UNIVERSIDADE FEDERAL DO CEARÁ CENTRO DE CIÊNCIAS DA SAÚDE
DEPARTAMENTO DE PATOLOGIA E MEDICINA LEGAL PROGRAMA DE PÓS-GRADUAÇÃO EM PATOLOGIA
MARIA JURACY SOLON PETROLA
PERFIL DO ESTRESSE OXIDATIVO EM PACIENTES PORTADORES DE LEUCEMIA MIELÓIDE CRÔNICA
MARIA JURACY SOLON PETROLA
PERFIL DO ESTRESSE OXIDATIVO EM PACIENTES PORTADORES DE LEUCEMIA MIELÓIDE CRÔNICA
Dissertação apresentada ao Programa de Pós-Graduação em Patologia daUniversidade Federal do Ceará, como requisito parcial à obtenção do título de mestre em patologia.
Orientadora: Profa. Dra. Romélia Pinheiro Gonçalves Coorientadora: Profa. Dra. Silvia Ma Meira Magalhães
Dados Internacionais de Catalogação na Publicação Universidade Federal do Ceará
Biblioteca de Ciências da Saúde P591p Petrola, Maria Juracy Solon.
Perfil do estresse oxidativo em pacientes portadores de leucemia mieloide crônica / Maria Juracy Solon Petrola. – 2011.
79f. : il. color., enc. ; 30 cm.
Dissertação (Mestrado) – Universidade Federal do Ceará, Faculdade de Medicina, Departamento de Patologia e Medicina Legal, Programa de Pós-Graduação em Patologia, Mestrado em Patologia, Fortaleza, 2011.
Área de concentração: Patologia Tropical.
Orientação: Profa. Dra. Romélia Pinheiro Gonçalves. Co-orientação: Profa. Dra. Silvia Maria Meira Magalhães
1. Leucemia Mielogênica Crônica BCR – ABL Positiva. 2. Estresse Oxidativo. I. Título.
MARIA JURACY SOLON PETROLA
PERFIL DO ESTRESSE OXIDATIVO EM PACIENTES PORTADORES DE LEUCEMIA MIELÓIDE CRÔNICA
Dissertação apresentada ao Programa de Pós-Graduação em Patologia da Universidade Federal do Ceará, como requisito parcial à obtenção do título de mestre em patologia.
Aprovada em: ______/ ______/ 2011
BANCA EXAMINADORA
Profa. Dra. Romélia Pinheiro Gonçalves (Orientadora) Universidade Federal do Ceará-UFC
Profa. Dra. Maria da Silva Pitombeira Universidade Federal do Ceará-UFC
Prof. Dr. Jose Ajax Nogueira Quieiroz Universidade Federal do Ceará-UFC
______________________________________________________________________________________________ Profa. Dra. Margarida Maria de Lima Pompeu
Aos meus pais Luis Ubaldo Solon (in memoriam) e Maria Isabel Neves Solon.
Aos meus irmãos Ubaldo, Silvia, Ricardo, Silvana e Roberto.
Ao Petrola meu marido.
AGRADECIMENTO
.
Agradeço com carinho e emoção: À minha família, meu porto seguro;
À minha orientadora, Profa. Dra. Romélia Pinheiro Gonçalves, em especial, pelo constante apoio, amizade e colaboração na realização deste trabalho.
À Profa. Dra. Silvia Maria Meira Magalhães pela co-orientação no desenvolvimento deste trabalho:
À amiga Alana Jocelina Montenegro de Castro pelo carinho e companheirismo; À Profa. Dra. Margarida Maria Lima Pompeu pelo empenho e motivação contagiantes;
À Profa. Dra. Maria da Silva Pitombeira pelo incentivo e exemplo; Ao Prof. Dr. José Murilo Martins pelo exemplo e pioneirismo;
Aos professores e funcionários do Departamento de Patologia da UFC pela acolhida fraterna;
Aos alunos do Laboratório de Hemoglobinopatia do curso de Farmácia, em especial à Maritza pela inestimável ajuda;
Aos médicos do serviço de Hematologia do HUWC; Aos colegas do mestrado;
RESUMO
A Leucemia mielóide crônica (LMC) é caracterizada pela expansão clonal de células progenitoras hematopoéticas, resultante da translocação (9:22). O oncogene de fusão BCR-ABL,
no cromossomo Ph, é transcrito e traduzido numa proteína de fusão BCR/ABL. A tirosina quinase (TK) ABL na proteína de fusão é constitutivamente ativada sendo necessária para os eventos leucemogênicos iniciais da LMC e sua atividade induz a produção de espécies reativas de oxigênio (EROs). De particular relevância para LMC é o fato de que um aumento de EROs pode ter consequências, facilitando a instabilidade genômica podendo contribuir para a progressão da doença. O objetivo do estudo foi determinar o perfil oxidativo em pacientes com LMC, em acompanhamento ambulatorial no Hospital Universitário Walter Cantídio (HUWC). Trata-se de um estudo transversal constituído de 30 pacientes adultos, com diagnostico clínico e laboratorial de LMC, em tratamento com inibidores de (TK) de 1ª e 2ª geração. As concentrações de malonaldeído (MDA) e de nitrito (NO2-) foram realizadas por método espectofotométrico. As atividades das enzimas Glutationa peroxidase (GSH-Px) e catalase (CAT) foram determinadas no hemolisado, por kit Glutathione Peroxidase Cellular Activity Assay® (Sigma-Aldrich) e por
espectofotometria, respectivamente. Glutationa total, glutationa reduzida (GSH reduzida), glutationa oxidada (GSSG) foram determinadas por kit Total Glutathione Activity® (Assay
Designs, Inc) e calculada a relação GSH/GSSG. Para a análise estatística de dados não
paramétricos foi utilizado o ANOVA® e o teste de múltiplas comparações de Tukey. Foi considerado o nível mínimo de significância de 5%. As concentrações média de MDA e de NO2- foram aumentadas nos pacientes com LMC em relação ao controle, independente da atividade da doença. O perfil antioxidante foi caracterizado pela diminuição da CAT e aumento da GSH-Px também independente da atividade da doença. A GSH reduzida se apresentou diminuída, a GSSH, aumentada e a relação GSH/GSSG diminuída. Os pacientes em uso de inibidores de TK de 2ª geração apresentaram parâmetros do estresse oxidativo significativamente elevados em relação ao grupo controle. Conclui-se que os pacientes com LMC estão sob estresse oxidativo e com atividade antioxidante comprometida.
Palavras-chave: Leucemia Mielogênica Crônica BCR-ABL positiva; Estresse Oxidativo;
ABSTRACT
Chronic myeloid leukemia (CML) is characterized by clonal expansion of hematopoietic progenitor cells, result from the translocation (9:22). The oncogene BCR-ABL, in the Ph chromosome, is transcribed and translated into a fusion protein BCR / ABL. The ABL tyrosine kinase (TK) in the fusion protein is constitutively activated and is needed for the initial leukemogenic event of CML and its activity induces production of reactive oxygen species (ROS). Of particular relevance to CML is the fact that an increase of ROS can have consequences, facilitating genomic instability may contribute to disease progression. The aim of this study was to determine the oxidative status in patients with CML, in attendance at a university hospital (HUWC). This is a cross-sectional study consisted of 30 adult patients of both sexes with clinical and laboratory diagnosis of CML on treatment with inhibitors (TK) 1st and 2nd generation. The concentrations of malondialdehyde (MDA) and nitrite (NO2-) were performed by spectrophotometric method. The activities of enzymes glutathione peroxidase (GSH-Px) and catalase (CAT) were determined in hemolysate by Glutathione Peroxidase Cellular Activity Kit ® Assay (Sigma-Aldrich) and spectrophotometry, respectively. Total glutathione, reduced glutathione (reduced GSH), glutathione (GSSG) were determined by Total Glutathione Activity Kit ® (Assay Designs, Inc) and calculated the ratio GSH / GSSG. For statistical analysis of nonparametric data, It was used ANOVA® and the Tukey test for multiple comparisons. It was considered the minimum level of significance of 5%. The average concentrations of NO2- and MDA were increased in CML patients compared to control, regardless of disease activity. The antioxidant profile was characterized by decreased CAT and GSH-Px increased also independent of disease activity. The reduced GSH is presented decreased, the GSSH, increased and GSH / GSSG decreased. It was observed that patients using protease inhibitors of TK 2nd generation of oxidative stress parameters were significantly elevated compared to controls. In the analysis of patients on imatinib were not detected significant changes in oxidative status. We conclude that patients with CML are under oxidative stress and impaired antioxidant activity.
