1
Identification and characterization of novel splice variants of human
1
farnesoid X receptor
2
Enni-Kaisa Mustonena, Serene M. L. Leeb, Hanno Nießb, Matthias Schwaba,c, Tatu Pantsard,e, Oliver 3
Burka*
4
aDr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart and University of Tübingen, 5
Tübingen, Germany 6
bBiobank of the Department of General, Visceral and Transplantation Surgery, University Hospital LMU 7
Munich, Munich, Germany 8
cDepartments of Clinical Pharmacology, and Pharmacy and Biochemistry, University of Tübingen, 9
Tübingen, Germany 10
dDepartment of Pharmaceutical and Medicinal Chemistry, Institute of Pharmaceutical Sciences, University 11
of Tübingen, Tübingen, Germany 12
eSchool of Pharmacy, Faculty of Health Sciences, University of Eastern Finland, Kuopio, Finland 13
*Corresponding author:
14
Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Auerbachstrasse 112, D-70736 Stuttgart, 15
Germany.
16
E-mail address: [email protected] 17
2
Abbreviations
18
AD, activation domain; BSEP, bile salt export pump; CAR, constitutive androstane receptor;
19
cDNA, complementary DNA; CDCA, chenodeoxycholic acid; DBD, DNA binding domain; EMSA, 20
electrophoretic mobility shift assay; ER, everted repeat; FXR, farnesoid X receptor; FXRE, FXR 21
response element; IR, inverted repeat; LBD, ligand binding domain; LBP, Ligand binding pocket;
22
MD, molecular dynamics; NASH, non-alcoholic steatohepatitis; NCOA1, nuclear receptor 23
coactivator 1; RID, receptor interaction domain; RXR, retinoid X receptor; SHP, small heterodimer 24
partner; SV, splice variant; UTR, untranslated region 25
3
Abstract
26
Farnesoid X receptor (FXR, NR1H4) is a ligand-activated nuclear receptor, which regulates bile 27
acid, lipid and glucose metabolism. Due to these functions, FXR has been investigated as a 28
potential drug target for the treatment of liver diseases, such as primary biliary cholangitis and 29
non-alcoholic steatohepatitis. Based on the previously described four splice variants, it has been 30
suggested that alternative promoter usage and splicing may have an impact on total FXR activity 31
as a result of encoding functionally diverse variants. Here we aimed for a systematic analysis of 32
human hepatic FXR splice variants. In addition to the previously described FXRα1–4, we identified 33
four novel splice variants (FXRα5–8) in human hepatocytes, which resulted from previously 34
undetected exon skipping events. These newly identified isoforms displayed diminished DNA 35
binding and impaired transactivation activities. Isoform FXRα5, which suppressed the 36
transactivation activity of the functional isoform FXRα2, was further characterized as deficient in 37
heterodimerization, coactivator recruitment and ligand binding. These findings were further 38
supported by molecular dynamics simulations, which offered an explanation for the behavior of 39
this isoform on the molecular level. FXRα5 exhibited low uniform expression levels in nearly all 40
human tissues. Our systematic analysis of FXR splice variants in human hepatocytes resulted in 41
the identification of four novel FXR isoforms, which all proved to be functionally deficient, but one 42
novel variant, FXRα5, also displayed dominant negative activity. The possible associations with 43
and roles of these novel isoforms in human liver diseases require further investigation.
44
45 46 47 48 49 50 51 52
Keywords: Farnesoid X receptor, Alternative splicing, Dominant negative protein, Transactivation, DNA 53
binding, Nuclear receptor 54
55
4
1. Introduction
56
Farnesoid X receptor (FXR, NR1H4) is a ligand-activated transcription factor that belongs to the 57
nuclear receptor family. FXR was initially identified in rat and mouse liver, and the first recognized 58
FXR agonists were farnesol metabolites. A few years later, bile acids were identified as 59
endogenous ligands of FXR; hence, it is also known and sometimes referred to as bile acid 60
receptor (for a review see [1]). Similar to many other nuclear receptors, FXR activates its target 61
genes mainly by binding to FXR response elements (FXRE) in regulatory chromatin regions as a 62
heterodimer with retinoid X receptor alpha (RXRα). Usually, these FXREs consist of inverted 63
repeats of the canonical nuclear receptor hexamer motif separatedby one nucleotide (IR1) [2].
64
However, genome-wide analyses of FXR chromatin binding in human and mouse liver and/or 65
intestine additionally identified everted repeats separated by two nucleotides (ER2) and combined 66
IR1/ER2 motifs as prominent FXR binding sites [3,4].
67
FXR is most highly expressed in liver, small intestine and colon [5,6]. The receptor plays a vital 68
role in bile acid homeostasis by upregulating the expression of bile salt export pump (BSEP, 69
ABCB11) and small heterodimer partner (SHP, NR0B2) [7, 8]. Additionally, FXR contributes to 70
hepatic lipid and glucose metabolism [9], as well as to amino acid metabolism and ureagenesis 71
[10]. Consequently, FXR activation is discussed as an approach for the treatment of liver diseases 72
[11]. The FXR agonist obeticholic acid was recently approved for the treatment of primary biliary 73
cholangitis, and it has been investigated in clinical trials for the treatment of non-alcoholic 74
steatohepatitis (NASH) [12,13,14]. Besides affecting metabolic liver diseases, FXR has also a 75
role in liver and gastrointestinal tumorigenesis [15].
76
To date, four distinct human FXR splice variants (SV), resulting from alternative promoter and 5’
77
donor splice site usage, have been identified [5], which are named FXRα1–4 here (Fig. 1), 78
according to [1]. FXRα1 and FXRα2 transcripts originate by transcription initiation at the promoter 79
upstream of exon 1. FXRα3 and FXRα4 utilize an alternative internal promoter upstream of exon 80
3a, which results in a different N-terminus of the encoded isoforms (Fig. 2A). In addition, use of 81
an alternative splice donor site in intron 5 leads to the in-frame insertion of 12 nucleotides at the 82
3`end of exon 5 (FXRα1, FXRα3), encoding the inserted amino acids MYTG. While FXRα1 and 83
FXRα2 display highest expression in liver and adrenal gland, FXRα3 and FXRα4 are expressed 84
most abundantly in intestine and kidney [5,6]. Differential target gene activation by the encoded 85
FXR isoforms has been observed [5,6,16]. In general, FXRα2 activates target genes most 86
strongly. However, some genes, such as the organic solute transporter β (SLC51B), are activated 87
to the same extent by all isoforms [6]. Consequently, the reported modulation of FXR splicing by 88
5
physiological stress, which resulted in a change of FXRα1/α2 ratio [16], is expected to impact on 89
the expression of distinct FXR target genes.
90
To the best of our knowledge, no systematic approach for identification and characterization of 91
human FXR SVs has been reported. Thus, we conducted here an analysis of FXR SVs in human 92
hepatocytes. As a result, four novel FXR SVs resulting from previously unknown exon skipping 93
events were identified. We characterized the encoded novel FXR isoforms with respect to their 94
DNA binding and transactivation properties and compared their function to the previously 95
described isoforms FXRα1–4. All newly identified FXR isoforms demonstrated loss-of-function, 96
with FXRα5 further demonstrating dominant negative activity. Our data demonstrate that exon 97
skipping produces functionally defective isoforms that could impair total FXR activity.
98 99
2. Material and methods
100
2.1. Chemicals, oligonucleotides and cell culture reagents
101DMSO and chenodeoxycholic acid (CDCA) were purchased from Sigma Aldrich (Taufkirchen, 102
Germany). GW4064 was provided by Tocris Bioscience (Bristol, UK). Oligonucleotide primers 103
were purchased from Biomers (Ulm, Germany). Minimum essential medium (MEM) and trypsin- 104
EDTA were purchased from Thermo Fisher Scientific (Waltham, MA). L-glutamine and penicillin- 105
streptomycin mixture were obtained from Biozym (Hessisch Oldendorf, Germany). Biowest 106
(Nuaillé, France) provided the fetal bovine serum (FBS).
107 108
2.2. Cloning of FXR isoforms and plasmids
109The open reading frame of FXR variants with the start codon in exon 3 (position 477-1895 of 110
NM_005123.3) was amplified by PCR from oligo(dT)-primed cDNA samples of primary human 111
hepatocytes, which were derived from two donors, with primer pair F1/R1 (Table 1). Primer pair 112
F2/R1 (Table 1) was used to amplify correspondingly the open reading frame of FXR variants with 113
the start codon residing in exon 3a (position 29-1477 of NM_001206992.1). The forward primers 114
F1 and F2 both introduced KpnI sites and Kozak consensus sequences, while the common 115
reverse primer R1 introduced NotI site. Respective KpnI/NotI digested PCR products were cloned 116
into appropriately digested eukaryotic expression vector pcDNA3. The inserts of resulting clones 117
were sequenced and clones corresponding to the known isoforms FXRα1–4 were selected, 118
yielding expression plasmids pcDFXRα1–4. Clones harboring cDNA inserts, corresponding to 119
previously unknown FXR isoforms due to novel exon skipping events (see Fig. 1), were 120
consequently called FXRα5–8, with expression plasmids named pcDFXRα5–8.
