enzymes and purified. Agarose gel 1% concentration, stained with Sybr Safe and run at 90 V. Legend: M – 1 kb ladder. ... 53
Figure 6 – Double digestion of pQE-30 cloning vector in agarose gel 0.7% concentration in order to promote better separation of digested plasmid The gel was stained with Sybr Safe and run at 90 V. Legend: M – DNA 1 kb ladder. ... 54
Figure 7 – Representative illustration pQE-30 vector cloned with ply500 (A) and ply511 (B) using Vector NTI software. The ligation generates two plasmids with different lengths – 4039 bps and 4465 bps, respectively. Both genes are cloned between BamHI and SalI restriction sites. ... 55
xxviii
Figure 8 – Colony-PCR results after “heat-shock” transformation using NZY5α E. coli competent cells. Only pQE-30 + ply500 ligation were assembled by competent cells. Agarose gel 1% concentration, stained with Sybr Safe run 90 V. Legend: M – DNA 1 kb ladder. ... 56
Figure 9 – PCR of amplified endolysins sequences derived from co-evolved (wells 1-5) and evolved (wells 6-10) adapted phages. Amplified products were visualized by 1% agarose gel, stained with Syber Safe, at 90 V. The marker (M) is 1 kb ladder. ... 57
Figure 10 – Example of sequencing chromatogram of adapted plyP100 endolysin derived from co-evolutionary adapted phage. The putative mutations are highlighted. PlyP100CE1_Fw and PlyP100CE_Rv are the primers used for gene sequencing. ... 59
xxix
List of tables
Chapter 2
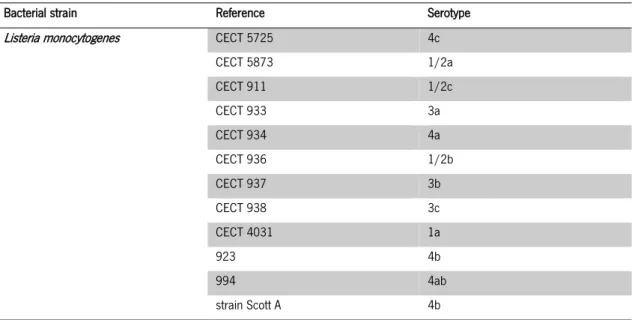
Table 1 - L. monocytogenes pathogen strains, references and serovars used for phage isolation. . 40
Table 2 - Designed primers for endolysins sequences amplification. ply500 (primers 1 e 2) e ply511 (primers 3 e 4). Underlined are shown the restriction sites for enzymes BamHI and SalI. Melting temperatures also are present in this table. ... 26
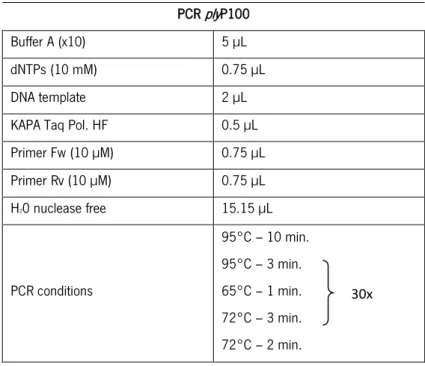
Table 3 – PCR conditions for amplification of ply500 and ply511 endolysins sequences in Error-Prone PCR and Colony-PCR. The volumes were calculated for 50 µL of final reaction volume. .... 34
Table 4 – Used primers for amplification of the PlyP100 endolysins derived from phage adapted assays. This primers were already available in the group primers collection. Underlined are shown the restriction sites for enzymes NcoI and BamHI, respectively.. ... 39
Table 5 – PCR conditions for amplification of plyP100 from phage adapted endolysins. The volumes were calculated for 30 µL of final reaction volume. The initialization step at 95°C during 10 minutes was used in order to promote the release of phage DNA. ... 40
Chapter 3
Table 1 – Putative binding and catalytic domains for Ply500 and Ply511 endolysins. ... 47
Table 2 – Amplified plyP100 sequences of the 5 different phage plaques isolated from Co-Evolution (CE) and Evolution (E) populations. Once amplified the sequences were aligned and compared to the original plyP100. Point mutations are observable at specific positions. Legend: A – adenine, T – thymine, G – guanine, C – cytosine, * – absence of nucleotide. ... 58
Chapter 1
Introduction and
Background
CHAPTER 1 INTRODUCTION AND BACKGROUND
3
1.
Listeria monocytogenes
and listeriosis
The genus Listeria is an important group of Gram-positive and anaerobic bacteria with low G+C content and it is closely related to Bacillus, Clostridium, Enterococcus, Streptococcus and Staphylococcus [1]. It can be isolated from various sources, such as soil, water, effluents and human and animal faeces [2]. Currently it is divided into six species: L. monocytogenes, L. ivanovii, L. seeligeri, L. innocua, L. welshimeri, and L. grayi.
L. monocytogenes it is classified as psychrotolerant organism as its optimal growth temperature can vary between 30 and 37ᵒC. It is considered to be highly pathogenic causing listeriosis, a foodborne illness that can induce death (20-30% of fatality rate) especially in risk groups such as elderly, pregnant women and other people with impaired immune system. In this case invasive listeriosis can be triggered causing meningitis, meningoencephalitis, endocardiditis,septicemia, premature births, neonatal listeriosis, stillbirths or misbirths [3][4][5] . Non-invasive listeriosis can cause gastro-intestinal infections and mainly affects healthy individuals.