LISTA DE ILUSTRAÇÕES
Figura 1 Distribuição dos grupos quanto à medicação em uso 52
Figura 2 Distribuição dos pacientes quanto ao tempo de doença e ao nível de atividade da doença
53
Figura 3 Distribuição dos pacientes estratificados pelo nível de atividade da doença quanto ao tempo médio de doença
54
Figura 4 Valores médios de MDA e Nitrito nos grupos 54
Figura 5 Níveis médios de GSH-Px e CAT nos grupos em estudo 55
Figura 6 Níveis dos tióis não protéicos e relação GSH/GSSG nos grupos 57
Figura 7 Valores de MDA e Nitrito nos grupos estudados 58
Figura 8 Valores de GSH-Px e CAT nos grupos estudados 59
Figura 9 Valores de tióis não protéicos e da relação GSH/GSSG nos grupos controle e de pacientes em atividade estratificados pela medicação em uso
61
Figura 10 Valores de MDA e Nitrito nos grupos controle e de pacientes em uso de imatinibe estratificados pelo nível de doença.
62
Figura 11 Valores de GSH-Px e CAT nos grupos controle e de pacientes em uso de imatinibe estratificados pelo nível de doença
63
Figura 12 Valores de tióis não protéicos e da relação GSH/GSSG nos grupos controle e de pacientes em uso de imatinibe estratificados pelo nível de doença
64
Figura 13 Níveis médios de MDA e Nitrito em pacientes estratificados pelo tempo de doença
65
Figura 14 Níveis médios de CAT e GSH-Px em pacientes estratificados pelo tempo de doença.
66
Figura 15 Níveis médios de tióis não protéicos e relação GSH/GSSG em pacientes
estratificados pelo tempo de doença
LISTA DE TABELAS
Tabela 1 Distribuição dos pacientes quanto ao sexo 50
Tabela 2 Distribuição dos pacientes por faixa etária 50
Tabela 3 Distribuição dos pacientes com LMC de acordo com a fase
da doença ao diagnóstico. 51
LISTA DE ABREVIATURAS
Abi-1 Abl interactor proteins 1
Abi-2 Abl interactor proteins 2
ARG Abelson Relacion Gene
ATP trifosfato de adenosina
BAD Bcl-2 antagonist of the cell death
BAX Bcl-2 associated X protein
Bcl-2 B-cell CLL/Lynphoma 2
bcl-x Bcl-2 related gene
BCR-ABL Oncogene Breakpoint Cluster Region/Abelson
BCR-ABL Protein Breakpoint Cluster Region/Abelson
Bus Bussulfan
Ca Cálcio
CAT Catalase
c-Kit stem cell factor receptor
Cu, Zn-SOD Cobre,Zinco-Superóxido Dismutase
DNA Ácido Desoxirribonucléico
ERNs Espécies Reativas de Nitrogênio
EROs Espécies Reativas de Oxigênio
EUA Estados Unidos da América
FISH Fluorescence in situ hybridization
G2 Crescimento 2 (ciclo celular antes da divisão celular) GSH REDUZIDA Glutationa Reduzida
GSH TOTAL Glutationa Total
GSH-Px Glutationa Peroxidase
GSSG Glutationa Oxidada
HNE 4 hidroxi 2 nonenal
HU Hidroxiuréia
HUWC Hospital Universitário Walter Cantídio
IFN-α Interferon alfa
IRIS International Randomized Study of Interferon and STI571
K Potássio
LMC Leucemia Mielóide Crônica
LOG Logarítmo
LPHDGH Laboratório de Pesquisa em Hemoglobinopatias e Doenças Genéticas Hematológicas
M Mitose
MBCR Major breakpoint cluster region
MDA Malonaldeído
Mg Magnésio
Mn-SOD Manganês-Superóxido dismutase
Na Sódio
NaCl Cloreto de sódio
NADPH Fosfato de nicotinamida adenina dinucleotídeo NEED Cloridrato de N-(1-naftil)-etilenediamina
NO- anion nitroxil
NO Óxido Nítrico
NO+ Cátion Nitrosônio
NOS Óxido Nítrico Sintetase
O2- Radical superóxido
O2- Oxigênio Molecular
OH- Radical hidroxila
OMS Organização Mundial de Saúde
ONOO- Peroxinitrito
P190BCR-ABL Proteína BCR-ABL com peso molecular 190 kD
P210BCR-AB Proteína BCR-ABL com peso molecular 210 kD
PDGF alfa Fator de crescimento derivado das plaquetas alfa PDGF beta Fator de crescimento derivado das plaquetas beta
Ph Filadélfia
RCgC Resposta Citogenética Completa
RHC Resposta Hematológica Completa
RMoC Resposta Molecular Completa
RNAm Ácido Ribonucléico Mensageiro
RT-PCR reverse transcription - Polimerase chain reaction
S Síntese
SAME Serviço de Arquivo Médico
SH3 Src homology 3–tyrosine kinase
SOD Superóxido dismutase
STI171 Specific Tirosine Kinase Inibitor 171
TBARS Ácido tiobarbitúrico
TCLE Termo de Consentimento Livre e Esclarecido
TK Tirosina Kinase
TKF Tirosina Kinase de Fusão
TMO Transplante de Medula Óssea
SUMÁRIO
1 INTRODUÇÃO
1.1 Leucemia mielóide crônica...15
1.2 Estresse Oxidativo ... 24
1.3. Estresse oxidativo e câncer ... 29
1.3 Estresse oxidativo e LMC ... 31
2. OBJETIVOS ... 33
2.1 Objetivo geral ... 33
2.2 Objetivos específicos ... 33
3. CASUÍSTICA E MÉTODOS ... 34
3. 1 Casuística ... 34
3.1.1. Grupo de Estudo ... 34
3.1.3. Obtenção dos dados ... 35
3.2. Estratificação do Grupo de estudo ... 35
3.2.1. Estratificação em relação à atividade ou não da doença ... 35
3.2.2 Estratificados de acordo com o tipo de inibidor de TK ... 37
3.2.3. Estratificados de acordo com o tempo de doença ... 37
3.3. Local do Estudo ... 37
3.4 Aspectos Éticos ... 37
3.5 Métodos ... 38
3.5.1 Coleta e processamento das amostras ... 38
3.5.2 Análises do Perfil oxidativo ... 38
3.6 Descarte do Material Biológico ... 40
3.7 Análise Estatística ... 40
4.1 Características sócio demográficas dos pacientes ... 41
4.2 Caracterização clínica dos pacientes ao diagnóstico ... 41
4.3 Caracterização clínica atual dos pacientes ... 42
4.4 análise dos parâmetros do perfil oxidativo nos pacientes estratificados de acordo com a atividade da doença ... 44
4.4.1 Marcadores do estresse oxidativo ... 44
4.4.2 Enzimas antioxidantes ... 45
4.4.3 Tióis não protéicos ... 46
4.5 Perfil oxidativo dos pacientes com doença em atividade estratificados pela medicação em uso 47 4.5.1 Marcadores do estresse oxidativo. ... 48
4.5.2 Enzimas antioxidante ... 48
4.5.3 Tióis não protéicos ... 49
4.6 Análise dos parâmetros do perfil oxidativo nos pacientes em uso de imatinibe, estratificados pela atividade da doença ... 50
4.6.1 Marcadores do estresse oxidativo. ... 51
4.6.2 Enzimas antioxidantes ... 52
4.6.3 Tióis não protéicos ... 52
4.7 Análise dos parâmetros do perfil oxidativo nos pacientes estratificados pelo tempo de doença54 4.7.1 Marcadores do estresse oxidativo. ... 54
4.7.2 Enzimas antioxidantes ... 55
4.7.3 Tióis não-protéicos ... 56
5. DISCUSSÃO ... 57
6. CONCLUSÕES ... 63
REFERÊNCIAS ... …64
1
INTRODUÇÃO
1.1 Leucemia Mielóide Crônica
A Leucemia Mielóide Crônica (LMC) é uma neoplasia da medula óssea resultante da expansão clonal de uma célula precursora hematopoética. LMC é provavelmente a doença maligna humana mais extensivamente estudada, tendo sido a primeira a ser associada com uma alteração genética específica, o cromossomo Philadelphia (Ph) (NOWELL, HUNGERFORD,
1960), em células primordiais e suas descendentes (FARDEL, KANTARJIAN, TALPAZ, 1999). Essa anormalidade genética característica da LMC resulta da translocação recíproca e equilibrada entre os braços longos dos cromossomos 9 e 22 [t(9;22)(q34;q11)] (ROWLEY, 1973). A translocação cria dois novos genes: BCR-ABL no cromossomo 22q- ou cromossomo Ph,
e o recíproco ABL-BCR no cromossomo 9q+ (DE KLEIN, et al.,1982; HEISTERKAMP, et al.,
1983).
A anormalidade genética característica da LMC, gene BCR-ABL, é observada em células
das linhagens eritróide, granulocítica, megacariocítica e linfóide (KURZROCK, GUTTERMAN, TALPAZ, 1988). O gene quimérico BCR-ABL, por sua vez é transcrito e traduzido na
oncoproteína BCR-ABL que apresenta como característica principal uma atividade tirosina kinase (TK) descontrolada (BEN-NERIAH, 1986; McLAUGHLIN, CHIANESE, WITTE, 1987; DALEY, VAN ETTEN, BALTIMORE, 1990).
A atividade da proteína BCR-ABL é necessária e suficiente para a atividade oncogênica da fase inicial da LMC: principalmente a superprodução de células da série granulocítica (DALEY, VAN ETTEN, BALTIMORE, 1990; PASTERNAK, HOCHLAUS, SCHUTEIS, 1998; FARDEL, KANTARJIAN, TALPAZ, 1999; GORDON, DAZZI, MARLEY, 1999).