121
6
The expression plasmid pcDFXRα5-FLAG, encoding FXRα5, which was tagged at its carboxy 122
terminus with the FLAG epitope, was constructed by cloning the PCR-amplified open reading 123
frame of FXRα5 in frame with the 8 amino acid DYKDDDDK-FLAG peptide into a respectively 124
modified pcDNA3 vector (pcDNA3-FLAG) [17]. The eukaryotic expression plasmid 125
pcDhuRXRα(orf) encoding human RXRα has been described [18]. Promega (Madison, WI) 126
provided the Renilla luciferase expression plasmid pRL-CMV.
127
A 251 bp fragment of the human BSEP promoter/exon 1 region (positions 1279-1529 of 128
AF190696.1), ranging from -170 to +81 (numbering refers to the transcriptional start site) was 129
amplified by PCR from genomic DNA of Huh7 cells using appropriate primers, which introduced 130
KpnI and HindIII sites, respectively. The KpnI/HindIII-digested PCR fragment was cloned into 131
correspondingly digested pGL3-Basic (Promega) to generate pGL3-BSEP(-170/+81). Identity of 132
the cloned fragment was verified by sequencing.
133
The GAL4-DNA-binding domain (DBD)-dependent firefly luciferase promoter/reporter gene 134
plasmid pGL3-G5 and expression plasmids encoding fusion proteins of the GAL4-DBD and the 135
ligand-binding domain (LBD) of human RXRα (residues 226-462) or the receptor interaction 136
domain (RID) of human nuclear receptor co-activator (NCOA) 1 (residues 583-783) have been 137
described before [19]. Expression plasmids encoding fusion proteins of the VP16-activation 138
domain (AD) and the LBDs of FXRα2 (residues 241-472) or FXRα5 (residues 241-434) were 139
constructed by cloning the respective fragments, generated by PCR using appropriate primers 140
and respective expression plasmids as templates, into vector pVP16 (Takara Bio Clontech, 141
Mountain View, CA). PCR-derived fragments were verified by sequencing.
142 143
2.3. In vitro protein synthesis
144Human FXR protein isoforms were synthesized in vitro using the respective expression plasmids 145
and the TNT T7 Quick Coupled Transcription/Translation System (Promega). For radiolabeling of 146
the proteins, L-methionine was replaced by L-35S-methionine with specific activity > 1000 Ci/mmol 147
(Hartmann Analytic, Braunschweig, Germany).
148 149
2.4.
Electrophoretic mobility shift assays (EMSA) 150Unlabeled, in vitro synthesized human FXR isoform proteins and human RXRα protein were used.
151
Annealing of complementary single-stranded oligonucleotides generated the respective double- 152
stranded oligonucleotides, which harbored the indicated FXR binding motifs. The sequences of 153
the single-stranded oligonucleotides are shown in Supplementary Table S1. Double-stranded 154
oligonucleotides were radiolabeled by filling 5’ protruding ends using Klenow enzyme and α-32P- 155
7
dCTP with specific activity of 3000 Ci/mmol (Hartmann Analytic), and purified as described before 156
[18]. Binding reactions were set up and gel electrophoresis was done, as described previously 157
[20]. After drying, the gels were exposed to BAS-IP MS2325 imaging plates (Fuji, Kanagawa, 158
Japan), which were read after exposure with the CR35 Bio radioluminography laser scanner 159
(Raytest, Straubenhardt, Germany). In antibody supershift experiments, in vitro synthesized 160
FLAG-tagged FXRα5 protein and monoclonal anti-FLAG® M2 antibody (Sigma-Aldrich, cat.-no.
161
F3165; RRID: AB_259529) were applied.
162 163
2.5. Limited proteolytic digestion
164Limited proteolytic digestion assays were performed as described previously [21]. Briefly, 165
radiolabeled FXRα2 or FXRα5 proteins were pre-incubated with GW4064 or solvent DMSO and 166
then subjected to proteolytic digestion by trypsin. Reactions were separated on 12% SDS 167
polyacrylamide protein gels, which, after the run, were stained with Coomassie, dried and 168
exposed to BAS-IP MS 2325 imaging plates. Input FXR proteins and protected proteolytic 169
fragments were visualized by scanning the imaging plates with CR35 Bio radioluminography laser 170
scanner.
171 172
2.6. Cell culture
173HepG2 cells (HB-8065, lot number 58341723, ATCC, Manassas, VA) were cultivated at 37°C, 174
5% CO2 in MEM, which was supplemented with 10% FBS, 2 mM glutamine, 100 U/ml penicillin 175
and 100 µg/ml streptomycin. HepG2 cells were used in the experiments for 25 passages. In 176
chemical treatments, regular FBS was replaced by dextran-coated charcoal-treated FBS. HepG2 177
cells were routinely checked for contamination with mycoplasma by PCR (VenorGeM Classic, 178
Minerva Biolabs, Berlin, Germany).
179 180
2.7. Transient transfections, mammalian two hybrid and reporter gene assays
181HepG2 cells were seeded at 1.5 x 105 cells per well into 24-well plates one day prior to 182
transfection. Transient transfections were conducted using jetPRIME transfection reagent 183
(Polyplus, Illkirch, France). Per well, a mixture containing 0.3 µg pGL3-BSEP(-170/+81) luciferase 184
reporter gene plasmid, 0.01 µg Renilla luciferase expression plasmid pRL-CMV and 0.02 µg 185
respective FXR isoform expression plasmid or empty expression vector pcDNA3 was prepared.
186
Similarly, 0.02 µg or 0.01 µg of pcDFXRα2 was co-transfected with either 0.02 µg or 0.04 µg of 187
pcDNA3, pcDFXRα1 and pcDFXRα5–α8 expression plasmids.
188
8
In mammalian two hybrid assays, a mixture containing 0.3 µg pGL3-G5 luciferase reporter gene 189
plasmid, 0.01 µg pRL-CMV and 0.08 µg expression plasmids encoding VP16-AD/FXRα2- or 190
FXRα5-LBD and 0.02 µg GAL4-DBD/RXRα-LBD or GAL4-DBD/NCOA1-RID fusion proteins was 191
used per well.
192
The total amount of DNA was adjusted to 0.5 µg per well with pUC18 and diluted with jetPRIME 193
buffer to achieve a volume of 50 µl. 1 µl of jetPRIME transfection reagent was added, mixed and 194
incubated at room temperature for 10 minutes. Then the reaction mixture was added onto cells.
195
After incubation for 22-24 h, cells were treated with 0.1% DMSO, 50 µM CDCA or 1 µM GW4064 196
for 24 h before cell lysis with 150 µl of passive lysis buffer (Promega). Firefly luciferase activity 197
was measured from 20 µl of lysate with automatically injected 300 µl of firefly luciferase assay 198
solution [20] using the AutoLumat Plus LB953 (Berthold Technologies, Bad Wildbad, Germany).
199
Similarly, Renilla luciferase activity was measured from 10 µl of lysate with injection of 100 µl of 200
Renilla assay solution [22].Results were normalized by dividing firefly luciferase activity by Renilla 201
luciferase activity. Each transfection was done five times independently in technical triplicates.
202 203
2.8. PCR analysis of alternative spliced exon 10 in human tissues
204A panel of human tissue total RNA samples (Human Total RNA Master Panel II) was obtained by 205
Takara Bio Clontech, and supplemented with RNA samples from adult human breast (Stratagene- 206
Agilent, La Jolla, CA), and human colon and small intestine, which have been described 207
previously [23]. Additionally, total RNA samples of the pregnane X receptor–expressing human 208
colon adenocarcinoma cell line LS174T, treated with 0.1% DMSO or 10 µM rifampin, were further 209
used.
210
250 ng of total RNA was reverse transcribed in a 25 µl reaction mixture consisting of 1.25 U/µl 211
MultiScribe reverse transcriptase, 2.5 mM random hexamers, 5.5 mM MgCl2, 1x TaqMan RT 212
buffer, 0.5 mM each dNTP and 0.4 U/µl RNase inhibitor according to the manufacturer’s standard 213
protocol (Thermo Fisher Scientific).
214
Expression of FXR transcripts with and without exon 10 was analyzed with primer pair F3/R3 as 215
listed in Table 1. PCR was conducted in a total volume of 25 µl, according to manufacturer’s 216
protocol. The reaction mixture contained cDNA corresponding to 25 ng of RNA, 0.625 units One 217
Taq Hot Start DNA Polymerase (New England Biolabs, Ipswich, MA), 0.2 µM each primer, 200 218
µM each dNTP in 1x One Taq Standard reaction buffer. Thermal cycling parameters consisted of 219
initial denaturation at 94°C for 30 s, followed by 30 cycles at 94°C for 15 s, 53°C for 30 s and 220
68°C for 30 s and after that the final extension at 68°C for 5 min. PCR products were analyzed 221
on 1.5% agarose gels and visualized with ethidium bromide staining.