It is associated with the ingestion of contaminated food mainly in a wide variety of ready-to-eat foods such as milk, seafood, fish products, meats and meat products [6]. However, the consumption of soft cheese and seafood is the main responsible for the most human listeriosis cases reported [7]. Since 1960’s decade the listeriosis has become more widespread which has been correlated to the increased use of refrigerators, consumption of processed foods and extension of shelf-life food products. According to most recent reports from the European Food Safety Authority (EFSA) and the European Centre Disease Prevention and Control (ECDC) the number of cases with listeriosis infections in Europe is on the increase from 0.1 cases per 100.000 in 2000 to 0.3 cases per 100.000 in 2006. It also refers to Germany, Ireland, Lithuania, the Netherlands, Spain and United Kingdom (UK) as the countries with highest number of infections in the same period [8].
However the increasing demand of consumers for clean-label products in most recent years also has led to a greater scientific investigation, which consequently increased the characterization of this pathogen becoming a well-studied model.
Not all strains of L. monocytogenes are equally capable to cause disease in humans and only four serovars – 1/2a, 1/2c, 1/2b and 4b – of the 13 identified for this strain are responsible for 98% of the reported listeriosis cases. The strains with 4b serovar are responsible for most food-borne outbreaks of listeriosis and for the sporadic cases which can suggest that this serovar can possess unique mechanisms of virulence [9].
CHAPTER 1
INTRODUCTION AND BACKGROUND
4
1.1.
Adaptation mechanisms
In food industry usually low temperatures, low pH conditions and high salt concentrations are the common conditions in order to preserve food products. However, L. monocytogenes has the ability to generate adaptation mechanisms to maintain its activity, stability and growth, making it difficult to control this pathogen in the food industry [10]. At low temperatures the mechanisms consists mainly in changing the membrane composition increasing unsaturated and shortening fatty acids chains [11], production of cold shock proteins [11][12] and accumulation of solutes as cryoprotectants [13][14]. For survival under acidic stress the induction of specific proteins (GroEL, ATP synthase and transcriptional regulators) [15], mechanism of pH homeostasis achieved by proton transport chain, the glutamate decarboxylase system (GAD) [16] and the two-component regulatory system LisR/ LisK [17] are the main mechanisms of adaptation in low pH conditions. The pathogen also possess osmoadapation mechanisms such as the induction of salt shock proteins, accumulation of compatible solutes such as glycine betaine, proline betaine, acetyl carnitine, carnitine, ϒ- butyrobetaine as osmoprotectants. Furthermore, this bacteria has the ability to change the expression levels of transcription genes under adverse conditions through the association of the alternative sigma factor with RNA polymerase core [10].
1.2.
Antimicrobial resistance of
L. monocytogenes
The current treatment for listeriosis for immunocompromised patients is based on the application of high doses of β-lactam antibiotics (ampicillin or penicillin) alone or in association with aminoglycoside (gentamicin). For allergic people to β-lactam antibiotics the treatment consists in the combination of trimethoprim with sulfonamide [18].
Usually L. monocytogenes is susceptible to a wide range of antibiotics, however, some recent studies reported an increasing antimicrobial resistance to one or more relevant antibiotics in environmental isolates [19][20][21]. In animals the use of antimicrobials can lead to new potential development of antimicrobial-resistant zoonotic foodborne bacterial pathogens, such as L. monocytogenes, that can subsequent be transmitted to humans as food contaminants.
Furthermore, spontaneous mutations of this foodborne pathogen agent or gene transfection between antibiotic-resistant bacteria and L. monocytogenes cells may contribute to the increasing of antimicrobial resistance and subsequent spreading in environment and food.
CHAPTER 1 INTRODUCTION AND BACKGROUND
5
2. Bacteriophages as a potential means to battle listeriosis
Bacteriophages or phages are viruses that infect specific bacterial cells and represent one of the most abundant biological entities in nature and have been recognized by their potential use as therapeutic agents in alternative to antibiotics [22]. The history of phages started over a century ago (1986) when Ernest Hankin observed bacteriocidal activity against Vibrio cholera from filtered and collected water from Ganges and Jumma River, in India. Almost 20 years after the first observations by Hankin, two investigators, Frederick Twort, an English bacteriologist, and the French-Canadian microbiologist Felix d’Herelle reintroduced the subject and independently identified filterable and transmissible viral particles responsible for bacterial lysis. However, only d’Herelle continue to pursuit this findings and named the viral particles as bacteriophages [23][24].
Phages are omnipresent and accidentally consumed through the ingestion of food or water, they are presumed to be safe as undesirable effects have not been detected. Their different genome sizes, from 17 kpb to 0.5 Mbp and the high frequency of novel genes found in newly characterized phage genomes proof that bacteriophages are genetically extremely diversified. This genetic diversification represent a great advantage in infection role as the ability of phages to evolve and circumvent the defense mechanisms of the host is higher [25].
Bacteriophages are constituted by a protein or lipoprotein coat, called capsid, with different shapes and sizes that encloses nucleic acid genome which can be single-stranded (ss) or double-stranded (ds), circular or linear, DNA or RNA. The International Committee on the Taxonomy of Viruses (ICTV) is responsible for the phage classification that is based on the properties of the virion as morphology and type of nucleic acid form. Currently, over 5,500 phages are known, divided and recognized by ICTV into one order and 14 families, among which 96% are tailed (belongs to Caudovirales order) and possess double-stranded DNA (dsDNA) enclosed in icosahedral symmetry heads and three different tails lengths subdivided in Myoviridae (25%) with contractile tails, Siphoviridae (61%) with noncontractile and long tails and Podoviridae (15%) with short tails [26]. The remaining phages are “cubic”, filamentous or pleomorphic and possess ssDNA, ssRNA or dsDNA as genome and represent 4% of phage population.