A ativacão do proto-oncogene c-Abl ocorre como resultado da adição de éxons do gene
BCR e quebra do primeiro éxon do gene ABL. Variação na quantidade de éxons do gene BCR
resulta em diferenças funcionais entre as proteínas de fusão de pesos moleculares diferentes (LUGO et al. 1990).
Dependendo do ponto de quebra break cluster region (bcr) no gene BCR, três principais
tipos de genes BCR-ABL podem ser formados, levando à geração de proteínas de fusão de
Proteína 190BCR-ABL (p190BCR-ABL) tem uma atividade TK mais alta e está associada
com o desenvolvimento de um fenótipo de leucemia aguda mais agressiva (LUGO et al., 1990).
O gene híbrido predominante na LMC clássica é derivado da quebra na principal região de quebra (Mbcr) com a geração de uma proteína de fusão citoplasmática com peso molecular de 210kd (P210 BCR-ABL), que está associado a uma doença com evolução mais indolente ( LUGO, 1990; MELO, 1996; KANTARJIAN, et al, 2000).
LMC é uma síndrome mieloproliferativa, termo primeiramente utilizado por Damesheck, em 1951, modificado para Neoplasia Mieloproliferativa Crônica, na 4a edição da classificação da Organização Mundial de Saúde (OMS) de Tumores Hematopoéticos e Tecidos Linfóides. Nesta categoria encontram-se mais outras sete doenças hematológicas, a saber: Leucemia Neutrofílica Crônica, Policitemia Vera, Mielofibrose Primária, Trombocitemia Essencial, Leucemia Eosinofílica Crônica, Mastocitose e Neoplasia Mieloproliferativa não classificável (SWERDLON
et al., 2008).
A LMC é uma doença progressiva e geralmente evolui através de três fases distintas caracterizadas por piora do quadro clínico e laboratorial. São elas: fase crônica, fase acelerada e crise blástica (FARDEL, KANTARJIAN, TALPAZ, 1999; MELO; HUGHES; APPERLEY, 2000).
A maioria dos pacientes é diagnosticada na fase crônica, sendo o diagnóstico incidental. Durante esta fase ocorre uma expansão clonal maciça de células mielóides, que mantém a capacidade de diferenciação (KANTARJIAN, TALPAZ, 1993; FARDEL, KANTARJIAN, TALPAZ, 1999; SAWYERS, 1999).
A medula óssea é hipercelular, com intensa hiperplasia granulocítica, diferenciação preservada, e uma relação mielóide: eritróide acima de 10:1. O número de blastos é inferior a 10%. Hiperplasia megacariocítica pode ser observada. A biópsia óssea ratifica a hipercelularidade e é importante para avaliar a fibrose e a proliferação de fibras de reticulina (FARDEL, et al.,
1999).
Quando os sintomas aparecem, eles são relacionados à expansão das células do clone leucêmico e consistem de fadiga, perda de peso e desconforto devido à esplenomegalia. A leucocitose pode levar ao surgimento de sinais e sintomas relacionados à hiperviscosidade tais como hemorragia retiniana, priapismo, acidente vascular, confusão e estupor (FARDEL, KANTARJIAN, TALPAZ, 1999).
Em pacientes não tratados e em uso de terapêutica baseada em drogas citorredutoras, após em média três a cinco anos, o clone leucêmico perde a capacidade de diferenciação e a doença progride. O reconhecimento da progressão da doença da fase crônica para as fases transformadas (fase acelerada e fase blástica) é importante para o prognóstico e tratamento (KANTARJIAN, et
al.,1988).
O diagnóstico de fase acelerada, segundo a OMS, pode ser feito na presença de qualquer um dos seguintes parâmetros: 1 - persistente aumento dos leucócitos ou do baço; 2- persistente trombocitose não responsiva à terapia; 3 - persistente trombocitopenia; 4- evolução citogenética clonal (KANTARJIAN, et al.,1988) 5 – basofilia acima de 20% no sangue periférico; 6 – 10% a
19% de mieloblastos no sangue periférico ou na medula óssea. O surgimento de anormalidade clonal adicional à presença do cromossomo Ph é suficiente para caracterização da fase acelerada (SWERDLOW et al., 2008).
A fase blástica, é definida pela presença de infiltração extramedular blástica (CORTES, TALPAZ, KANTARJIAN, 1996; FARDEL, KANTARJIAN, TALPAZ, 1999), ou mais que 20%
de blastos no sangue periférico e/ou medula óssea (SWERDLOW, et al., 2008).
Em 1/3 dos pacientes na fase aguda os blastos expressam marcadores linfóides e 2/3 dos pacientes apresentam fenótipo de leucemia mieloblástica aguda ou indiferenciada (KANTARJIAN, et al, 1988). A linhagem dos blastos pode ser difícil de determinar pela
nervoso central. A expectativa de vida é de 3 a 6 meses após o inicio da crise blástica (QUINTÁS-CARDAMA, CORTES, 2006).
A análise citogenética é o teste padrão ouro para o diagnóstico de LMC, é também uma ferramenta valiosa na detecção de anormalidades cariotípicas adicionais que ocorrem com a resistência ao tratamento e a evolução da doença (MITELMAN, 1993).
O cromossomo Ph é observado nas células nucleadas do sangue periférico e da medula óssea em 90% dos pacientes com LMC. Nos pacientes nos quais a citogenética falha em detectar o cromossomo Ph (10% dos casos), a análise molecular (FISH ou PCR qualitativo) irá detectar o rearranjo BCR-ABL em até 50% destes casos (DEININGER, GOLDMAN, MELO, 2000; BRANFORD, et al., 2004).
O estudo molecular não deveria substituir a citogenética tradicional uma vez que anormalidades cromossômicas adicionais nas células Ph+ poderiam não ser detectadas (DEININGER, GOLDMAN, MELO, 2000; DEININGER, 2008).
Embora possua uma origem molecular comum caracterizada pelo cromossomo Ph, o curso clínico da LMC é variável, com alguns pacientes progredindo rapidamente enquanto outros permanecem na fase crônica por um longo tempo (CORTES, TALPAZ, KANTARJIAN, 1996).
O prognóstico da LMC pode variar significativamente mesmo entre pacientes que se encontram na mesma fase da doença. Estratégias para estratificação de risco são importantes como um guia de prognostico e para o manuseio do paciente (SOKAL et al., 1984).
Há 2 grupos de fatores prognósticos a serem considerados em pacientes com LMC: aqueles que podem ser identificados antes do tratamento e os que são identificados durante o tratamento. Os principais fatores iniciais são a fase da doença e o risco relativo (RR).
A fase da doença influencia fortemente a resposta, a duração da respostas e a sobrevida total sendo observados melhores resultados na fase crônica que na fase acelerada e na fase acelerada que na crise blástica (QUINTAS-CARDAMA, CORTES, 2006).
Várias estratégias de estratificação de risco têm sido desenvolvidas na tentativa de orientar o manejo do paciente (SOKAL et al., 1984; GRATWOHL, HERMANS,
NIEDERWIESER, 1993; HASFORD, PFIRRMANN, HEHLMAN, 1998).
grupos prognósticos com taxas de risco de menos que 0,8; 0,8 a 1,2 e mais que 1,2 com médias de sobrevida de aproximadamente 4,5; 3,5 e 2,5 anos, respectivamente (SOKAL et al.,1984).
Apesar deste sistema de estratificação ter sido criado a partir da observação de pacientes tratados com drogas que atualmente não se encontram mais em uso tais como bussulfan, e esplenectomia, estudos mais recentes tem demonstrado sua aplicabilidade em séries mais modernas (BACCARANI et al., 2009).
Embora alguns fatores prognósticos tenham perdido seu valor preditivo com o uso de inibidores de TKs, o escore de Sokal ainda é capaz de identificar pacientes com diferentes probabilidades de responder ao imatinibe apesar de que os pacientes com escores de alto risco apresentam agora uma melhor resposta (QUINTAS-CARDAMA, KANTARJIAN, TALPAZ, 2005).
O RR, baseado no sistema de Sokal consegue predizer a resposta citogenética. Os fatores de significância prognóstica no sistema de Sokal são idade superior a 60 anos, tamanho do baço, plaquetas acima de 700.000, o número de basófilos e de blastos no sangue periférico e medula óssea (SOKAL et al., 1984).
Outros sistemas de escore propostos são o escore de Hasford, 1998 e o de Gratwohl, 1993 que foram desenvolvido para serem utilizado em pacientes tratados com IFN-α e submetidos a
transplante de células tronco, respectivamente.
A LMC tem uma incidência anual em todo o mundo de 1 a 2 casos por 100.000 habitantes. Dos 44.240 novos casos de leucemias diagnosticadas nos EUA, em 2007, aproximadamente 4.600 foram LMC, representando em torno de 10% de todas as leucemias, com incidência maior no sexo masculino (JEMAL, et al., 2007).