222
9 223
2.9. Molecular dynamics simulations and analysis
224Molecular modelling was conducted with Maestro Small-Molecule Drug Discovery Suite 2019-1, 225
(Schrödinger, New York, NY) and with OPLS3e force field [24,25]. The FXRα5 models were built 226
from the high resolution (1.8 Å) FXR-LBD structure (PDB ID: 5Q0K [26]). First, the 5Q0K structure 227
was prepared with Protein Preparation Wizard [27]: waters were removed, preprocessed with 228
adding the missing side chains and loops with Prime [28,29], H-bond optimization and 229
minimization were conducted with default settings. Next, the NCOA1 peptide was deleted together 230
with the 38 residue-long fragment (GISDEYITPMFSFYKSIGELKMTQEEYALLTAIVILSP;
231
residues 356–393 in FXRα2) that is not present in FXRα5 (see Fig. 2A). Then the helix-9 (H9) 232
was manually reorientated on the place of the deleted helix-7 (H7) (see Fig. 8A) and D394 233
connected to S355 (FXRα2 residue numbering). The new spatial orientation of H9 was conducted 234
while respecting of the original H7 residue characteristics. For instance, position of the P364 from 235
H7 was replaced with P410 of the shifted H9. After this, the obtained FXRα5-LBD model structure 236
was prepared and minimized with Protein Preparation Wizard using the same procedure as before 237
[27].
238
Molecular dynamics (MD) simulations were conducted with Desmond [30]. The prepared FXRα5- 239
LBD models were solvated in a cubic box (edges >15 Å from the protein) and system was 240
neutralized with counterions using the end salt concentration 0.15 M of NaCl. The water 241
molecules were described with TIP3P model [31]. The total number of atoms in the final FXRα5- 242
LBD system was 55,830 atoms. The default Desmond system relaxation protocol was conducted 243
before the 1000 ns production simulations. These were conducted in NpT ensemble in 310 K and 244
1.01325 bar, using Nosé-Hoover chain thermostat and Martyna-Tobias-Klein barostat. The 245
default timestep (2 fs) and cutoff for Coulombic interactions (9.0 Å radius) were used. A different 246
random seed was used for each of the replica simulations 1–3.
247
The control simulation for the conserved FXR-LBD (the intact LBD observed in all other FXR 248
isoforms, except in FXRα5 and α7) was conducted for the prepared 5Q0K structure, from where 249
the NCOA1 was deleted, with the same simulation settings as with FXRα5. The final FXR-LBD 250
system consisted of 59,147 atoms.
251
Collapsing of the ligand binding pocket was evaluated from the end conformations of the 252
simulations with Sitemap [32,33] using default settings.
253
The PyMOL Molecular Graphics System, Version 2.0 (Schrödinger) was used for the visualization 254
of the final structures and the supplementary movies.
255 256
10
2.10. Data analysis
257
Data are presented as means ± SD of five independent experiments. Multiple comparisons were 258
done using one-way ANOVA with post-tests as mentioned in the respective figure legends.
259
Statistical analyses were performed with GraphPad Prism 8.3.0 (GraphPad software, San Diego, 260
CA). If the coefficient of variation of technical triplicates in transfection experiments exceeded 261
20%, the outlier was omitted.
262 263
11
3. Results
264
3.1. Identification of human hepatic FXR SVs
265The open reading frame part of human FXR cDNAs was cloned from primary human hepatocytes, 266
using primer pairs F1/R1 and F2/R1 (Table 1). Subsequent sequencing of 22 and 24 clones, 267
respectively, resulted in identification of eight different FXR SVs (Fig. 1). Four of these SVs were 268
identical to the previously described FXRα1 (6 clones), FXRα2 (13 clones), FXRα3 (10 clones) 269
and FXRα4 (10 clones). In addition to these, four novel FXR SVs, hereafter named FXRα5 (1 270
clone), FXRα6 (1 clone), FXRα7 (1 clone) and FXRα8 (4 clones), were identified. FXRα5 271
demonstrated skipping of exon 10, whereas both FXRα6 and FXRα8 lacked exon 5. In contrast 272
to the other novel SVs, FXRα8 was derived from the alternative intronic promoter. The exon 273
skipping resulted in the in-frame deletion of 38 residues in FXRα5 and 47 amino acids in FXRα6 274
and FXRα8 (Fig. 2A). Thus, FXRα5 demonstrated deletion of part of the LBD, whereas FXRα6 275
and FXRα8 experienced deletion of the carboxyterminal half of the DBD. FXRα7 showed deletion 276
of exon 8, which resulted in a frame shift, the presence of a premature stop codon and thus a 277
truncated LBD with 13 distinct carboxy-terminal amino acids (Fig. 2A). To assess the effect of 278
these splicing events on FXR protein expression, in vitro protein synthesis was performed, using 279
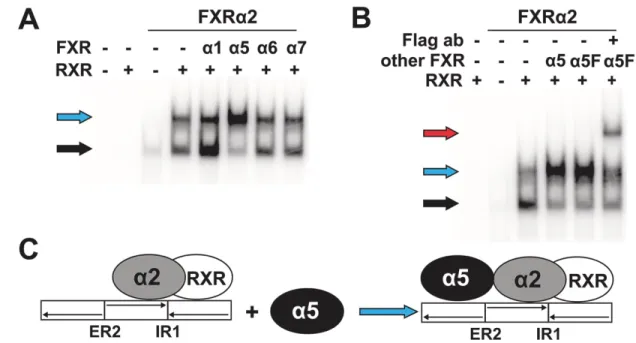
the respective expression plasmids in radiolabeling in vitro transcription/translation reactions.
280
Protein gel electrophoresis demonstrated expression of proteins with the expected molecular 281
weights (Fig. 2B). A second smaller molecular weight species was observed in the in vitro 282
synthesis of isoforms, which possess the exon 3a-encoded amino-terminus, most likely generated 283
by the use of an internal ATG in the exon 3a-specific amino-terminus as a start codon. None of 284
the alternative splicing events affected the in vitro synthesis and stability of the respective FXR 285
protein isoform.
286 287
3.2. In vitro DNA binding of FXR protein isoforms
288Genome-wide analyses of FXR chromatin binding sites in mouse liver and intestine and primary 289
human hepatocytes revealed that the top-ranked binding sites consisted of single IR1 motifs or 290
combined IR1/ER2 motifs, which share a common central half site [3,4]. Thus, we analyzed DNA 291
binding activities of FXR protein isoforms with EMSA, using consensus IR1 and ER2 motifs.
292
Additionally, we used the combined IR1a/ER2-FXRE of the human BSEP promoter, which has 293
been shown to mediate specific and strong transactivation by FXRα2 [34]. With the consensus 294
IR1 motif, isoforms FXRα1–5 showed comparable binding activities as monomers and 295
homodimers (Fig. 3A). With addition of RXRα, monomer binding was no longer observed and 296
12
strong heterodimer binding appeared. However, FXRα5 binding was weaker compared to 297
FXRα1–4. No binding was observed for FXRα6 and FXRα7, while FXRα8 demonstrated very 298
weak binding. With the consensus ER2 motif, FXRα1 and FXRα3 displayed exclusively monomer 299
and heterodimer binding with RXRα, while FXRα2 and FXRα4 showed strong homodimer binding, 300
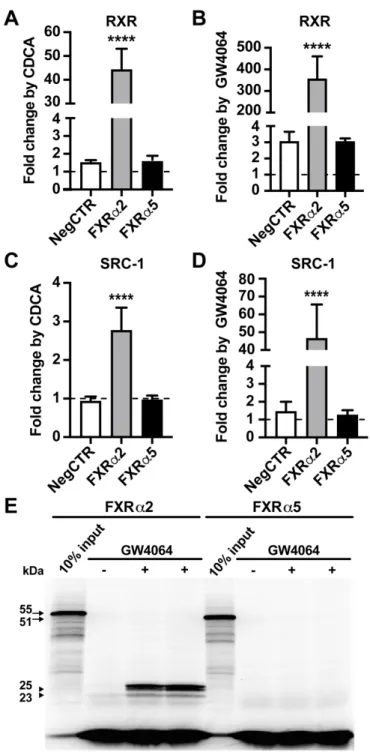
which was largely reduced by addition of RXRα, indicating that these heterodimers could not bind 301
efficiently to ER2 motifs in vitro (Fig. 3B). FXRα5 showed only weak homodimer binding, which 302
was not altered by the addition of RXRα. Similarly, FXRα1 and FXRα3 showed exclusive 303
heterodimer binding at the BSEP-FXRE, while FXRα2 and FXRα4 showed homodimer binding 304
and, in the presence of RXRα, increased dimer binding, which most likely is caused by 305
heterodimers (Fig. 3C). Interestingly, addition of RXRα also resulted in the appearance of a higher 306
order complex. Among the newly identified isoforms, only FXRα5 showed very weak homodimer 307
binding. Overall, the newly identified isoforms FXRα5–8 exhibited strongly reduced or even 308
absent in vitro DNA binding.