As a virus, phages are obligatory parasites and needs to infect the host cells. To be able to infect host cells, phages attach to the cell through specific receptors in membrane surface using mechanisms that depends of the virion morphology. The most usual mechanism consists in tail contraction and enzymatic degradation of small portion of cell membrane allowing injection of genetic material. Using the replication mechanisms of the host the synthesis of new phage particles occurs
CHAPTER 1
INTRODUCTION AND BACKGROUND
6
which are released by the action of produced lytic proteins responsible for cell lysis (lytic cycle). Phage infection follows the lysogenic cycle, when phage infection leads to the integration of genetic information into the chromosome of the bacterial host without cell death. In lysogenic cycle the phage genome will assume a quiescent state called prophage coexisting in a stable form with the host. The two possible life cycles of phages can be observed in Figure 1.
Fig. 1 – Phage life cycle: lytic and lysogenic. Phage attaches to host and injects genetic material. In lytic cycle the genomic material is transcripted and replicated by replication and protein synthesis mechanisms of the host cell used to assemble new phage particles and subsequent release by cellular lysis. In the lysogenic cycle the phage genome integrates chromosomal DNA of the host without cell death Adapted from Campbell et al., (2003) [27].
CHAPTER 1 INTRODUCTION AND BACKGROUND
7
2.1.
Listeria
phages
The first reports of specific L. monocytogenes phages date from 1940 and 1960. Currently more than 500 Listeria specific phages have been isolated and characterized mainly in the course of phage typing studies.
Usually the described Listeria-specific bacteriophages possess dsDNA genomes with sizes between 30-65 kb but a few possess larger genomes (124-140 kb). All Listeria phages belong to Caudovirales order featuring long noncontractile tails (Siphoviridae family) or with complex contractile tails (Myoviridae family). However, no Listeria specific podoviruses have ever been isolated which can be related to the low diversity of morphological structure of this bacterial cells [28] [29].
Listeria bacteriophage genomes contain one module responsible for the encoding of structural proteins, another responsible for encoding DNA functions as recombination, replication and repair. It also contain a lysis cassette which contains holin and endolysin genes. A lysogeny control region with the integrase gene is present in case of the temperate phages, which is responsible for mediate integration of the phage genome into the chromosomal host genome and excision in induction conditions. In fact many Listeria phages are temperate and most of them are capable of generalized transduction [28].
There is already some application of Listeria phages in food industry although this application as biocontrol of bacteria require some specific characteristics: phage must be strictly virulent, feature a broad host-range, unable to perform generalized transduction, does not affect the pathogenicity or virulence of the host and also does not integrate its genome on the genetic material of the host [30]. The well characterized Listeria phage P100 is already used as a biocontrol agent product and it acquired the Generally Recognized as Safe (GRAS) status by U.S. Food and Drug Administration (FDA). Furthermore P100 phage has been showing some interesting properties, displaying high efficacy by removing Listeria contaminations from fish and cooked ham and Listeria biofilms in steel surfaces [31][32][33].
There are also other FDA-approved anti-Listeria products such as a six-phages cocktail. This cocktail was allowed to reduce Listeria occurrence in food production facilities of fresh cut produce and melons [34]. Some other studies also showed interesting lytic properties of A511 phage against Listeria contaminations on various ready-to-eat food samples providing up to 5 log reduction of this foodborne pathogenic cells [35].
Despite showing many advantages, the use of lytic phages as biocontrol agents in food industry remains unclear, especially in European countries where it remains uncertain whether phages can
CHAPTER 1
INTRODUCTION AND BACKGROUND
8
be considered as processing aids or as decontaminants or additives [36]. Furthermore, the lack of studies about phage application in food industry performed in specific preparation, processing and storage conditions is considered to be an obstacle for its application in this field [22].
An alternative way to phage application consists in the use of its recombinant encoded peptidoglycan hydrolases (endolysins) which can be easier to approve than virus-based food additive. Endolysins have no effect on the original organoleptic and texture properties on food and act as an innocuous substance for human consumption, making these type of enzymes a good candidate for control of foodborne pathogens [37].
3. Endolysins
The name endolysin was first coined in 1958 with the aim to designate a proteinaceous lytic substance synthesized inside bacterial cells during phage multiplication and infection stage [38]. Endolysins are dsDNA bacteriophage encoded lytic enzymes that target the integrity of the cell wall and degrade its main constituent, peptidoglycan (PG), causing cell lysis and the consequent release of the bacteriophage progeny [39][40].
During phage infection lysins are produced and accumulated in the cytoplasm, however as they do not have signal sequences to be translocated through the cell membrane this movement is controlled by a holin. The holin is typically expressed in the late stages of the lytic infection cycle, forming a pore in the cell membrane allowing cytoplasmic lysins to access the peptidoglycan, causing cell lysis [41][42][43].
Endolysins are also capable to digest the cell wall when applied exogenously, especially in Gram-positive organisms, such as L. monocytogenes, and can lyse the cell wall of healthy and uninfected cells, originating the “lysis from without” phenomenon [39][44]. It proved its ability to be used as alternative antimicrobial agents [40].
Numerous studies have been demonstrating the potential application of phage derived endolysins as biocontrol agents in foods which has been considered a good alternative over other antimicrobial agents. Their high specificity for the target pathogen leaves the natural and desired flora of the food products untouched. Due to their proteinacious nature endolysins are noncorrosive and biodegradable [45].
These applications of lysins as biocontrol agents against foodborne diseases have been recently reviewed [46]. The first and more obvious approach in control strategy consists in the application of purified endolysins to food products.
CHAPTER 1 INTRODUCTION AND BACKGROUND
9
Some studies have reported the staphylococcal phage endolysin LysH5 [47] rapidly showed great lytic activity in pasteurized milk, reducing bacterial numbers below the detection level within 4 h. Also streptococcal lysins B30 [48] and Ply700 [49] and the Clostridium butyricum phage ΦCTP1 [50] lysin reported to be high lytic activity in milk and milk products. The three Listeria phage endolysins Ply118, Ply511 and PlyP35 exhibit high thermoresistance which is a very important characteristic for products that undergo heat treatment such as pasteurized milk products [51].