A média de idade de apresentação é de 45 a 55 anos, porém pode ocorrer em qualquer idade (QUINTÁS-CARDAMA; CORTES, 2006). Kalidas, Kantarjian, Talpaz (2001) observaram a idade média de diagnóstico entre 50 e 60 anos e o percentual de pacientes diagnosticados acima de 60 anos variava de 12 a 30%. Em revisão de Redaelli e colaboradores, 2004, foi observado que a faixa etária mais comum de apresentação da LMC era dos 40 a 60 anos. Pasquini e colaboradores (2010) verificaram que a média de idade do diagnóstico variava de acordo com a região estudada sendo mais elevada nos Estados Unidos (53 anos) e mais baixa na Ásia, 41 anos.
atômica, em 1945 no Japão (MALONEY, LANGE, 1954; TANAKA et al, 1989) e em expostos
a radiação ionizante (DEININGER, et al, 1998) como radiologistas e pacientes tratados com
radioterapia (QUINTÁS-CARDAMA; CORTES, 2006).
Na LMC, a molécula de ácido ribonucléico mensageiro (RNAm) transcrita do gene híbrido, BCR-ABL, contem uma das duas junções, designadas e13a2 (formalmente b2a2) e e14a2 (b3a2). Ambos os RNAs são traduzidos em uma oncoproteína de peso molecular de 210 kD
(QUINTAS-CARDAMA; CORTES, 2006) necessária e suficiente para induzir as alterações observadas na fase crônica da LMC (DALEY, VAN ETTEN, BALTIMORE, 1990; DALEY, VAN ETTEN, BALTIMORE, 1991; PEAR, et al., 1998).
O potencial leucemogênico da p210BCR-ABL reside no fato de que a atividade TK ABL,
normalmente regulada, é constitutivamente ativada pela justaposição da seqüência estranha do
BCR. O BCR age promovendo dimerização da oncoproteina que fosforila as moléculas
BCR-ABL adjacentes. A atividade quinase descontrolada da BCR-BCR-ABL usurpa a função fisiológica da enzima ABL normal, através da interação com uma variedade de proteínas efetoras (WILSON-RAWS et al., 1996; MELO, HUGHES, APPERLEY, 2003 ou 2000).
Quatro principais mecanismos tem sido implicados na transformação maligna pelo
BCR-ABL:
Adesão alterada ao estroma e matriz extracelular – a regulação da hematopoese normal envolve a interação entre as células do estroma e as células progenitoras hematopoéticas. A LMC é clinicamente caracterizada pela prematura liberação de células progenitoras da medula óssea, um fenômeno que pode ser atribuído a defeitos nas propriedades de adesão destas células. Alguns eventos leucemogênicos como observados na LMC produzem seletiva vantagem de crescimento pela diminuição da capacidade das células de aderir ao estroma da medula óssea (GORDON, et al.,1987; VERFAILLIE et al., 1997).
Sinalização mitogênica ativada – O BCR-ABL pode revogar a dependência aos
fatores de crescimento. Vários mecanismos podem ser operativos, incluindo ativação de moléculas sinalizadoras intracelulares (ILARIA JR, VAN ETTEN, 1996), interação de receptores de fatores de crescimento (por exemplo, receptores para interleucina-3 e fator de células tronco) (WILSON-RAWLS et al., 1996; DONATO et al., 2001) e o aumento da expressão dos próprios
Reduzida apoptose – a expressão da oncoproteína BCR-ABL prolonga a sobrevida das células progenitoras mielóides e granulócitos maduros através da inibição da apotose. Este mecanismo pode permitir às células acumularem alterações genéticas secundárias que tem um papel importante na expansão clonal, progressão tumoral e resistência à terapia citotóxica (BEDI
et al., 1994).
Degradação de proteínas inibitórias da Abl TK – o domínio SH3 (Src-homology 3) tem
um papel critico na regulação da atividade TK da proteína Abl. a oncoproteína BCR-ABL induz a degradação das Abl interactor proteins 1 and 2 (Abi-1 e Abi-2) as quais ativam a função
inibitória do domínio SH3. Portanto tal degradação é outra maneira através da qual BCR-ABL induz transformação celular (DAI, et al.,1998).
Uma vez confirmado o diagnóstico de LMC, o tratamento está indicado em todos os pacientes. Pacientes diagnosticados atualmente podem esperar ter uma sobrevida substancialmente maior que pacientes diagnosticados há 20 ou mesmo 10 anos atrás em vista das novas abordagens terapêuticas (CORTES, KANTARJIAN, 2005).
O tratamento da LMC acompanhou a evolução do conhecimento sobre a patogênese desta doença conforme descrito por Geary, 2000, em sua revisão histórica da LMC. No século XIX, era utilizado arsênico. A irradiação esplênica foi utilizada como opção terapêutica a partir do final do século XIX até meados do século XX (GEARY, 2000; TEFFERI, 2008).
O Bussulfan (Bus) foi introduzido em 1959, dez anos mais tarde hidroxiurea (Hu) se tornou disponível para o tratamento de LMC com demonstrado aumento da sobrevida em relação ao Bus (HEHLMANN, et al., 1993). Tais agentes são capazes de normalizar rapidamente as
elevadas contagens de leucócitos na fase crônica da doença, porém não induzem remissão citogenética, portanto não estão associados a mudanças no curso natural da doença (HEHLMANN et al., 1993).
Na década de 70, transplante de medula óssea (TMO) e regimes de tratamento com interferon alfa (INF-α) demonstraram ser mais eficazes que a quimioterapia convencional para o tratamento da LMC, sendo potencialmente capazes de modificar a história natural da doença, porém associados com diferentes taxas de riscos e benefícios (GRATWOHL, HERMANS, NIEDERWIESER, 1993).
taxa de morbidade e mortalidade (GRATWOHL, HERMANS, NIEDERWIESER, 1993). O
IFN-α era a terapia padrão para os casos de LMC na fase crônica até a descoberta do mesilato de imatinibe (MI) (KANTARJIAN, TALPAZ, GUTTERMAN, 1987).
As pesquisas objetivando o tratamento da leucemia através da inibição do oncogene se iniciaram na década de 90 (BURKE JR, et al, 1992) e culminaram em 1996 com a síntese do
composto GCP571148B (imatinibe) que inibe a Abl e várias outras TKs (BUCHDUNGER et al,
1996), tais como c-Kit, fator de crescimento derivado das plaquetas alfa e beta (PDGF alfa e beta) e gene relacionado ao Abl (ARG) (DRUKER et al., 1996; BERAN et al., 1998; DRUKER
et al., 2001).
O oncogene BCR-ABL funciona como um ativador natural da proteína TK. Esta proteína
liga-se ao ATP e transfere fosfato do adenosina trifosfato (ATP) para resíduos de tirosina em proteínas específicas. Estas proteínas, agora fosforiladas, tornam-se responsáveis por uma série de etapas que levam aos defeitos fisiopatológicos observados na LMC. Por essa razão, sendo a ligação do ATP com essa tirosina bloqueada, todas as etapas envolvidas também serão bloqueadas. O conhecimento desse mecanismo foi essencial para o desenvolvimento de uma terapia efetiva e seletiva para a LMC (DEININGER, GOLDMAN, MELO, 2000).
Estudos clínicos estabeleceram a ação do composto em pacientes com LMC (DRUKER et
al.,1996), e em 1998, Imatinibe foi introduzido no arsenal de drogas para o tratamento da LMC
(DEININGER, GOLDMAN, MELO, 2000).
A partir do ano 2000, Imatinibe se tornou o tratamento inicial para LMC, conduta que foi respaldada por estudos populacionais International Randomized Study of Interferon and STI571
(DRUKER et al.,2001; DRUKER et al.,2006). A introdução do imatinibe iniciou a era da terapia
molecular, com resultados marcantes, incluindo completa resposta citogenética em até 90% dos pacientes e com resposta molecular na maioria (CORTES, KANTARJIAN, 2005).
O Imatinibe também induz resposta em uma significante percentagem de pacientes que se encontram nas fases acelerada e blástica. Esta resposta é, porém, transitória. Alguns pacientes com LMC, particularmente, aqueles em estado avançado, poderão apresentar resistência primária ou secundária ao imatinibe (DRUKER, et al., 2006; MELO,CHUAH, 2007; BACCARANI, et
Novos inibidores das TKs Abl tem sido desenvolvidas com aumento da potência e menor necessidade de ligações estritas, e em alguns casos com ação inibitória sobre outras quinases envolvidas no mecanismo de resistência ao imatinibe (MELO; CHUAH, 2007).
Os agentes que bloqueiam BCR-ABL de uma maneira não ATP-competitiva podem
representar uma alternativa para paciente que desenvolveram mutações, tornando-os insensíveis ao imatinibe (MELO; CHUAH, 2007).
Nilotinibe (AMN107) - É estruturalmente não relacionado ao imatinibe, ele inibe membros da família de quinase Src. O Nilotinibe é 20 a 30 vezes mais potente que o imatinibe. Este aumento de potencia se deve provavelmente a um melhor ajuste ao domínio da Kinase Abl.