309 310
3.3. Ligand-dependent transactivation by human FXR protein isoforms
311To analyze ligand-dependent transactivation activities of FXR isoforms, transient co-transfections 312
of the BSEP promoter/reporter gene construct, harboring the above analyzed FXRE, and 313
expression plasmids encoding FXR isoforms were executed in HepG2 cells. Strong ligand- 314
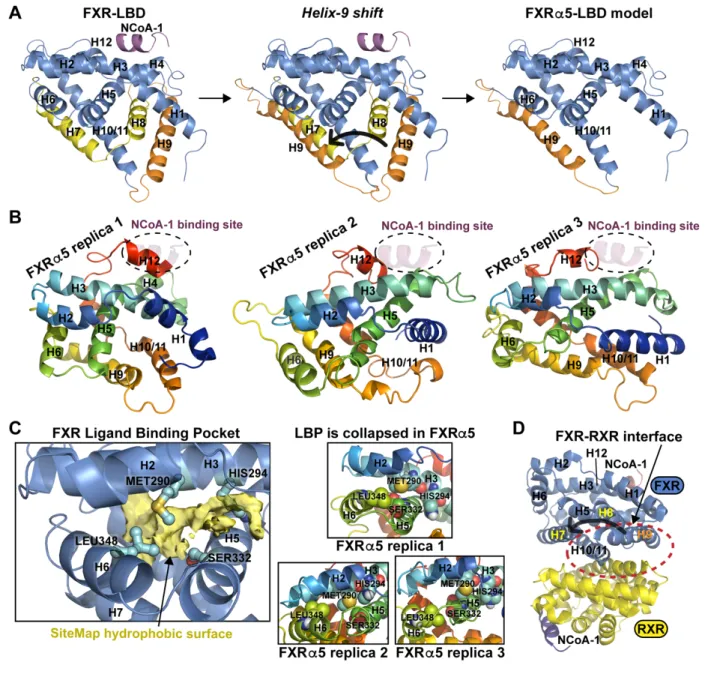
dependent transactivation by FXRα2 and FXRα4 were observed in the presence of agonists 315
CDCA (Fig. 4A) and GW4064 (Fig. 4B). While induction of reporter activity was much weaker by 316
ligand-stimulated FXRα1 and FXRα3, the novel isoforms FXRα5–8 demonstrated loss-of- 317
function.
318 319
3.4. FXRα5 displays dominant negative activity
320Among the novel loss-of-function isoforms, FXRα5 was distinguished as it showed residual in 321
vitro DNA binding activity. Thus, we further investigated whether this isoform could interfere with 322
the activity of the functional isoform FXRα2. EMSA analyses revealed that the addition of FXRα5 323
strongly increased the higher order complex at the BSEP-FXRE, while the FXRα2/RXRα 324
heterodimer complex was diminished, thereby suggesting binding of FXRα5 to the FXRα2/RXRα- 325
heterodimer/DNA complex (Fig. 5A). In contrast, FXRα1 and the novel FXR isoforms α6 and α7 326
did not affect the appearance of the higher order complex. As expected, addition of FXRα1 327
resulted in increased heterodimer binding. Using FLAG-tagged FXRα5 and anti-FLAG antibody 328
in antibody supershift EMSA experiments, we could demonstrate the actual presence of the 329
FXRα5 protein in the higher order complex (Fig. 5B). Figure 5C schematically presents the 330
13
proposed model for the generation of increased amounts of the higher order complex by binding 331
of FXRα5 to the FXRα2/RXR heterodimer complex with the BSEP-FXRE. Binding of FXRα5 to 332
the ER2 motif is suggested by the disappearance of the higher order complex when ER2 was 333
mutated in the BSEP-FXRE. Participation of the FXRα2/RXR heterodimer, bound at the IR1, in 334
recruitment of FXRa5 is suggested by the disappearance of all binding, except residual weak 335
FXRα2 homodimer binding, at BSEP-FXRE with mutated IR1 (Supplementary Fig. S1).
336
To investigate a potential effect of FXRα5 on FXRα2 activity, we transiently transfected HepG2 337
cells with the BSEP promoter/reporter gene construct, together with FXRα2 and FXRα5 at 338
different molar ratios. At equal ratio, FXRα5 did not affect ligand-dependent transactivation by 339
FXRα2 (Fig. 6A). However, four-fold excess of FXRα5 reduced transactivation by FXRα2 by 43%
340
(Fig. 6B). On the contrary, co-transfection of FXRα1 or of the other novel FXR isoforms neither 341
affected transactivation by FXRα2 at equal ratio (Fig. 6A) nor at four-fold excess (Fig. 6B).
342 343
3.5. Ligand-dependent protein interactions and ligand binding of FXRα2 and FXRα5
344Because FXRα5 displayed a dominant negative effect on transactivation by FXRα2, we were 345
interested in its ligand-dependent protein-protein interactions. Fig. 7A shows that FXRα2 346
displayed strong ligand-dependent interaction with RXRα, while FXRα5 did not. Similarly, only 347
FXRα2, but not FXRα5, recruited coactivator NCOA1 in the presence of the ligands CDCA or 348
GW4064 (Fig. 7B).
349
To demonstrate directly that FXRα5 is defective in ligand binding, as suggested by the deletion 350
of part of its LBD, limited proteolytic digestion assays were executed. These assays make use of 351
the conformational change, which is induced by ligand binding to the LBD of nuclear receptors 352
[35]. Consequently, the accessibility of proteolytic cleavage sites is altered. The high affinity FXR 353
ligand GW4064 was able to bind to FXRα2 resulting in the protection of proteolytic fragments with 354
sizes of 25 and 23 kDa (Fig. 7C). Respective fragments were not observed with FXRα5, thereby 355
clearly indicating that this isoform is defective in ligand binding.
356 357
3.6. Molecular dynamics simulations of FXRα5-LBD
358As FXRα5 appeared functionally defective and displayed dominant negative activity, we decided 359
to further investigate this isoform on the molecular level. To this end, we built a model of the 360
FXRα5-LBD and conducted microsecond timescale all-atom molecular dynamics (MD) 361
simulations with three independent replicates (Fig. 8A). All the simulated systems stabilized and 362
were considered suitable for further analysis (Supplementary Fig. S2A–C; Supplementary Movies 363
S1–3).
364
14
According to our MD simulations, the deletion of the 38 amino acids fragment from the canonical 365
FXR-LBD in FXRα5-LBD had several consequences for the LBD’s functionality. Firstly, the spatial 366
orientation of helix-12 (H12) was shifted, resulting in a conformation where it occupied the 367
coactivator binding site: fully in replica 1 and partially in replica 2 and 3 (Fig. 8B). This obtrusion 368
would impair the coactivator-FXR interaction. Secondly, the ligand binding pocket (LBP) collapsed 369
in all the three replicates (Fig. 8C). Obviously, this compromises ligand binding to the LBP in 370
FXRα5. Thirdly, the RXR interaction site (H9, H10/11) was totally distorted in the FXRα5-LBD 371
(Fig. 8B, D), and this would compromise the FXRα5–RXR interaction. All these observed events 372
are in agreement with our experimental data: FXRα5 displayed no interaction with the coactivator 373
NCOA1; it showed no ligand binding; dimerization between FXRα5 and RXRα was strongly 374
impaired. Thus, our simulations provide an explanation on the molecular level for the loss-of 375
function of FXRα5. Importantly, we also conducted a control simulation with the canonical FXR- 376
LBD. With canonical FXR only the coactivator binding site was partially occupied by H12 (which 377
is natural with apo structure as ligand induces coactivator binding), but the dimerization interface 378
and the LBP remained fully intact (Supplementary Fig. S2D–E).
379 380
3.7. Alternative splicing of exon 10 in human tissues
381As FXRα5 is generated by the skipping of exon 10, we analyzed the occurrence of this splicing 382
event in human tissues by qualitative PCR. The use of primer pair F3/R3 allowed for the 383
discrimination of transcripts with and without exon 10, displayed as fragments of 322 and 208 bp, 384
respectively. Transcripts with exon 10 were most prominently detected in adrenal gland, colon, 385
small intestine, kidney and liver (Fig. 9). In contrast, transcripts without exon 10 were expressed 386
in nearly all human tissues, albeit at exceptionally low, uniform levels, with the single exception 387
of the adrenal gland.
388 389
3.8. Hepatic expression levels of FXRα5
390To determine the expression levels of FXRα5 transcripts, we designed a specific TaqMan assay 391
and quantified expression of FXRα5 in primary human hepatocytes, which were derived from 25 392
donors. In addition, we measured total FXR gene expression levels to assess the proportion of 393
FXRα5 in comparison to total FXR. Overall, the expression levels of FXRα5 were extremely low.
394
The median percentage of FXRα5 from total FXR was 0.29% (0.22–0.35 95% CI) (Supplementary 395
Fig. S3).