Another approach is the production and release of this endolysins in starter organisms in fermentation processes. This alternative has been reported for derived Listeria and Clostridium phage endolysins, although no studies of application in food has been yet demonstrated [52][53][54]. In conclusion, phage endolysins represent a good alternative for the control of foodborne pathogens. However some tests are required to verify their stability on other food products and consumer safety. Physiochemical conditions where these type of biocontrol agents act, must be first evaluated as well. Furthermore, it has to be economically attractive possible to justify an investment of it application in food industry [55].
3.1.
Different types of endolysins
The cell wall is responsible for maintaining the shape and physical integrity, protecting against mechanical damage and osmotic rupture. Peptidoglycan is the main constituent and it is composed of several chains of N-acetylmuramic acid and N-acetylglucosamine residues, linked together by β-1,4-glycosidic bonds, connected to a short stem tetrapeptide side chains [40][55].
Adjacent tetrapeptides may be cross-linked by an interpeptide bond (in Gram-negative bacteria) or by an interpeptide bridge (in Gram-positive bacteria) [40].
In Gram-negative bacteria the cell wall is relatively thin (10 nm) and is composed by single layer of PG surrounded by an outer membrane. In these organisms, lactyl ether connects the glycan backbone to a peptide side chain that contains mostly L- and D-amino acids [56].
In Gram-positive bacteria, the cell wall is thick (15-80 nanometers), consisting of several layers of PG. Running perpendicular to the PG sheets is a group of molecules called teichoic acids which are unique for these organisms. In this case the cell wall it is considerably more diverse in length and composition coinciding with many different peptide arrangements among PG.
Peptidoglycans may vary the amino in position 3 of the tetrapeptide, defining two types of PGs: meso-2,6-diaminopimelic acid type (mDAP-type) in Gram-negative and some Gram-positive species (i.e. L. monocytogenes) and a l-Lysine type (Lys-Type) typical for Gram-positive species [55][57].
CHAPTER 1
INTRODUCTION AND BACKGROUND
10
Therefore, endolysins can be divided in five classes according to their enzymatic specificity. Glycosidases (Fig.2, target 1) cleave the glycan component at the reducing end of N-acetylglucosamine (GlcNAc) [58] or at the reducing end of N-acetylmuramic acid (MurNAc) [59]. Muramidases/lysozymes (Fig. 2, target 2) share the same glycan target as the lytic transglycosidases (Fig. 2, target 3), however both form different products. The 1,6-anhydro bond is formed instead of the muramic acid residue, due to absence of water that is important for correct function of transglycosidase activity [60]. N-acetylmuramoyl-L-alanine amidases (Fig. 2, target 4) cut the amide bond between N-acetylmuramic acid residues and L-amino acid residues. These types of endolysins are responsible for the strongest destabilization effect in the peptidoglycan layer. Endopeptidases (Fig. 2, target 5, 6, 7 and 8) cleave the peptides moieties attacking LD- and DD-bonds.
3.2.
Endolysin structure
Generally, the sizes of phage endolysins are between 25 and 45 kDa. The only exception is the 114 kDa streptococcal bacteriophage C1 endolysin, PlyC, as it is a multimeric lysin with globular structure [61]. Endolysins can vary in their molecular structure, modular or globular, and in their domain orientations.
Fig. 2 - Representation of peptidoglycan differences between Gram-positive (left) and Gram-negative (right) cells and different type of endolysins mode of action. 1) Glycosidases; 2) Muramidases; 3) Transglycosidases; 4) Amidases; 5), 6), 7) e 8) Endopeptidases. Withdrawn from Oliveira et al., (2012) [55]
CHAPTER 1 INTRODUCTION AND BACKGROUND
11
The typical feature for all Gram-positive phage endolysins is their two-domain structure also called modular structure which is composed by an enzymatic catalytic domain (ECD) at the N-terminal and the cell wall binding (CBD) domain at the C-terminal [39][41][44]. A few endolysins which have a modular organization display an inverted structure, with the ECD at the C-terminal and CBD at the N-terminal.
Gram-negative derived phage endolysins usually presents a globular structure with only ECD. The evolutionary explanation for the occurrence of CBD domains only in Gram-positive endolysins is based on previous studies by Fischetti et al., (2008) [41] and Loessner et al., (2002) [62] which say that CBD domain binds irreversibly to the cell wall preventing the lysis of surrounded cells before the new phage particles release. Thus it can be said that phages are induced to produce right amounts of enzymes to induce the cell lysis. This phenomenon is not verified in Gram-negative bacteria because the presence of the protective outer membrane inhibits the external lysin treatment. The ECD function consists in the cleavage of the bacterial cell wall while the CBD is responsible for the substrate recognition after cell binding. The main advantage of modular organization is that it enables the specificity binding and the enzyme activity in an independent way, enabling the substitution of either domain with other domain from another enzyme [63][64]. Furthermore, some interesting studies have demonstrated that some endolysins acquired more than a single ECD, such as endopeptidase/amidase lysin from the phage phi11 and endopeptidase/muramidase from Streptococcus agalactiae bacteriophage B30 Hermoso (2003) [65] and Pritchard (2004) [59].
4. Strategies for endolysins improvement
Natural evolution results in a large number of enzyme variants which exhibit their specific function and efficiency adjusted only to perform under physiological activities. Therefore, naturally occurring enzymes often lack features necessary for biotechnology industry applications. In this field this study pretends to accelerate the evolutionary process of a well-adapted endolysin to the most adversal conditions in food industry more specifically at low temperatures and high salt and low pH conditions. The strategies consists in phage adaptation at successively low temperatures, also known as cryodrilling and one of the most used random mutagenesis technique, error-PCR. However some other methods for directed evolution are presented in this section in order to individually compare the advantages of each technical approach.