O Nilotinibe inibe a atividade TK da maioria dos mutantes BCR-ABL clinicamente importantes
(O’HAREet al., 2005).
O dastinibe (354825) é um agente com capacidade inibitória contra outras kinases, é um
ATP competitivo, inibidor duplo específico para Src e Abl-kinase, com potencia 100 a 300 vezes
maior que o imatinibe. Assim como o nilotinibe, ele não inibe o mutante T315I (LOMBARDO et
al., 2004; O’HARE et al., 2005).
É possível que só a inibição da BCR-ABL não seja suficiente para erradicar as células da
LMC, particularmente as células tronco quiescentes que são imatinibe insensíveis (BARNES, MELO, 2006; QUINTAS-CARDAMA, CORTES, 2006).
Eventos imune-mediados têm papel importante na supressão do clone de LMC. O uso de vacinas que elicitam resposta imune específica dirigida a antígenos tumorais restritos à LMC estão em estudo (PINILLA-IBARZ, , KORONTSVIT, 2000; CATCHART , et al, 2004;
BOCCHIA, et al., 2005;. A imunomodulação também poderia eliminar pequena quantidade de
doença residual (BARNES; MELO, 2006).
Estudos têm demonstrado que a inibição da formação de EROs através do uso concomitante de antioxidantes N acetilcisteina e vitamina E diminui a taxa de mutagênese e a frequência de resistência ao imatinibe (KOPTIRA et al., 2006).
Algumas questões permanecem a serem respondidas, porém ‘’A despeito de todas as
incertezas, as perspectivas de tratamento para pacientes com LMC nunca foram tão claras”
1.2 Estresse Oxidativo
Oxigênio é uma molécula essencial para todos os organismos aeróbios e tem papel predominante na geração de ATP. Espécies reativas são geradas, como subproduto, durante este processo. Um desequilíbrio entre a produção de radicais livres e metabólitos reativos, e sua eliminação pelos mecanismos protetores, os antioxidantes é definido como estresse oxidativo (GUTTERIDGE, 1995).
A produção de espécies reativas ou radicais livres é gerada durante o metabolismo normal de toda célula eucarionte em diversas localizações celulares. As mitocôndrias são consideradas as principais fontes de geração intracelular destas espécies, ocorrendo também no retículo endoplasmático, no citosol e nos peroxissomas (GUTTERIDGE, 1995).
Os oxidantes são também gerados por diferentes tipos de radiação com a irradiação X gerando radical hidroxila, irradiação com luz ultravioleta gerando estados eletronicamente excitados com subseqüente formação de radicais. Ultrassom e radiação de micro-ondas podem também gerar EROs (SIES, 1997). Os radicais mais relevantes na regulação biológica são:
Os formados durante o processo de redução do O2 à água, também conhecidos como espécies reativas do oxigênio (EROs): O ânion superóxido (O2-), o peróxido de Hidrogênio (H2O2) e o radical hidroxila HO- (SIES, 1997);
O Óxido Nítrico (NO), produzido pela oxidação de um dos átomos terminais de nitrogênio da L-arginina, processo catalizado pela enzima Óxido Nítrico Sintetase (NOS). Dependendo do meio ambiente, NO pode ser convertido a várias outras espécies reativas de nitrogênio (ERNs) tais como o cátion nitrosônio (NO+), o anion nitroxil (NO-) ou peroxinitrito (ONOO-) que podem modificar macromoléculas incluindo proteínas lipídios e ácidos nucléicos (MARLETTA, 1994). Os radicais têm uma capacidade de reagir de maneira indiscriminada podendo lesar qualquer componente celular. Para proteger os componentes celulares das lesões induzidas por estes radicais, existem defesas antioxidantes (SIES, 1997).
Na definição de Halliwell e Gutteridge, 1997, “antioxidante é qualquer substância que,
quando presente em baixas concentrações comparado com a concentração do substrato oxidável, significantemente diminui ou inibe a oxidação daquele substrato”.
1 – Prevenção - A primeira linha de defesa contra as espécies reativas de Oxigênio é a proteção contra a sua formação. A quelação de metais, principalmente cobre e ferro, é o principal mecanismo de controle da peroxidação lipídica e fragmentação do DNA. Portanto a ligação das proteínas ferritina, transferrina, ceruloplasmina e outras metalotioninas aos metais, são de importância central no controle de rações potencialmente geradoras de radicais.
2 – Interceptação - este é o domínio dos antioxidantes propriamente ditos. Os radicais lesivos, uma vez formados, são interceptados para prevenir as suas ações deletérias.
3 - Reparo e eliminação de moléculas com danos excessivos – Os processos de prevenção e a interceptação não são completamente efetivos. Produtos de lesões oxidativas podem se acumular causando lesão no DNA na forma de bases lesadas, quebras de fitas de DNA simples ou dupla; danos na membrana e lesões protéicas e em outros compostos. Existem múltiplos sistemas enzimáticos envolvidos no reparo do DNA, enzimas lipolíticas e proteolíticas capazes de restituir funções ou mesmo substituir compostos extremamente lesados (SIES,1993).
A função de interceptar os radicais lesivos pode ser realizada pelos antioxidantes não-enzimáticos e os antioxidantes não-enzimáticos.
1 - Os antioxidantes não enzimáticos estão envolvidos nos processos de desativação, isto é, a formação de não radicais e produtos finais não reativos a partir de compostos radicais ou a transferência de radicais livres de sítios mais sensíveis para compartimentos celulares no qual o estresse oxidativo possa ser menos deletério. Em geral, isto significa transferir os agentes oxidantes de fases hidrofóbicas para fases aquosas, isto é, da membrana para o citosol ou da lipoproteínas para o meio aquoso do plasma (YOUNG, WOODSIDE, 2001).
Os Antioxidantes não enzimáticos, em sua maioria são exógenos, ou seja, necessitam ser absorvidos pela alimentação apropriada. Entre os antioxidantes não enzimáticos podemos citar as vitaminas Lipossolúveis (vitamina A, vitamina E, beta-caroteno) e Vitaminas Hidrossolúveis (vitamina C, vitaminas do complexo B) os oligoelementos (Zinco, cobre, selênio, magnésio etc.), os bioflavonóides (derivados de plantas) e os tióis antioxidantes (Glutationa, Thioredoxina e Ácido Lipóico) que são radicais hidrofílicos decompositores (BECKMAN, AMES, 1998).
As principais funções protetoras da glutationa contra o estresse oxidativo são: agir como cofator para várias enzimas detoxificantes; participar do transporte de aminoácidos através da membrana plasmática e decompor radicais hidroxila (MEISTER, ANDERSON, 1983).
Elevados níveis de GSH são observados em vários tipos de células cancerosas e tumores sólidos tornando estas células mais resistentes à quimioterapia. CALVERT, et al., 1998;
BALENDIRAN, DABUR, FRASER, 2004; ESTRELA, ORTEGA, OBRADOR, 2006).
2 - Antioxidantes enzimáticos – todas as células eucarióticas possuem poderosas enzimas antioxidantes. As enzimas antioxidantes catalizam a quebra dos radicais livres principalmente no meio intracelular (SIES, 1993).
As três principais classes de enzimas antioxidantes são:
Superóxido dismutase (SOD) que representam o principal sistema antioxidante enzimático intracelular: Cobre e Zinco superóxido dismutase (Cu, Zn-SOD) no citoplasma e manganês Superóxido Dismutase (Mn-SOD) na mitocôndria, rapidamente e especificamente reduzem HO- a H2O2 (MARKLUND, 1982; MATES, PEREZ-GOMEZ, DE CASTRO, 1999);
Glutationa peroxidase (GSH-Px) catalisa a oxidação da glutationa através da conversão do hidroxiperóxido que pode ser peróxido de hidrogênio ou outra espécie tais como hidroxiperóxido lipídico. A glutationa peroxidase parece ser o principal mecanismo de decomposição do peróxido de hidrogênio no citosol (MEISTER, ANDERSON, 1983);
Catalases (CAT) cataliza os dois estágios de conversão do peróxido de hidrogênio a água e oxigênio. Está localizada dentro das células, nos peroxissomos, que também contem a maioria das enzimas capazes de gerar peróxido de hidrogênio. (KIRKMAN, GALIANO, GAETANI, 1987).
Alem disso existem enzimas com função antioxidante indireta; Glutationa S redutase redutase) que repõe a GSH a partir da GSSG; e Glutationa S transferase (GSSG-transferase) responsável pelo transporte e eliminação de compostos reativos e pelo sistema de transporte para os conjugados S-glutationa (SIES, 1997).
Quando há um desequilíbrio entre espécies reativas e antioxidantes dentro da célula, nas membranas e no espaço extracelular, com aumento das espécies reativas pode ocorrer lesão de importantes biomoléculas e células, com potencial impacto em todo o organismo (REUTER et
As principais ações das espécies reativas nos sistemas biológicos são: peroxidação lipídica, oxidação protéica e dano ao DNA.