396
15
4. Discussion
397
Alternative mRNA splicing is an important mechanism for increasing the diversity of proteins from 398
a single gene. Almost 95% of human multi-exon genes are estimated to generate more than one 399
mRNA transcript [36]. For instance, alternative splicing of only 48 human nuclear receptor genes 400
could possibly result in over 1000 mRNA transcripts [37]. Splice variants have been reported for 401
several other nuclear receptors besides FXR, such as liver X receptor alpha [38] and constitutive 402
androstane receptor (CAR) [19].
403
In this study, we identified and characterized alternatively spliced FXR variants in human liver, 404
originating from alternative promoter usage and mRNA splicing events. The cloning with two 405
primer pairs covering FXR coding regions and concomitant sequencing revealed eight FXR SVs.
406
Four of these, FXRα1–4, have been described previously [5,6,16]; however, to the best of our 407
knowledge, this is the first report to describe four novel FXR SVs, FXRα5–8. Among these, only 408
FXRα6 can be found in public gene and protein sequence databases, such as RefSeq 409
(NM_001206978) or UniProt (Q96RI1-5). While FXRα5–7 are transcribed from the same 410
promoter as FXRα1 and FXRα2, FXRα8 is transcribed from the alternative internal promoter 411
upstream of exon 3a, similarly to FXRα3 and FXRα4. All novel SVs were derived from exon 412
skipping events. FXRα5 demonstrated skipping of exon 10, resulting in the loss of 38 amino acids 413
in the LBD. Exon 5 is deleted from both FXRα6 and FXRα8, causing a 47 amino acid deletion in 414
the DBD. Thus, exons 5 and 10 are cassette exons, because their inclusion or exclusion does not 415
change the reading frame and finally the protein expression. In addition to exons 5 and 10, exons 416
4, 7 and 9 also represent possible FXR cassette exons [37]. However, we did not detect the 417
skipping of these exons in FXR open reading frame clones, which simply may be due to the limited 418
number of sequenced clones. In contrast, the deletion of exon 8 in FXRα7 introduces a frame 419
shift and premature stop codon, resulting in a truncated protein. It is also possible that FXRα7 420
would not be expressed as a protein in vivo because of nonsense-mediated decay [39].
421
The PCR primers that we used for cloning covered only the coding regions and did not cover the 422
untranslated regions (UTR). Therefore, mRNA transcripts containing altered UTRs remain 423
undetected here. Nevertheless, all the protein isoforms, which are based on the known translation 424
initiation sites in exons 3 and 3a, should have been detected. However, SVs were analyzed in 425
hepatic cells only; therefore, additional isoforms may exist that are exclusively expressed in 426
tissues other than liver.
427
The known isoforms FXRα1–4 differ considerably in their transactivation activities and potency to 428
activate target gene expression [5,6,16], which is confirmed here. FXRα2 and FXRα4 exhibited 429
higher transactivation activities compared to their counterparts with insertion of the four amino 430
16
acids MYTG (FXRα1/FXRα3). Another example highlighting the difference between FXRα1/α3 431
and FXRα2/α4 was reported by Ramos Pittol et al. [40], who recently demonstrated that FXRα2 432
and FXRα4 occupied almost 90% of FXR binding sites. In addition, while FXRα1–4 showed 433
binding to IR1, only FXRα2 and FXRα4 demonstrated binding to ER2 [40]. Similarly, we observed 434
here that FXRα2 and FXRα4 bind strongly and specifically to ER2 as homodimers, while FXRα1 435
and FXRα3 only displayed comparatively weak heterodimer binding.
436
The LBD of nuclear receptors is not only important for ligand recognition, but also plays a crucial 437
role in dimerization and binding of coregulators, whereas the DBD mediates the binding to the 438
DNA response elements in the target gene’s regulatory chromatin regions [41]. Typically, both 439
domains have to be intact for a functional nuclear receptor. As the novel FXR isoforms either 440
exhibit truncated LBD or DBD, the loss-of-function phenotype observed here is perhaps not 441
surprising. In contrast to a complete loss of transactivation activity, which was shown by all novel 442
isoforms, residual DNA-binding activity was observed, especially for FXRα5. This isoform 443
displayed substantial in vitro DNA binding as part of a higher order complex together with the 444
FXRα2/RXRα heterodimer at the BSEP-FXRE, and further demonstrated inhibition of FXRα2 445
transactivation activity in the BSEP promoter/reporter gene assay. Furthermore, FXRα5 does not 446
interact in a ligand-dependent manner with heterodimerization partner RXRα and coactivator 447
NCOA1, or bind ligands. Our MD simulations of the FXRα5-LBD, provided a putative explanation 448
for these observations at the molecular level. Overall, the deletion of helices H7 and H8 had a 449
dramatic impact on the LBD configuration and functionality. The actual mechanism behind the 450
dominant negative effect, however, remains largely unresolved. In general, it could be possible 451
that binding of FXRα5 to the FXRα2/RXRα complex impairs for instance co-activator recruitment.
452
Further research is needed to elucidate the respective molecular mechanism.
453
It may be speculated that the dominant negative effect of FXRα5 could affect total FXR activity 454
and thus target gene expression, even if it is expressed at only low levels in healthy tissues.
455
Several scenarios that may result in elevated FXRα5/α2 ratios are conceivable. Firstly, high 456
FXRα5 expression may be specific for pathological conditions, such as tumorigenesis. For 457
instance, exclusive expression of a dominant negative variant of PPARγ is observed in colorectal 458
cancer samples but not in normal colon tissue [42]. Secondly, the frequency of exon 10 skipping 459
may be increased by physiological stimuli. It has been shown previously that FXR splicing is 460
influenced by fasting and exercise [16]. Thirdly, the FXRα5/α2 ratio will automatically rise with 461
decreasing FXRα2 expression, or due to a decrease in total FXR expression, which has been 462
observed in non-alcoholic liver disease, and hepatocellular and colorectal cancer [43-45].
463
Therefore, it could be hypothesized that diseases and other conditions may alter the FXR splicing 464
17
resulting in increased expression or proportion of FXRα5. Analyzing FXRα5 expression and FXR 465
splice patterns in disease and physiological stress situations is required to investigate this 466
hypothesis. Lastly, transcripts without exon 10, i.e. FXRα5, are weakly but uniformly expressed 467
in nearly all tissues. Our data indicate comparable weak expression of transcripts with and without 468
exon 10 in nearly all tissues besides liver, kidney, small intestine, colon and adrenal gland (see 469
Fig. 9). In the former, the extent of the dominant negative activity of FXRα5 may thus be sufficient 470
to counteract the low-level FXR activity due to the small amounts of functional FXR, especially as 471
the sum of exon 10-containing transcripts also comprises further non-functional variants. Hence, 472
the dominant negative activity of FXRα5 may play a part in restricting FXR activity to tissues with 473
a substantial expression of functional FXR transcripts. The apparent lack of FXRα5 expression in 474
the adrenal gland may indicate tissue-specific splicing of exon 10. Elucidation of the respective 475
molecular mechanism will require further studies.
476
In conclusion, we have identified four novel FXR SVs (FXRα5–8) and characterized the functional 477
properties of the encoded isoforms. They all appear to be functionally defective. Among these 478
novel isoforms, the most peculiar is the dominant negative FXRα5 with its unique LBD. This 479
systematic analysis of FXR SVs provides new information about the diversity of human FXR 480
proteins. Further research, however, is required to elucidate the full biological relevance of these 481
novel SVs.
482
18 Funding
483
This work was supported by the Robert-Bosch Foundation, Stuttgart, Germany (EKM, OB), and 484
by the Interfaculty Center for Pharmacogenomics and Pharma Research of the University of 485
Tübingen, Germany (EKM, OB). TP acknowledges the Orion Research Foundation sr for financial 486
support and funding from the European Union`s Horizon 2020 research and innovation program 487
under the Marie Sklodowska-Curie grant agreement [Grant No 839230].
488 489
Declaration of competing interests 490
None.
491 492
Author contributions 493
Enni-Kaisa Mustonen: Conceptualization, Methodology, Formal analysis, Investigation, Writing 494
– original draft, Writing – review & editing, Visualization. Serene M. L. Lee: Methodology, 495
Investigation, Resources. Hanno Nieß: Methodology, Investigation, Resources. Matthias 496
Schwab: Writing – review & editing. Tatu Pantsar: Conceptualization, Methodology, 497
Investigation, Resources, Writing – original draft,Writing – review & editing, Visualization. Oliver 498
Burk: Conceptualization, Methodology, Formal analysis, Investigation, Resources, Writing – 499
original draft, Writing – review & editing, Visualization, Project administration, Funding acquisition.
500
501
Acknowledgements 502
We appreciate the expert technical assistance of K. Abuazi Rincones. Human Tissue and Cell 503
Research Foundation, a non-profit foundation regulated by German civil law, which facilitates 504
research with human tissue through the provision of an ethical and legal framework, provided liver 505
tissue for the preparation of primary human hepatocytes. M. Demmel and colleagues kindly 506
prepared primary human hepatocytes. The authors wish to acknowledge CSC – IT Center for 507
Science, Finland, for computational resources.
508 509
Appendix A. Supplementary data 510
Supplementary data to this article can be found online at …..