CHAPTER 1
INTRODUCTION AND BACKGROUND
12
4.1.
Phage adaptation – “Cryodrilling”
Interactions in many host-parasites are influenced by biology and environment conditions. The pathogenic parasites impose selection for resistant hosts which in turn impose selection for infective parasites resulting in rapid antagonist coevolution.
According to the Red Queen hypothesis these biotic interactions give rise to continual natural selection for adaptation and counter-adaptation playing an important role in molecular evolution and consequently in population dynamic [66].
There are two types of antagonistic coevolution: the “arms race” dynamics, leading to broader resistance range in the host against a greater number of parasite genotypes and an increased variability of host allowing the infection of most host genotypes; and the fluctuating selection dynamics, favoring hosts that resists to the most common parasite phage genotypes, not contributing for directional evolution of host range.
Coevolutionary dynamics between hosts and parasites is increasingly investigated using experimental evolution. The most used model for antagonistic coevolutionary studies are bacteria and their lytic phages.
Pseudomonas fluorescens SBW25 and its lytic phage ϕ2 is the most studied model of bacteria-phage evolution given deeper knowledge about this process and coevolution interactions. Although it is important to diversify this coevolutionary studies to other host-parasite dynamics with particularly importance in human health as they can be applied as biocontrol agents [67][68][69].
4.2 Directed evolution: site-directed mutagenesis Vs. random mutagenesis
Two contradictory tools can be used on a molecular level: rational protein design and directed evolution. The first method requires extensive information about relationships between sequence structures, mechanisms and function of the enzyme. Directed evolution concept was first introduced in 1967 [70]. It is a laboratory in vitro process responsible for the generation of proteins, enzymes, metabolic pathways and entire genomes with desired properties under different conditions, such as extreme temperatures, pH and salinity [71]. Directed evolution implements an iterative Darwinian optimization process, whereby the fittest variants are selected from an ensemble of random mutations [72]. Despite the advances to date on the industrially enzymes, the most challenging part of the directed evolution experiment consists in the development of generic screening or selection tools, identifying novel enzymes activities more efficiently [73][74].
CHAPTER 1 INTRODUCTION AND BACKGROUND
13
Our understanding of protein function has been enhanced significantly since the early 1980s, due to the advent of site-directed mutagenesis (SDM) and random mutagenesis (RM).
The use of SDM for the generation of mutations at specified sites within a known polynucleotide sequence is an extremely common process nowadays, providing a very powerful tool for the manipulation of sequences and structures.
However, there are certain limitations associated with this technique such as lack of sufficient structural information or the need to know which sites within a molecule should be investigated in detail.
At that point, RM has proved to be very useful to identify functional sites within a protein or nucleic acid sequences. Moreover, RM can provide the opportunity to screen proteins, which has previously been determined, for structurally important residues or sites, without introducing the selective bias typical of directed approach. RM also allows the study of regulatory nature and structural features of nucleic acid sequences, such as promoters, enhancers, ribosome binding sequences, transcription termination sequences, structures and functions of transfer and ribosomal RNA, the processes involved in viral assembly, in the primary transcript splicing and the initiation of nucleic acid replication. When combining RM technique with later measurements large and useful amounts of important information can be obtained that suggest targets in order that a site-specific or random oligonucleotide approach can be posteriorly directed [75].
4.3 Site-directed mutagenesis
SDM has a variety of applications and is an important tool in molecular biology that has revolutionized the study of gene regulation and protein structure-function relationship [76]. The SDM techniques can be grouped into two major categories: polymerase chain reaction (PCR)-based or non-PCR-based [77].
The first, also known as single-primer, uses an oligonucleotide complementary to part of single-strand DNA. The primer contain an internal mismatch to promote the desired mutation. However, because of the several limitations of this method, low and variable mutants were obtained, which forced the use of suitable screening approaches to identify mutants [75].
The second strategy, denominated by “cassette mutagenesis” refers to the replacement of a region of interest with a synthetic mutant fragment generated by annealing complementary oligonucleotides [78] or by hybridization and ligation of a number of oligonucleotides [79]. The main disadvantages of this method is that in most of the cases, it needs at least two rounds of primer-based mutagenesis
CHAPTER 1
INTRODUCTION AND BACKGROUND
14
to introduce suitable restriction enzyme sites into templates and it is not appropriate for routine mutagenesis [80].
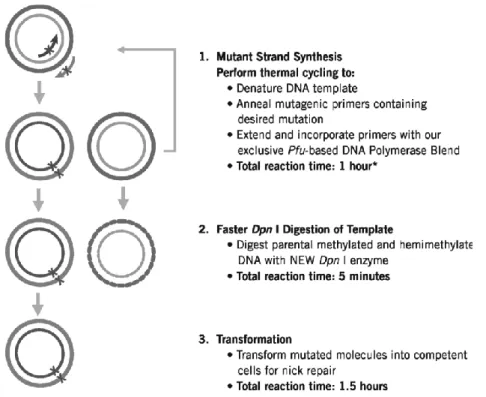
Several approaches of SDM technique have been published. However this method generally are labor intensive or technically difficult. To overcome this difficulties QuickChange Lightning Site-Directed Mutagenesis (Fig. 3) is a commercial Kit by Stratagene used that allows site-specific mutations. The main advantage of its usage is that eliminates the need for subcloning and for single-stranded DNA rescue. Furthermore, does not require specialized vectors, unique restriction sites, multiple transformations or in vitro methylation treatment steps. Also a single kit allows rapid, efficient and accurate mutagenesis of small and large plasmids.