A peroxidação lipídica um processo através do qual as EROs agridem os ácidos graxos polinsaturados dos fosfolipídeos das membranas das células, desintegrando-as e permitindo, desta feita, a entrada dessas espécies nas estruturas intracelulares, resultando na ruptura das membranas celulares (bombas Na/K e Ca/Mg); Mutações do DNA; Oxidação dos lipídios insaturados; Formação de resíduos químicos como o malonaldeído; Comprometimento dos componentes da matriz extracelular, proteoglicanos, colágeno e elastina (GUTTERIDGE,1995).
A modificação protéica determinada pelas EROs é iniciada principalmente por reações com radical hidroxila (OH-). Coletivamente os radicais formados neste processo podem levar a oxidação de resíduos de aminoácidos das cadeias laterais, formação de ligações cruzadas entre proteínas e oxidação do arcabouço resultando na fragmentação protéica (BERLETT, STADTMAN, 1997).
A oxidação de resíduos de aminoácidos que contem enxofre, são particularmente sensíveis a ação das EROs porem são os únicos que podem converter os seus resíduos oxidados à sua forma não modificada. Portanto são as únicas modificações oxidativas protéicas passíveis de reparação.
A clivagem oxidativa de proteínas leva à formação de derivados carbonil que podem ser usado como marcador de oxidação protéica mediada por EROs. O nível intracelular de proteína oxidada reflete o equilíbrio entre a taxa de oxidação protéica e a taxa de degradação de proteína oxidada que também é dependente de muitas variáveis, incluindo a concentração de proteases cuja ação lítica pode gerar inúmeros fatores que, por sua vez, podem afetar sua atividade proteolítica (BERLETT, STADTMAN, 1997).
A observação do aumento do estresse oxidativo em condições patológicas tem sugerido o uso de biomarcadores de estresse oxidativo/nitrosativo para o desenvolvimento de novas estratégias diagnósticas, terapêuticas e preventivas para impedir ou retardar o desenvolvimento de complicações.
Produtos da peroxidação lipídica tem sido comumente utilizado como biomarcadores do estresse e dano oxidativo. A peroxidação lipídica gera uma variedade de produtos finais de decomposição relativamente estáveis. Os reativos de aldeído alfa beta insaturados, tais como malonaldeido (MDA), 4 hidroxi 2 nonenal (HNE) e 2-propenal (acroleína) e isoprostano podem ser medidos no plasma e urina como um índice indireto de estresse oxidativo (DALLE-DONNE,
et al, 2006).
A técnica mais largamente utilizada para avaliar a peroxidação lipídica é a análise do TBARS que inclui MDA. MDA é um cetoaldeido produzido pela decomposição peroxidativa de lipídios insaturados (DALLE-DONNE, et al, 2006). TBA reage com o MDA e produz um
cromógeno que pode ser quantificado pelo espectofotômetro (DRAPER, HADLEY,1990). Autores tem medido as concentrações de GHS e GSSG no sangue total ou hemácias, comparando os resultados obtidos em indivíduos saudáveis com os encontrados em pessoas afetadas por várias doenças, reportando a correlação entre a doença e as alterações nas concentrações de GSSG, GSH ou GSH/GSSG (ISIDÓRIO, SILVEIRA, 2008)
Na inativação de um agente oxidante ocorre produção de GSSG e depleção de GSH. Em situações em que o sistema redox está íntegro, haverá recuperação da GSH via glutationa redutase na presença de NADPH (ISIDÓRIO, SILVEIRA, 2008).
A formação de compostos carboniol é o mais largamente utilizado marcador de severa oxidação protéica (DALLE-DONNE et al, 2006).
1.3 Estresse oxidativo e câncer
A superprodução de EROs ou a supressão da habilidade celular de eliminar as EROs , pode resultar num aumento de espécies reativas no meio intracelular, levando a lesão celular incluindo peroxidação lipídica, modificações oxidativas do DNA, oxidação protéica e inativação enzimática (REUTER et.al, 2010).
O acúmulo de lesão oxidativa tem sido implicada na formação de vários tipos de doenças, inclusive câncer (KLAUNIG et.al, 1998).
Os efeitos biológicos do estresse oxidativo no câncer são múltiplos e não lineares (FANG, SEKI, MAEDA, 2009).
A lesão oxidativa aguda pode produzir morte celular seletiva e aumento compensatório da proliferação celular que pode resultar na expansão clonal seletiva de células preneoplásicas latentes. Níveis baixos ou intermediários de estresse oxidativo, lesão oxidativa subletal, são mais efetivos em produzir lesões do DNA causando mutação, reação inflamatória e finalmente induzindo à carcinogênese (MARTINDALE; HOLBROOK, 2002; FANG, SEKI, MAEDA, 2009).
Espécies reativas de oxigênio tem um potencial carcinogênico pois elas facilitam a mutagênese, a promoção e progressão tumoral (DREHER, JUNOD,1996). EROs tem sido consideradas como uma classe endógena de carcinógeno visto que desencadeia mutação nas células (GUYTON, KENSLER, 1993; CERUTTI, 1994; FANG; SEKI; MAEDA, 2009).
O estágio inicial envolve uma mutação somática e não letal que confere vantagem de crescimento. Espécies reativas podem mediar esta ativação Inicial através da formação de bases de DNA hidroxiladas, uma lesão oxidativa comum considerada um importante evento na carcinogênese, pois interfere com o crescimento celular normal causando mutações genéticas e alterando a transcrição gênica normal.
As próprias células iniciadas, e as células inflamatórias são fontes de EROs. Estas múltiplas fontes de EROs podem contribuir para o persistente estresse oxidativo que resulta em alterações fisiopatológicas e permitem o crescimento destas células preneoplásicas (KLAUNIG
et.al, 1998).
Altos níveis de espécies reativas podem alterar as vias de sinalização através de lesão oxidativa da membrana celular, de mudanças na atividade enzimática e/ou de ativação dos fatores de transcrição, que são proteínas de baixo peso molecular que se ligam à região promotora do gene e regulam a transcrição dos genes envolvidos no desenvolvimento, crescimento e envelhecimento celular (VELLANOWETH; SUPRAKAR; ROY, 1994).
O estresse oxidativo está envolvido no processo de regulação da localização subcelular dos fatores de transcrição, do citoplasma para o núcleo. A localização nuclear dos fatores de transcrição é um dos primeiros passos para que tais fatores iniciem suas atividades (REUTER et
al, 2010).
No estágio de progressão tumoral, o estresse oxidativo tem um papel direto no desenvolvimento das características das células malignas, tais como crescimento descontrolado e instabilidade genômica.
Através do processo de carcinogênese surgem células com genes supressores de tumor deletados e oncogenes ativados e capazes de produzir seus próprios fatores de crescimento (TOYOKUNI et al, 1995).
Inúmeros fatores de estresse celular, inclusive EROs são responsáveis por características das células tumorais tais como sobrevida, proliferação, invasão, quimioresistência, radioresistênca e autorenovação que fornece indesejável vantagem às células malignas (TOYOKUNI et.al, 1995). Células cancerosas possuem mecanismos antioxidantes super
regulados que irão protegê-las contra as espécies reativas de oxigênio ou utilizá-las a seu favor (TOYOKUNI et.al, 1995).
1.4 Estresse oxidativo e LMC
Estresse oxidativo, radiação, químicos genotóxicos e ou estresse de replicação podem induzir quebras de dupla fita de DNA. O reparo infiel destas quebras pode fazer surgir translocações cromossômicas recíprocas. Os genes que codificam tirosinas kinases (TK) são alvo destes mecanismos de erros genéticos, resultando na geração de genes quiméricos que codificam tirosinas kinase de fusão (TKF). As TKF apresentam atividade kinase descontrolada que levam ao desenvolvimento de neoplasias (PENSERGA, SKORSKI, 2007).
A proteína BCR-ABL, resultado da translocação (9:22), é uma TKF associada à LMC e a
um grupo de LLA. A constitutiva atividade kinase desta oncoproteína é responsável pelo aumento de leucócitos, característica da fase crônica da LMC. Além da proliferação desregulada, na ausência de fatores de crescimento as TKFs protegem as células malignas contra a apoptose e levam também a desregulação da diferenciação e da adesão (BEDI et al., 1994; GORDON et
al.,1987; WILSON-RAWLS, et al., 1996; DONATO, et al., 2001; PENSERGA; SKORSKI,
2007).
As TKFs tem um papel importante na modulação da resposta a lesões no DNA, aumentando a habilidade das células malignas de sobreviver ao estresse genotóxico e oxidativo que normalmente induziriam resposta apoptótica em células não leucêmicas (AMARANTE et.al.,
1998).
As principais linhas de defesa elucidadas pela atividade kinase BCR-ABL para dotar às
células fenotipicamente anormais de mecanismos de vantagem de crescimento são:
Estimulo de proteínas antiapoptóticas tais como BCL2 e BCL-X (AMARANTE, et.al, 1998)e
inibição das proteínas proaptóticas BAD e BAX como foi demonstrado por Yoshida e colaboradores, 2007.
Prolongamento das fases S e G2/M do ciclo celular, estendendo o tempo de reparo bem como ativando os mecanismos de reparo (SLUPIANEK et al., 2006; PENSERGA, SKORSKI, 2007).