511
Original raw trajectories of MD simulations are freely available at 512
https://doi.org/10.5281/zenodo.3974151.
513 514
19 References
515
[1] S. Modica, R.M. Gadaleta, A. Moschetta, Deciphering the nuclear bile acid receptor FXR 516
paradigm, Nucl. Recept. Signal. 8 (2010) e005. https://doi.org/10.1621/nrs.08005.
517 [2] B.A. Laffitte, H.R. Kast, C.M. Nguyen, A.M. Zavacki, D.D. Moore, P.A. Edwards, 518
Identification of the DNA binding specificity and potential target genes for the farnesoid 519
X-activated receptor, J. Biol. Chem. 275 (2000) 10638–10647.
520
https://doi.org/10.1074/jbc.275.14.10638.
521
[3] A.M. Thomas, S.N. Hart, B. Kong, J. Fang, X.-B. Zhong, G.L. Guo, Genome-wide tissue- 522
specific farnesoid X receptor binding in mouse liver and intestine, Hepatology 51 (2010) 523
1410–1419. https://doi.org/10.1002/hep.23450.
524
[4] L. Zhan, H.-X. Liu, Y. Fang, B. Kong, Y. He, X. Zhong, J. Fang, Y.-J.Y. Wan, G.L. Guo, 525
Genome-wide binding and transcriptome analysis of human farnesoid X receptor in 526
primary human hepatocytes, PLoS One 9 (2014) e105930.
527
https://doi.org/10.1371/journal.pone.0105930 528
[5] R.M. Huber, K. Murphy, B. Miao, J.R. Link, M.R. Cunningham, M.J. Rupar, P.L.
529
Gunyuzlu, T.F. Haws, A. Kassam, F. Powell, G.F. Hollis, P.R. Young, R. Mukherjee, T.C.
530
Burn, Generation of multiple farnesoid-X-receptor isoforms through the use of alternative 531
promoters, Gene. 290 (2002) 35–43. https://doi.org/10.1016/S0378-1119(02)00557-7.
532
[6] J. Vaquero, M.J. Monte, M. Dominguez, J. Muntané, J.J.G. Marin, Differential activation 533
of the human farnesoid X receptor depends on the pattern of expressed isoforms and 534
the bile acid pool composition, Biochem. Pharmacol. 86 (2013) 926–939.
535
https://doi.org/10.1016/j.bcp.2013.07.022.
536
[7] M. Ananthanarayanan, N. Balasubramanian, M. Makishima, D.J. Mangelsdorf, F.J.
537 Suchy, Human bile salt export pump promoter is transactivated by the farnesoid X 538
receptor/bile acid receptor, J. Biol. Chem. 276 (2001) 28857–28865.
539
https://doi.org/10.1074/jbc.M011610200.
540
[8] B. Goodwin, S.A. Jones, R.R. Price, M.A. Watson, D.D. McKee, L.B. Moore, C. Galardi, 541 J.G. Wilson, M.C. Lewis, M.E. Roth, P.R. Maloney, T.M. Willson, S.A. Kliewer, A
542 regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid 543
biosynthesis, Mol. Cell. 6 (2000) 517–526. https://doi.org/10.1016/s1097- 544 2765(00)00051-4.
545 [9] T. Claudel, B. Staels, F. Kuipers, The Farnesoid X receptor: a molecular link between 546 bile acid and lipid and glucose metabolism, Arterioscler. Thromb. Vasc. Biol. 25 (2005) 547
2020–2030. https://doi.org/10.1161/01.ATV.0000178994.21828.a7.
548 [10] V. Massafra, A. Milona, H.R. Vos, R.J.J. Ramos, J. Gerrits, E.C.L. Willemsen, J.M.
549
Ramos Pittol, N. Ijssennagger, M. Houweling, H.C.M.T. Prinsen, N.M. Verhoeven-Duif, 550 B.M.T. Burgering, S.W.C. van Mil, Farnesoid X Receptor Activation Promotes Hepatic 551 Amino Acid Catabolism and Ammonium Clearance in Mice, Gastroenterology 152 (2017) 552
1462-1476.e10. https://doi.org/10.1053/j.gastro.2017.01.014.
553
[11] C.Y. Han, Update on FXR Biology: Promising Therapeutic Target?, Int. J. Mol. Sci. 19 554 (2018) 2069. https://doi.org/10.3390/ijms19072069.
555
[12] FDA, Ocaliva Summary Review 2016, https://www.accessdata.fda.gov. Accessed April 556
30, 2020.
557
[13] R. Neuschwander-Tetri, A. Loomba, A.J. Sanyal, J.E. Lavine, M.L. Van Natta, M.F.
558
Abdelmalek, N. Chalasani, D. Srinivasan, A.M. Diehl, Hameed, K.V. Kowdley, A.
559
McCullough, N. Terrault, J.M. Clark, J. Tonascia, E.M. Brunt, D.E. Kleiner, E. Doo, 560
Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic 561
steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial - The Lancet, 562
Lancet. 2015 (2015) 956–965. https://doi.org/10.1016/s0140-6736(14)61933-4.
563
20
[14] Z.M. Younossi, V. Ratziu, R. Loomba, M. Rinella, Q.M. Anstee, Z. Goodman, P.
564 Bedossa, A. Geier, S. Beckebaum, P.N. Newsome, D. Sheridan, M.Y. Sheikh, J. Trotter, 565
W. Knapple, E. Lawitz, M.F. Abdelmalek, K.V. Kowdley, A.J. Montano-Loza, J. Boursier, 566 P. Mathurin, E. Bugianesi, G. Mazzella, A. Olveira, H. Cortez-Pinto, I. Graupera, D. Orr, 567 L.L. Gluud, J.-F. Dufour, D. Shapiro, J. Campagna, L. Zaru, L. MacConell, R.
568 Shringarpure, S. Harrison, A.J. Sanyal, Obeticholic acid for the treatment of non- 569
alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo- 570
controlled phase 3 trial, The Lancet. 394 (2019) 2184–2196.
571 https://doi.org/10.1016/S0140-6736(19)33041-7.
572
[15] R.M. Gadaleta, M. Cariello, C. Sabbà, A. Moschetta, Tissue-specific actions of FXR in 573
metabolism and cancer, Biochim. Biophys. Acta. 1851 (2015) 30–39.
574
https://doi.org/10.1016/j.bbalip.2014.08.005.
575
[16] J.C. Correia, J. Massart, J.F. de Boer, M. Porsmyr-Palmertz, V. Martínez-Redondo, L.Z.
576
Agudelo, I. Sinha, D. Meierhofer, V. Ribeiro, M. Björnholm, S. Sauer, K. Dahlman- 577
Wright, J.R. Zierath, A.K. Groen, J.L. Ruas, Bioenergetic cues shift FXR splicing towards 578
FXRα2 to modulate hepatic lipolysis and fatty acid metabolism, Mol. Metab. 4 (2015) 579
891–902. https://doi.org/10.1016/j.molmet.2015.09.005.
580
[17] J. Jeske, A. Bitter, W.E. Thasler, T.S. Weiss, M. Schwab, O. Burk, Ligand-dependent 581
and -independent regulation of human hepatic sphingomyelin phosphodiesterase acid- 582
like 3A expression by pregnane X receptor and crosstalk with liver X receptor, Biochem.
583
Pharmacol. 136 (2017) 122–135. https://doi.org/10.1016/j.bcp.2017.04.013.
584
[18] M. Mathäs, O. Burk, H. Qiu, C. Nusshag, U. Gödtel-Armbrust, D. Baranyai, S. Deng, K.
585
Römer, D. Nem, B. Windshügel, L. Wojnowski, Evolutionary history and functional 586
characterization of the amphibian xenosensor CAR, Mol. Endocrinol. 26 (2012) 14–26.
587
https://doi.org/10.1210/me.2011-1235.
588
[19] K.A. Arnold, M. Eichelbaum, O. Burk, Alternative splicing affects the function and tissue- 589
specific expression of the human constitutive androstane receptor, Nucl. Recept. 2 590
(2004) 1. https://doi.org/10.1186/1478-1336-2-1.
591 [20] A. Geick, M. Eichelbaum, O. Burk, Nuclear receptor response elements mediate 592
induction of intestinal MDR1 by rifampin, J. Biol. Chem. 276 (2001) 14581–14587.
593
https://doi.org/10.1074/jbc.M010173200.
594
[21] J. Jeske, B. Windshügel, W.E. Thasler, M. Schwab, O. Burk, Human pregnane X 595 receptor is activated by dibenzazepine carbamate-based inhibitors of constitutive 596
androstane receptor, Arch. Toxicol. 91 (2017) 2375–2390.
597
https://doi.org/10.1007/s00204-017-1948-3.
598 [22] R. Piedade, S. Traub, A. Bitter, A.K. Nüssler, J.P. Gil, M. Schwab, O. Burk,
599 Carboxymefloquine, the major metabolite of the antimalarial drug mefloquine, induces 600 drug-metabolizing enzyme and transporter expression by activation of pregnane X 601 receptor, Antimicrob. Agents Chemother. 59 (2015) 96–104.