Fig. 3 - Overview of the QuikChange Lightning site-directed mutagenesis method.
4.3.1 Domain swapping
Domains are the most important functional units in proteins and a significant proportion of protein-protein interactions (PPI). Domain swapping is a mechanism for forming oligomeric assemblies. In domain swapping, a secondary or tertiary element of a monomeric protein is replaced by the same element of another protein. Domain swapping can range from secondary structure elements to whole structural domains [81]. It also represents a model of evolution for functional adaptation by oligomerisation of enzymes that have their active site at subunit interfaces [82].
CHAPTER 1 INTRODUCTION AND BACKGROUND
15
The modular architecture of phage lysins of Gram-positive background and the knowledge accumulated in recent years from available crystal structures and bioinformatics allows the creation of a protein with multiple modules of a single origin and also any desired amino acid sequence and function [83][84][85].
4.4 Random Mutagenesis for in vitro directed enzyme evolution
Random Mutagenesis (RM) is a powerful tool for generating enzymes, proteins and entire metabolic pathways or even entire genomes with improved properties. RM mechanisms can be divided into five different categories: (i) transitions, involving substitution of a purine by another, or a pyrimidine by a second pyrimidine, (ii) transversions, involving substitution of a purine nucleotide by a pyrimidine, or vice versa, (iii) deletions, in which one or more nucleotides are deleted from a gene, (iv) insertions, one or more extra nucleotides are incorporated into a gene, and (v) inversions, involving the 180º rotation of a double-stranded DNA segment [86].
There are many methods by random mutagenesis that can generate genetic diversity such as treating DNA or entire bacteria with chemical mutagens [87], passing cloned genes using mutator strains [88], error-prone PCR (ep-PCR) [89], site-saturating mutagenesis (SSM) [90] and finally by using rolling circle error-prone PCR (ep-RCA) [91].
4.4.1 Chemical mutagenesis
In vitro chemical mutagenesis is one of the most used techniques for the generation of random mutagenesis in DNA and it does not involve the introduction of heterologous DNA or the manipulation of interesting DNA by recombinant methods [86][87].
Despite of several chemicals have been tested, the majority of the compounds cannot be used due to damage caused in the cell [87]. Consequently only five of the best-understood treatments are currently widely employed in generating randomly distributed single-point mutations in vitro: nitrous acid, sulfurous acid, hydroxylamine, hydrazine and mil-acid hydrolysis [75]. The main advantage of chemical mutagenesis is associated with the simple usage and the low cost processing. On the other hand, the main disadvantage is its inefficient control of the mutation rate and limitation to amino acid substitutions [86].
CHAPTER 1
INTRODUCTION AND BACKGROUND
16
4.4.2 Mutator strains
Another useful random mutagenesis method is the bacterial mutator strain method that introduces random point mutations into whole genes. The most popular mutator strain is Escherichia coli XL1-Red which lacks the DNA repair pathways, MutS, MutD and MutT resulting in a ≈ 5000-fold random mutation rate higher than the wild type strain [91]. Transformation of the mutator strain and recovery of the mutant from the transformant are the two main steps for the using mutator strain protocol. The main advantages of this method are its simplicity and absence of a ligation step, and it can incorporate a wide variety of mutations such as substitutions, deletions and frame-shift mutations. The several steps of the growth, plasmid isolation, transformation and re-growth, of this process originates a progressively sickness of the mutator strain. Additionally this method generates a low mutation frequency under standard conditions and requires a long cultivation period (longer than 24h) for introduce multiple mutations [86].
4.4.3 Site-saturating mutagenesis
Site-saturation mutagenesis (SSM) is another powerful technique for protein optimization due to its simplicity and efficiency in which a single amino acid can be substituted to any other 19 possible substituents [92][93]. As a result, the mutagenesis product is a collection of clones, each having a different codon in the targeted position, so it is called “saturated”. Whole plasmid single-round PCR it is a technique that makes SSM method more efficient [90]. This method uses two complementary primers containing a mutant codon with partial overlap in the 5’ region that improve the amplification efficiency by decreasing formation primers dimers. This amplification method requires the parental DNA which is then degraded by using methylation-dependent endonuclease digestion. As a result a circular, nicked and containing mutated gene vector is produced that can be transformed directly in E. coli cells [92].
The advantage of this procedure is that eliminates time-consuming subcloning, ligation and single-stranded DNA rescue. The main disadvantages refers to only two nucleotides can be replaced at a time and it does not work well with large plasmids (>10 kB).
This method was already successfully applied to generate improved activity of proteins under study [94][95][96].
CHAPTER 1 INTRODUCTION AND BACKGROUND
17
4.4.4 Error-prone PCR
Error-Prone PCR (ep-PCR) is the most used method to generate random mutagenesis into a defined DNA sequence that is too long to be chemically synthesized. It consists in reducing the fidelity of the DNA polymerase which can introduce random mutations during the PCR process [97]. In ep-PCR process the fidelity of the DNA polymerase is modulated by alteration of the conditions of reaction buffer, increasing the error rate. The conditions are the addition of Mn2+ for reducing base pair
specificity [98]; increase Mg2+ concentration for stabilizing no complementary base pairs [99];
unbalanced dNTP stoichiometry in order to achieve misincorporation; increase the concentration of DNA polymerase and altering the extension time [100].
After PCR, three steps are required to clone the library of genes into a host strain: digestion of the product with restriction enzymes, separation of the fragments by agarose gel electrophoresis and ligation into a vector, which makes it a very time consuming method [101].
In addition, the success of this method depends of the mutational bias of DNA polymerase, because most part of low-fidelity polymerases used nowadays show strong mutational preferences that can favor the substitution of certain nucleotides instead of others, causing the reduction of library diversity [86].