Aumento da atividade dos mecanismos de reparo de quebras de duplas fitas de DNA (TAKEDA, SHIBUYA, MARU, 1999).
avançadas (fase acelerada e crise blástica) e aquisição de resistência às moléculas inibidoras das TKFs, como o Imatinibe (HOCHHAUS et al., 2002; BURKE; CARROLL, 2010)
As células BCR-ABL positivas são fonte de EROs as quais causam lesão oxidativa no
DNA, contribuindo também para a geração de mutações. EROs trabalham em conjunto com
BCR-ABL para introduzir novas mutações na proteína de fusão, incluído aquelas que codificam
resistência ao imatinibe. A inibição da BCR-ABL associado à ação de antioxidantes poderia evitar
o processo cíclico de mutagênese (KOPTYRA et al., 2006).
2OBJETIVOS
2.1 Objetivo geral
Determinar o perfil oxidativo dos pacientes com LMC acompanhados no ambulatório de hematologia do Hospital Universitário Walter Cantídio (HUWC-UFC).
2.2 Objetivos específicos
Avaliar o perfil clínico (fase da doença e escore de Sokal) e demográfico (idade, sexo) dos pacientes com LMC, ao diagnóstico;
Comparar os pacientes em relação à atividade da doença com o uso de medicamentos e com o tempo de doença;
Comparar os parâmetros do perfil oxidativo dos pacientes portadores de LMC estratificados pela a atividade da doença;
Comparar o perfil oxidativo, dos pacientes com doença ativa estratificados pela inibidor TK em uso (1ª geração ou 2ª geração);
Comparar o perfil oxidativo dos pacientes com LMC em uso de imatinibe estratificados pela atividade da doença;
3CASUÍSTICAEMÉTODOS
3. 1 Casuística
3.1.1 Grupo de estudo
O estudo foi do tipo transversal, em 30 pacientes adultos, de ambos os sexos, com o diagnóstico de LMC segundo os critérios de classificação da OMS (SWERDLON et al., 2008),
acompanhados no ambulatório de hematologia do Hospital Universitário Walter Cantídio (HUWC). Os pacientes foram selecionados de forma aleatória obedecendo aos critérios de inclusão e exclusão definidos abaixo.
Critérios de Inclusão
Pacientes adultos de ambos os sexos, com diagnóstico clínico e laboratorial (hemograma, mielograma, citogenética e/ou molecular) de LMC, que estavam em uso de inibidores TK acompanhados no ambulatório de hematologia do HUWC-UFC /HEMOCE.
Critérios de exclusão
Pacientes com o diagnóstico de LMC na ausência de tratamento com inibidores de TK ou com avaliação citogenética ou molecular há mais de 6 meses, em relação a época do estudo. Pacientes que se recusaram a participar do estudo.
3.1. 2 Grupo controle
sangue do HEMOCE, não tabagistas e nem etilistas e que não estavam fazendo uso de suplementos vitamínico (Vitamina C e/ou vitamina E).
3.1.3 Obtenção dos dados
Foram obtidas informações, nos prontuários dos pacientes localizados no Serviço de Arquivo Médico-Hospitalar (SAME), do HUWC, e anotadas em ficha clínica padronizada (Apêndice 1, pag. 92), a qual continha a identificação dos pacientes, dados demográficos, clínicos e exames complementares, ao diagnóstico e durante o estudo.
3.2. Estratificação do Grupo de estudo
3.2.1 Estratificação em relação à atividade ou não da doença
Os pacientes com LMC foram estratificados em dois grupos, de acordo com as evidências de atividade da doença, definidas levando-se em conta: 1 - avaliação clínica; 2 - hemograma e os estudos citogenéticos e molecular realizado dentro do período de no máximo 6 meses antes da avaliação do perfil oxidativo de acordo com as recomendacões para monitoramento individual dos pacientes com LMC em uso de inibidores TK (BACCARANI et al., 2009).
Grupo 1 - Sem evidências de atividade da doença
Critérios clínicos e hematológicos
Baço impalpável
Leucócitos ≤ 10.000/mm3
Plaquetas ≤ 450.000/mm3
Ausência de células imaturas no sangue periférico
Obs.: pacientes que apresentavam contagem plaquetas abaixo de 150.000/mm3 porem igual ou superiora 100.000/mm3 não foram considerados como em atividade da doença sendo esta citopenia imputada à medicação (TALPAZ et al.,2006; KANTARJIAN, et al.,2006;
KANTARJIAN, et al.,2011; GNONI et al., 2011).
Critério Citogenético
AusênciadeCromossomo Ph nas metáfases analisadas
Critério Molecular
Queda no número de transcritos BCR-ABL da ordem de 3 ou mais Logs. Baseado no estudo IRIS que forneceu evidências que a redução de transcritos BCR-ABL de 3 logs ou mais
estava associada à maior sobrevida livre de doença (BACCARANI et al., 2009).
Grupo 2 - Com evidências de doença em atividade
Critérios clínicos e hematológicos (Qualquer um dos abaixo listados)
Baço palpável
Leucócitos > 10.000/mm3
100.000/mm3> Plaquetas > 450.000/mm3
Presença de células imaturas no sangue periférico
Basófilos ≥ 5%.
Critério Citogenético
Presença de Cromossomo Ph nas metáfases analisadas
Critério Molecular
Queda no número de transcritos BCR-ABL de 2 Logs ou menos. (BACCARANI
3.2.2 Estratificados de acordo com o tipo de inibidor de TK
Os pacientes foram estratificados quanto ao uso de Imatinib ou inibidores de 2ª geração de TK no momento do estudo.
3.2.3. Estratificados de acordo com o tempo de doença
Os pacientes foram estratificados de acordo com o tempo de doença, no momento do estudo, tendo como parâmetro a data do diagnóstico: 3 a 5 anos, 6 a 10 anos e > 10 anos de doença.
3.3. Local do Estudo
O estudo foi realizado no Laboratório de Pesquisa em Hemoglobinopatias e Doenças Genéticas Hematológicas (LPHDGH) do Departamento de Análises Clínicas e Toxicológicas do Curso de Farmácia da Universidade Federal do Ceará (UFC), no período de maio a outubro de 2010.
3.4 Aspectos Éticos
O presente trabalho foi submetido ao Comitê de Ética em Pesquisa do HUWC, tendo sido emitido parecer favorável para a execução da pesquisa, sob o protocolo No 042.05.10.
3.5 Métodos
3.5.1 Coleta e processamento das amostras
As amostras foram coletadas seguindo o procedimento padrão. Foram coletados 10 mL de sangue venoso em tubos de coleta a vácuo contendo heparina para a realização das dosagens dos marcadores do perfil oxidativo (MDA e NO2-) e das dosagens das enzimas antioxidantes (CAT e GSH-Px), e tiois: Glutationa Reduzida(GSH), glutationa oxidada(GSSH) e glutationa total.
As amostras foram encaminhadas para o laboratório de Hemoglobinopatias e Genética das Doenças Hematológicas (LHDGH), do Curso de Farmácia da UFC onde foram centrifugadas durante 10 minutos a 3.000 rpm. O plasma sobrenadante foi retirado, separado em alíquotas em triplicata, enumerados e armazenados a -80oC, para posterior análise. O hemolisado eritrocitário foi obtido da amostra com anticoagulante heparina após processo de remoção completa do plasma e da camada leucocitária por aspiração, de sucessivas lavagens com solução salina (NaCl 0,9%) e da hemólise por adição de água destilada 1:1. O hemolisado foi alicotado em triplicata, identificados e armazenado em freezer a -80o C até a análise. Ambas as amostras foram armazenadas até a análise, cerca de 2 meses após a coleta.
3.5.2 Análises do Perfil oxidativo
Protocolo para determinação da concentração de Nitrito (NO2-) (GREEN et.al., 1982)
Este método se baseia na detecção da presença de nitrito em amostras biológicas (urina, plasma, homogenado tecidual) através de uma reação de diazotização com formação de um cromóforo de cor rósea. O método consta da adição de 100 μL da amostra plasmática em 100 μL
reativo de Griess a 100 L do plasma e a absorbância foi medida em leitor de microplacas em 560 nm.
Protocolo para determinação da concentração de Malonaldeído (MDA) (DRAPER,
HADLEY, 1990)
O método mais empregado para determinação do MDA em amostras biológicas é baseado na sua reação com o ácido tiobarbitúrico -TBARS. Nessa reação, duas moléculas de TBARS reagem estequiometricamente com uma molécula de MDA para formar um cromóforo róseo que tem absorbância máxima em solução ácida em 532nm a 535nm. A quantificação foi realizada por espectrofotometria de acordo com o protocolo empregado, que consiste na incubação de 150μL
de plasma em banho-maria a 37º por 1 h, seguido por adição de 200μL de ácido perclórico a
35%, para precipitar as proteínas. Em seguida realizou-se a centrifugação a 14.000rpm por 10
min, após, foi adicionado ao sobrenadante, 100μL de uma solução de tiobarbiturato de Sódio a 0,6%. A amostra permaneceu em banho-maria aquecido entre 90 e 100oC por 30 min. Após resfriada, a absorbância foi medida em leitor de microplacas a 560nm.