602
https://doi.org/10.1128/AAC.04140-14.
603 [23] H. Tegude, A. Schnabel, U.M. Zanger, K. Klein, M. Eichelbaum, O. Burk, Molecular 604 Mechanism of Basal CYP3A4 Regulation by Hepatocyte Nuclear Factor 4α: Evidence for 605 Direct Regulation in the Intestine, Drug Metab. Dispos. 35 (2007) 946–954.
606
https://doi.org/10.1124/dmd.106.013565.
607
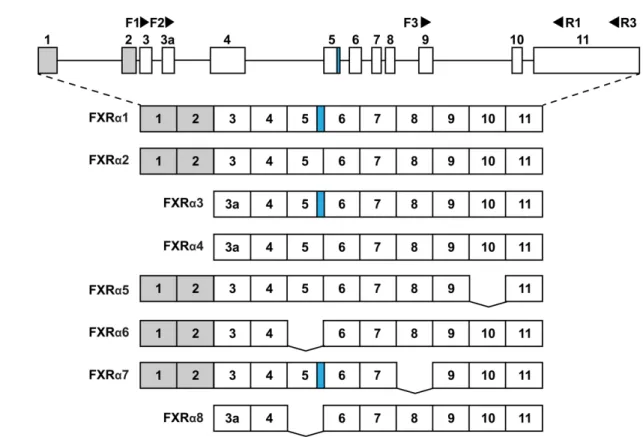
[24] E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J.Y. Xiang, L. Wang, D. Lupyan, M.K.
608 Dahlgren, J.L. Knight, J.W. Kaus, D.S. Cerutti, G. Krilov, W.L. Jorgensen, R. Abel, R.A.
609
Friesner, OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules 610
and Proteins, J. Chem. Theory Comput. 2016 (2016) 281–296.
611
https://doi.org/10.1021/acs.jctc.5b00864.
612
[25] K. Roos, C. Wu, W. Damm, M. Reboul, J.M. Stevenson, C. Lu, M.K. Dahlgren, S.
613
Mondal, W. Chen, L. Wang, R. Abel, R.A. Friesner, E.D. Harder, OPLS3e: Extending 614
21
Force Field Coverage for Drug-Like Small Molecules, J. Chem. Theory Comput. 15 615 (2019) 1863–1874. https://doi.org/10.1021/acs.jctc.8b01026.
616
[26] Z. Gaieb, S. Liu, S. Gathiaka, M. Chiu, H. Yang, C. Shao, V.A. Feher, W.P. Walters, B.
617 Kuhn, M.G. Rudolph, S.K. Burley, M.K. Gilson, R.E. Amaro, D3R Grand Challenge 2:
618 blind prediction of protein–ligand poses, affinity rankings, and relative binding free 619 energies, J. Comput. Aided Mol. Des. 32 (2018) 1–20. https://doi.org/10.1007/s10822- 620
017-0088-4.
621
[27] G.M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju, W. Sherman, Protein and ligand 622 preparation: parameters, protocols, and influence on virtual screening enrichments, J.
623
Comput. Aided Mol. Des. 27 (2013) 221–234. https://doi.org/10.1007/s10822-013-9644- 624
625 8.
[28] M.P. Jacobson, D.L. Pincus, C.S. Rapp, T.J.F. Day, B. Honig, D.E. Shaw, R.A. Friesner, 626
A hierarchical approach to all-atom protein loop prediction, Proteins. 2004 (2004) 351–
627 367. https://doi.org/10.1002/prot.10613.
628
[29] M.P. Jacobson, R.A. Friesner, Z. Xiang, B. Honig, On the Role of the Crystal
629 Environment in Determining Protein Side-chain Conformations, J. Mol. Biol. 320 (2002) 630
597–608. https://doi.org/10.1016/S0022-2836(02)00470-9.
631 [30] K.J. Bowers, E. Chow, H. Xu, Scalable Algorithms for Molecular Dynamics Simulations 632 on Commodity Clusters - IEEE Conference Publication (2006).
633
https://ieeexplore.ieee.org/document/4090217. Accessed August 2, 2020.
634
[31] W.L. Jorgensen, J. Chandrasekhar, J.D. Madura, R.W. Impey, M.L. Klein, Comparison 635 of simple potential functions for simulating liquid water, J. Chem. Phys. 79 (1983) 926–
636 935. https://doi.org/10.1063/1.445869.
637
[32] T. Halgren, New Method for Fast and Accurate Binding-site Identification and Analysis, 638
Chem. Biol. Drug Des. 69 (2007) 146–148. https://doi.org/10.1111/j.1747- 639
0285.2007.00483.x.
640
[33] T.A. Halgren, Identifying and characterizing binding sites and assessing druggability, J.
641
Chem. Inf. Model. 49 (2009) 377–389. https://doi.org/10.1021/ci800324m.
642
[34] X. Song, Y. Chen, L. Valanejad, R. Kaimal, B. Yan, M. Stoner, R. Deng, Mechanistic 643
insights into isoform-dependent and species-specific regulation of bile salt export pump 644
by farnesoid X receptor, J. Lipid Res. 54 (2013) 3030–3044.
645
https://doi.org/10.1194/jlr.M038323.
646
[35] G.F. Allan, X. Leng, S.Y. Tsai, N.L. Weigel, D.P. Edwards, M.J. Tsai, B.W. O’Malley, 647
Hormone and antihormone induce distinct conformational changes which are central to 648
steroid receptor activation, J. Biol. Chem. 267 (1992) 19513–19520.
649
[36] D. Sulakhe, M. D’Souza, S. Wang, S. Balasubramanian, P. Athri, B. Xie, S. Canzar, G.
650
Agam, T.C. Gilliam, N. Maltzev, Exploring the functional impact of alternative splicing on 651
human protein isoforms using available annotation sources, Brief Bioinform. 20 (2019) 652
1754-1768. https://doi.org/10.1093/bib/bby047 653
[37] A.J. Annalora, C.B. Marcus, P.L. Iversen, Alternative Splicing in the Nuclear Receptor 654
Superfamily Expands Gene Function to Refine Endo-Xenobiotic Metabolism, Drug 655
Metab. Dispos. 48 (2020) 272–287. https://doi.org/10.1124/dmd.119.089102.
656
[38] K. Endo-Umeda, S. Uno, K. Fujimori, Y. Naito, K. Saito, K. Yamagishi, Y. Jeong, H.
657
Miyachi, H. Tokiwa, S. Yamada, M. Makishima, Differential expression and function of 658
alternative splicing variants of human liver X receptor α, Mol. Pharmacol. 81 (2012) 800–
659 810. https://doi.org/10.1124/mol.111.077206.
660 [39] S. Kervestin, A. Jacobson, NMD: a multifaceted response to premature translational 661
termination, Nat. Rev. Mol. Cell Biol. 13 (2012) 700–712.
662
https://doi.org/10.1038/nrm3454.
663 [40] J.M. Ramos Pittol, A. Milona, I. Morris, E.C.L. Willemsen, S.W. van der Veen, E.
664 Kalkhoven, S.W.C. van Mil, FXR Isoforms Control Different Metabolic Functions in Liver 665
22
Cells via Binding to Specific DNA Motifs, Gastroenterology 159 (2020) 1853–1865.
666 https://doi.org/10.1053/j.gastro.2020.07.036.
667
[41] M. Pawlak, P. Lefebvre, B. Staels, General molecular biology and architecture of nuclear 668 receptors, Curr. Top. Med. Chem. 12 (2012) 486–504.
669 https://doi.org/10.2174/156802612799436641.
670 [42] L. Sabatino, A. Casamassimi, G. Peluso, M.V. Barone, D. Capaccio, C. Migliore, P.
671
Bonelli, A. Pedicini, A. Febbraro, A. Ciccodicola, V. Colantuoni, A novel peroxisome 672
proliferator-activated receptor gamma isoform with dominant negative activity generated 673 by alternative splicing, J. Biol. Chem. 280 (2005) 26517–26525.
674
https://doi.org/10.1074/jbc.M502716200.
675
[43] A.M. Bailey, L. Zhan, D. Maru, I. Shureiqi, C.R. Pickering, G. Kiriakova, J. Izzo, N. He, C.
676
Wei, V. Baladandayuthapani, H. Liang, S. Kopetz, G. Powis, G.L. Guo, FXR silencing in 677
human colon cancer by DNA methylation and KRAS signaling, Am. J. Physiol. - 678
Gastrointest. Liver Physiol. 306 (2014) G48–G58.
679
https://doi.org/10.1152/ajpgi.00234.2013.
680
[44] U. Deuschle, J. Schüler, A. Schulz, T. Schlüter, O. Kinzel, U. Abel, C. Kremoser, FXR 681
Controls the Tumor Suppressor NDRG2 and FXR Agonists Reduce Liver Tumor Growth 682
and Metastasis in an Orthotopic Mouse Xenograft Model, PLoS ONE. 7 (2012) e43044.