Studies by Vanherck et al., (2005) [102] combined to different low-fidelity DNA polymerases, Taq polymerase and Mutazyme, which exhibit opposite mutational spectra. This strategy should permit generating unbiased libraries or libraries with a specific degree of mutational bias by applying optimal mutagenesis frequencies through ep-PCR and controlling the concentration of template in the shuffling reaction while taking into account the GC content of the target gene.
Other studies by Asano et al., (2005) [103], Edmond et al., (2008) [104] and Maeda et al., (2008) [105] have used the efficiency of ep-PCR method in the modification of enzymes with the aim to increase their potential in industrial applications.
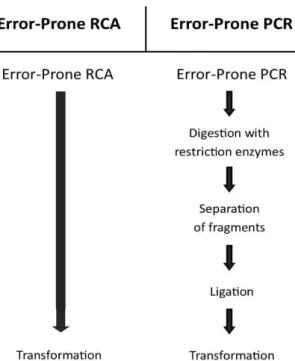
A variation of traditional ep-PCR is called error-prone rolling circle amplification (ep-RCA) and it is an in vitro amplification DNA method which amplifies circular DNA by a rolling circle mechanism, yielding linear DNA composed of tandem repeats of the circular DNA sequence [106]. This technique does not require specific primers because random hexamers can be used as a universal primer for any template and does not require a thermal-cycler because the amplification reaction proceeds at a constant temperature. Furthermore, ep-RCA products can be used directly to transform a host strain (Fig. 4).
CHAPTER 1
INTRODUCTION AND BACKGROUND
18
Ep-RCA consists of only one DNA amplification step followed by direct transformation of the host strain and producing mutants with an adequate mutation frequency, about 3–4 mutations per kilobase, for in vitro evolution experiments without using restriction enzymes, ligases, specific primers or special equipment such as a thermal-cycler are required. This method will enable random mutagenesis to become a more commonly used technique.
Fig. 4 - Diagram scheme comparing the most conventionally random mutagenesis procedures to ep-RCA evidencing the complexity and time-consuming of each method.
5 Advances in molecular engineering of endolysins
Despite that some endolysins have been optimized by their natural evolution and selection processes, in most of the cases there is still potential for improvement of their activity and stability, especially in complex environments such as certain food matrices [107].
Therefore, protein engineering based on directed evolution approaches can alter some endolysins properties for specific biotechnology applications.
In this section some studies and advances in molecular engineering of endolysins will be discussed using site-directed and random mutagenesis approaches
CHAPTER 1 INTRODUCTION AND BACKGROUND
19
5.1. Advances using site-directed mutagenesis
In this case, the enzymatic optimization is done through localized changes in amino acids and then the effect on lytic activity is examined.
The alteration of conserved amino acids in the streptococcal B30 endolysin CHAP and lysozyme domains resulted in a sequential loss of activity from each domain [59]. Afterward, Donovan et al., (2006) [48] analyzed this dual domain lysin on live bacteria and concluded that the B30 endolysin CHAP domain was the primary source of activity when lysing “from-without”. Site-directed mutagenesis and deletion analyses of the Bacillus anthracis phage lysin PlyG were essential in defining the binding domain and active site residues. These observations provided new knowledge about the mechanism of specific binding of lysin to B. anthracis and may be useful in establishing new methods for detection of B. anthracis [108].
Nelson et al., (2006) [61] also examined the streptococcal bacteriophage C1 lysin PlyC, using point mutations and concluded that subunit PlyCA is responsible for catalytic activity. The active-site residues were confirmed too. Similarly, site-directed mutations altering histidine codons in the staphylococcal glycyl-glycine PG hydrolase ALE-1 have been used to define essential amino acids in the M23 endopeptidase domain [109].
Low et al., (2011) [110] demonstrated the influence of a positive net charge in the peptidoglycan layer induced by site-directed mutagenesis. The increase of the positive net charge can be favorable for ECD function independently of its CBD through amino acid replacement. This can be important for fine-tuning enzyme activity, such as in cases where efficacy of an enzyme is limited by its size. The already described knowledge of binding domains and active site residues of these endolysins is very useful when constructing novel fusion constructs for the purpose of making a better antimicrobial agent.
5.1.1. Advances using domain-swapping
The earliest approaches to alter regulatory properties were created by exchange of the CBDs of pneumococcal autolysins (LytA) and phage endolysins Cpl-1 [111][112]. Studies by Lopes et al., (1997) [64] concluded that the fusion of lactococcal N-acetylmuramidase catalytic domains to choline-binding domains from pneumococcal endolysin CBD resulted in choline dependence of the chimeric enzyme. Croux et al., (1993) [113], made the reverse experiment by combining a clostridial CBD with LytA, thereby rendering the chimera active against choline-containing pneumococcal cell walls and abolishing its activity against clostridial cell walls.
CHAPTER 1
INTRODUCTION AND BACKGROUND
20
More recently, Donovan et al., (2006) [114], fused full-length and truncated versions of the Streptococcus agalactiae phage B30 endolysin to mature lysostaphin, yielding enzymes that were active against both streptococcal and staphylococcal cells.
Similar to the studies with protein chimeras of pneumococcal and clostridial origin, the exchange of Listeria phage CBDs of different serovar specificity also yielded enzymes with swapped lytic properties and enhanced activity, which constitute interesting antimicrobial candidates for control of the pathogen. In the same study it was also possible to combine the binding specificities of different single CBDs in heterologous tandem CBD constructs, which were then able to recognize the majority of Listeria strains. Manoharadas et al., (2009) [115], created a fusion of the insoluble S. aureus phage P68 endolysin with a minor coat protein of the same phage and demonstrated that in certain cases, modular engineering of endolysins may also solve solubility problems ensuring efficient production and purification of otherwise insoluble lytic proteins.