Protocolo para determinação da atividade enzimática da catalase (CAT) (AEBI, 1984)
A atividade da CAT nos eritrócitos foi determinada por método espectrofotométrico em ultravioleta a 240nm, baseado na monitorização da decomposição de H2O2. Os valores brutos
obtidos em Δa (delta/min) foram corrigidos por um fator de correção de decaimento da atividade
por diluição do hemolisado, divididos pela absortividade molar de H2O2 a 240nm (ε 0,0394 L mmo1-1mm-1).
Protocolo para determinação da atividade enzimática da glutationa peroxidase (GSH-Px)
A medida da GSH-Px nos eritrócitos foi determinada utilizando o kit Glutathione
Peroxidase Cellular Activity Assay® (Sigma-Aldrich). O kit utiliza método de determinação
Determinação das concentações de Glutationa
A determinação das concentrações de Glutationa Total, GSH e GSSG foram realizadas utilizando o método empregado no kit Total Glutathione Activity® (Assay Designs, Inc). A
quantificação da Glutationa Total utiliza um método otimizado de reciclagem enzimática. A GR
reduz a GSSG a GSG. O grupo sulfidril da GSH reage com o reagente de Ellman, o ácido 5,5’ -dithiobis-2-nitrobenzoico (DTNB), produzindo um ácido de cor amarela, o ácido 5,5-thio-2-nitrobenzoico (TNB), que é determinado espectrofotometricamente em uma absorbância entre 405 a 414 nm. A taxa de produção de TNB é diretamente proporcional à concentração de Glutationa total na amostra. A medição da absorbância do TNB fornece uma estimativa precisa da glutationa na amostra.
3.6 Descarte do Material Biológico
O descarte do material biológico foi realizado segundo a resolução da diretoria colegiada
– RDC 306, de 7 de dezembro de 2004 da Agência Nacional de Vigilância Sanitária.
3.7 Análise Estatística
4
RESULTADOS
4.1 características sócio demográficas dos pacientes
Participaram do estudo 30 pacientes com diagnóstico de LMC, sendo14 do sexo masculino e 16 do sexo feminino, todos classificados como pardos (Tabela 1).
Tabela 1 –Distribuição dos pacientes quanto ao sexo (n=30)
Fonte: Dados da pesquisa, 2011
A idade dos pacientes variou de 22 a 73 anos, com média de 44,13 anos. O pico de prevalência foi observado na faixa etária de 31 a 40 anos. (Tabela 2).
Tabela 2 - Distribuição dos pacientes por faixa etária
Faixa etária Pacientes n=30 %
22 a 30 anos 6 20
31 a 40 anos 10 33,3
41 a 50 anos 3 10
51 a 60 anos 6 20
61 a 70 anos 4 13,3
Acima de 70 anos 1 3,3
Fonte: Dados da pesquisa, 2011
4.2 Caracterização clínica dos pacientes ao diagnóstico
Ao diagnóstico, 83,3% dos pacientes se encontravam na fase crônica da doença como se observa pela tabela 3. Nenhum paciente do estudo foi diagnosticado em crise blástica.
Sexo Pacientes n=30 %
Masculino 14 47,7
Tabela 3 – Distribuição dos pacientes de acordo com a fase da doença ao diagnóstico. (n=30)
Fase da doença Pacientes n=30 %
Crônica 25 83,3
Acelerada 5 16,7
C. Blástica 0 0
Fonte: Dados da pesquisa, 2011
O prognóstico dos pacientes foi avaliado através do sistema de prognóstico de Sokal, conforme dados apresentados na tabela 4.
Tabela 4 –Distribuição dos pacientes quanto ao escore de Sokal (n=30)
Escore de Sokal Pacientes (n=30) (%)
Baixo 8 26
Intermediário 11 37
Alto 11 37
Fonte: Dados da pesquisa, 2011
4.3 Caracterização clínica atual dos pacientes
Figura 1 - Distribuição dos grupos quanto à medicação em uso
Fonte: Dados da pesquisa, 2011
Entre os pacientes com mais de 5 anos de diagnóstico havia 2 pacientes que estavam com mais de 10 anos de doença (19 e 24 anos), tais pacientes se apresentavam com doença em atividade (figura 2).
Figura 2 – Distribuição dos pacientes quanto ao tempo de doença e ao nível de atividade da doença.
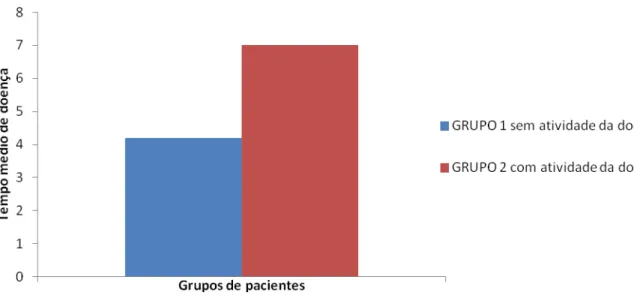
O tempo médio de diagnóstico da doença, nos pacientes do grupo 1 foi de 4,2 anos. O grupo 2 tinha em média 7 anos de doença (figura 3).
Figura 3– Distribuição dos pacientes estratificados pelo nível de atividade da doença quanto ao tempo médio de doença.
Fonte: Dados da pesquisa, 2011
4.4 análise dos parâmetros do perfil oxidativo nos pacientes estratificados de acordo com a atividade da doença
4.4.1 Marcadores do estresse oxidativo
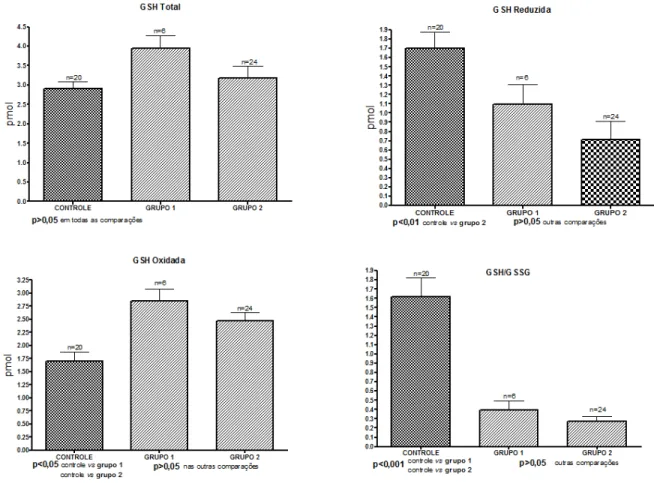
Os níveis de MDA se apresentaram elevados nos grupos de pacientes, em comparação com o grupo controle, observando-se diferença estatisticamente significante (p<0,05). Na comparação dos grupos de pacientes entre si não foi observado diferença significante embora tenha havido discreto aumento nos pacientes sem atividade em relação aos com atividade (Figura 4).
nitrito mais elevados que o grupo controle tal diferença não foi estatisticamente significante (Figura 4).
Figura 4 - Valores médios±desvio padrão de MDA e Nitrito no grupo controle (n=20) nos pacientes do grupo 1 sem atividade da doença (n=6) e no grupo 2 com atividade da doença (n=24)
Fonte: Dados da pesquisa, 2011
4.4.2 Enzimas antioxidantes
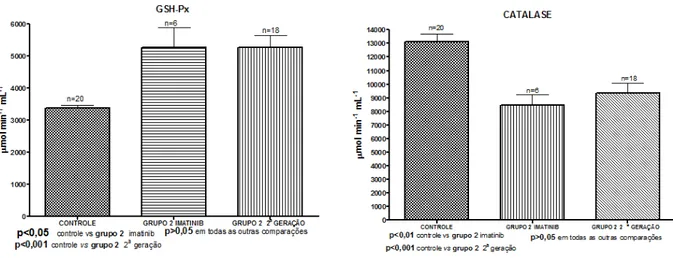
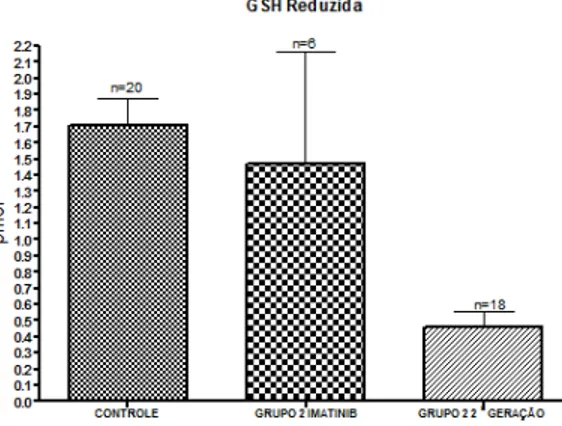
Como pode ser observada na figura 5 a atividade da GSH-Px foi significativamente mais elevada nos grupos de pacientes em comparação com o grupo controle.
Ao contrário, os níveis de CAT foram mais elevados no grupo controle que nos dos pacientes, com diferença significante entre o grupo controle e o grupo 2 (p<0,001).