5.2. Advances using random-mutagenesis
Random mutagenesis is another approach to improve lytic activity of phage endolysins. One of the first studies consisted in mutate the gene coding for the S. agalactiae phage endolysin PlyGBS using two DNA mutagenesis, E. coli mutator strain and ep-PCR. After repeated rounds of mutagenesis and screening a mutant with 28-fold activity was obtained comparatively to the parental enzyme. It also resulted in the incorporation of unpredicted sequences at the C-terminus of the generated mutant endolysin. The screening method in this study selected enzyme mutants with high diffusion capability such as the C-terminal truncation which is smaller than a molecule lacking a CBD. This methodology can be applied for identification of endolysin mutants with improved activity under various pH and salt conditions or certain food products [84]. However, the screening method has to be adapted for every individual condition.
Studies by Heselpoth et al., [116] involved PlyC endolysin thermostability improvement. In this study, the random mutagenesis approach was used to improve the thermal stability through the use of ep-PCR, followed by an optimized screening process. The results suggested the methodology generated PlyC mutants that retain high activities when compared to wild-type after elevated temperature treatment.
CHAPTER 1 INTRODUCTION AND BACKGROUND
21
6. Main goals of this work
In recent years, listeriosis has become more widespread due to the increasing consumption of processed foods and extension of shelf-life food products.
The phage derived endolysins are appointed as the best alternative to antibiotic and phage usage as biocontrol agents. Furthermore, endolysins have no effect on the original organoleptic and texture properties on food acting as an innocuous substance for human consumption making these type of enzymes a good candidate for control of foodborne pathogens such as L. monocytogenes.
However, naturally occurring enzymes often lack features necessary for biotechnology industry applications. Beyond that, little is known about endolysin function in food storage and processing conditions - low temperatures, low pH conditions and high salt concentrations. There is also a lack of knowledge aiming the improvement of derived Listeria phage endolysins using random mutagenesis techniques.
This study pretends to accelerate the evolutionary process, through the use of directed evolution strategies, aiming to generate well-adapted endolysins to the most adverse conditions in food industry such as low temperatures and also, high salt and low pH conditions.
Two different directed evolution strategies will be used in this work: error-PCR, the most used random mutagenesis technique and cryodrilling which mainly consists in biotic interaction of phage-host aiming its adaptation at successively low temperatures.
Therefore, this work will have two well-defined objectives:
The first objective is to improve endolysins activity at low temperatures, known as refrigerator temperatures.
The second objective is to reduce the usage of phage and chemical compounds application in food industry by using the improved endolysins.
Chapter 2
CHAPTER 1 INTRODUCTION AND BACKGROUND
25
1. Bacterial strains, endolysins and plasmids
Criovials of E. coli JM109 containing two different Listeria monocytogenes phage endolysins – pQE-30-ply500 (accession nº Q37979) and pQE-30-ply511 (accession nº Q38653) – and pQE-30 plasmid with CBD-P35 sequence were kindly provided by Doctor Mathias Schmelcher from Swiss Federal Institute of Technology Zurich – Department of Health Sciences and Technology.
The pQE-30 plasmid, present in Figure 1 was used for every cloning step.
E. coli JM109 strain belonging to the Centre of Biological Engineering of University of Minho (CEB) was used as the expression vector. Cultures of this strains, previously transformed with pQE-30, were done using Lysogeny Broth (LB, Liofilchem) supplemented with 100 µg/mL ampicillin (AppliChem). Overnight cultures E. coli BL21 (CEB collection) containing Lys68 sequence (plasmid PET28a-68gpLys) growth in LB supplemented with tetracycline marker (30 µg/mL).
After growth overnight at 37°C and 120 rpm agitation (Environmental Shaker incubator ES- 20/60), cultures were diluted 1:100 in fresh medium and incubated in the same conditions until reach exponential growth phase.
The induction for protein expression was accomplished by adding Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich).
For Listeria phage isolation twelve bacterial strains were used and are present in Table 1. All the strains were grown overnight in static incubator at 37°C in Tryptic Soy Broth (TSB) (Sigma-Aldrich) agar plates and incubated next day in TSB broth maintaining the growth conditions.
CHAPTER 2
MATERIALS AND METHODS
26
2.
Listeria
phage isolation
2.1. Non-lysogenic strains selection
In order to avoid the selection of temperate phages, lysogeny test was performed to verify if the strains used for phage detection did not possess lysogenic activity – prophages – responsible for bacterial lysis.
A double-layer agar (DLA) test was used to perform the lysogeny test [117]. This method consists in two LB broth medium layers supplemented with different agar concentrations, 1.2% and 0.6% for bottom layer and top agar respectively.
All bacteria, previously grown overnight, were resuspended in 100 µL of NaCl 0.1 mM and then gently homogenized in 5 mL of top-agar and poured into bottom agar layer of petri plate previously prepared. This procedure was done for all twelve L. monocytogenes strains listed in table 1. Ten µL of each Listeria strain in every bacterial lawn was placed. Bacterial lawns plates were dried at room temperature and incubated overnight.
Next day, by observing the plates, the strains that generate an inhibition halo should be discarded from phage isolation methodology.
Table 1 - L. monocytogenes pathogen strains, references and serovars used for phage isolation.
Bacterial strain Reference Serotype
Listeria monocytogenes CECT 5725 4c
CECT 5873 1/2a CECT 911 1/2c CECT 933 3a CECT 934 4a CECT 936 1/2b CECT 937 3b CECT 938 3c CECT 4031 1a 923 4b 994 4ab strain Scott A 4b