683
https://doi.org/10.1371/journal.pone.0043044.
684
[45] Z.-X. Yang, W. Shen, H. Sun, Effects of nuclear receptor FXR on the regulation of liver 685
lipid metabolism in patients with non-alcoholic fatty liver disease, Hepatol. Int. 4 (2010) 686
741–748. https://doi.org/10.1007/s12072-010-9202-6.
687
[46] N. Wang, Q. Zou, J. Xu, J. Zhang, J. Liu, Ligand binding and heterodimerization with 688
retinoid X receptor α (RXRα) induce farnesoid X receptor (FXR) conformational changes 689
affecting coactivator binding, J. Biol. Chem. 293 (2018) 18180–18191.
690
https://doi.org/10.1074/jbc.RA118.004652.
691
23 Tables
692 693
Table 1
694 Primers used for PCR.
695
Primer Location Sequence (5’ to 3’)
F1 Forward ata ggt acc acc ATG GGA TCA AAA ATG AAT CTC ATT GA
R1 Reverse ata gcg gcc gcT CAC TGC ACG TCC CAG ATT TCA
CAG
F2 Forward ata ggt acc acc ATG GTA ATG CAG TTT CAG GGG
TTA G
F3 Forward GCA TTC TGA CCT ATT GGA AGA AAG
R3 Reverse CAT CTC AGC GTG GTG ATG AT
Small letters indicate nucleotides, which have been added due to cloning (restriction enzyme 696
sites, underlined) or eukaryotic expression (Kozak consensus sequence, italics) 697
24 Figures
698 699
700
Fig. 1. Human FXR gene and mRNA transcripts of FXRα1–8. The FXR gene is located in 701
chromosome 12 at position 23.1 and contains 11 exons. Alternative splicing results in eight mRNA 702
transcripts (splice variants). Exons are shown as rectangles with the respective exon numbers.
703
Horizontal lines indicate introns. Grey color illustrates 5’-untranslated regions (5’-UTR). The use 704
of the alternative splice donor site in intron 5, resulting in the insertion of 12 nucleotides at the 705
end of exon 5 is depicted with blue. Exon deletions are displayed with gaps. Arrowheads illustrate 706
the location of indicated primers (Table 1). PCR for cloning was conducted with forward primers 707
F1 and F2 using the same reverse primer R1. F1 and F2 distinguish transcripts with different 708
translation initiation sites due to alternative promoter usage. Primer pair F3/R3 was used for the 709
detection of the SVs with alternatively spliced exon 10 from the tissue panel in Fig. 8.
710
25 711
Fig. 2. Structure and protein expression of FXR isoforms. (A) Schematic representation of the 712
structures of FXR isoforms. Numbers above schematics depict the first amino acid in each 713
corresponding domain. Vertical number at the end of schemes depicts the last amino acid in the 714
respective isoform. White color represents the N-terminal domain. Horizontal dashes at the end 715
of FXRα7 illustrate the distinct C-terminal compared to other isoforms. Light grey color shows the 716
DNA-binding domain. Black diagonal dashes illustrate the four amino acid insertion at the end of 717
DBD in isoforms FXRα1, α3 and α7. Dark gray area between DBD and ligand-binding domain 718
(black) represents the hinge region. Gaps depict deletions. (B) Representative protein gel of in 719
vitro translated FXR protein isoforms, which have been labelled with 35S-methionine. DBD, DNA 720
binding domain; LBD, ligand binding domain.
721
26 722
Fig. 3. In vitro DNA-binding of FXR isoforms. EMSA analyses of DNA binding activities of the 723
indicated FXR isoforms, in the absence (-) or presence (+) of 3-fold excess of RXRα, using (A) 724
consensus IR1 motif, (B) consensus ER2 motif and (C) FXR response element (FXRE) of 725
27
proximal BSEP promoter. Open arrow denotes free probe, filled arrows show specific complexes 726
(grey, monomers; black, hetero- and/or homodimers; blue, higher order complex). The sequences 727
of FXR binding motifs (upper strands only) are displayed.
728
729
Fig. 4. FXR isoforms differentially transactivate the human BSEP-promoter. HepG2 cells, which 730
were co-transfected with pGL3-BSEP(-170/+81) promoter reporter gene plasmid and respective 731
FXRα expression plasmids, were treated with either 0.1% DMSO, 50 µM CDCA (A) or 1 µM 732
GW4064 (B). pcDNA3 plasmid was used as negative control (NegCTR). Luciferase activities were 733
measured after 24 h treatment. Data is expressed as mean fold change (±SD) by ligand treatment 734
from five independent experiments with technical triplicates, relative to the activity of DMSO- 735
treated, respectively transfected cells, which was designated as 1 (dashed line). Statistically 736
significant differences are illustrated with asterisks. ****p<0.0001 compared to negative control 737
analyzed by one-way ANOVA with Dunnett’s multiple comparisons test.
738
739
28 740
Fig. 5. FXRα5 displays dominant negative activity by interacting with functional heterodimer. (A) 741
EMSA analysis of the effects of FXR isoforms on FXRα2/RXRα DNA binding to BSEP-FXRE.
742
Equal amounts of FXRα2 and the respective other FXR isoforms were used. (B) Antibody 743
supershift demonstrates presence of FXRα5 in the higher order complex of FXRα2/RXRα with 744
DNA. Filled black arrow shows homo-/heterodimers, filled blue arrow higher order complex, filled 745
red arrow illustrates antibody (Flag ab) supershift. α5F, FLAG-tagged FXRα5. (C) Scheme 746
depicting the proposed model for the increased appearance of the higher order complex by 747
additional binding of FXRα5 to the FXRα2/RXR heterodimer complex with the BSEP-FXRE.
748
749
750
29 751
Fig. 6 HepG2 cells were co-transfected with pGL3-BSEP(-170/+81) promoter reporter gene and 752
FXRα2 expression plasmids together with either pcDNA3 (NegCTR) or expression plasmids 753
encoding the indicated FXR isoforms in equal ratio (A) or 4-fold excess over FXRα2 (B). After 24 754
h treatment with 0.1% DMSO or 1 µM GW4064, luciferase activities were measured. Data is 755
expressed as mean fold change (±SD) by ligand treatment from five independent experiments 756
with technical triplicates, relative to the activity of DMSO-treated, respectively transfected cells, 757
which was designated as 1 (dashed line). Statistically significant differences are illustrated with 758
asterisks. *p<0.05 compared to FXRα2 + NegCTR analyzed by one-way ANOVA with Dunnett's 759
multiple comparisons test.
760
30 761
Fig. 7. FXRα5 displays impaired ligand-dependent protein interactions and ligand binding. (A–B) 762
Mammalian two hybrid assays were conducted in HepG2 cells, which were co-transfected with 763
expression plasmids encoding fusion proteins VP16-AD/FXRα2(241-472) or VP16- 764
AD/FXRα5(241-434) or with empty vector pVP16-AD as negative control (NegCTR) and 765
expression plasmids encoding fusion proteins GAL4-DBD/RXRα(226-462) (A) or GAL4- 766
DBD/NCOA1(583-783) (B). After 24 h treatment with 0.1% DMSO, 50 µM CDCA or 1 µM 767
31
GW4064, luciferase activities were measured. Data is expressed as mean fold change (±SD) by 768
ligand treatment from five independent experiments with technical triplicates, relative to the 769
activity of DMSO-treated, respectively transfected cells, which was designated as 1 (dashed line).
770
Statistically significant differences are illustrated with asterisks. ****p<0.0001 compared to 771
negative control analyzed by one-way ANOVA with Dunnett's multiple comparisons test. (C) 772
FXRα5 displays impaired ligand binding abilities. Limited proteolytic digestion analysis of FXRα2 773
and FXRα5, pre-incubated with 10 or 30 µM of GW4064 or 2.5% DMSO (-). Arrows show 55 kDa 774
input of FXRα2 and 51 kDa input of FXRα5 and arrowheads show protected 25 and 23 kDa 775
proteolytic fragments after limited digest with trypsin.
776
777
32 778
Fig. 8. Structural model of FXRα5 provides a putative explanation for its biological behavior. (A) 779
FXRα5-LBD model building from the FXR-LBD structure (PDB ID: 5Q0K, [26]). The 38 residue 780
fragment (yellow), including helices H7 and H8, missing from the FXRα5-LBD, was removed and 781
H9 was repositioned on the site of H7. (B) The end conformations (at 1000 ns) of the individual 782
FXRα5-LBD simulations (replica 1–3). In all simulations of the FXRα5-LBD, H12 occupies fully 783
(replica 1) or partially (replica 2 and 3) the NCOA1 binding site (transparent cartoon). (C) The 784
ligand binding pocket is collapsed in FXRα5. The SiteMap [32,33] calculated hydrophobic surface 785
in the FXR-LBP (PDB ID: 5Q0K) indicates the prominent ligand binding site (left panel; depicted 786
in yellow). The ligand binding pocket collapses in all FXRα5-LBD simulations (replicas 1-3) due 787
to the conformational rearrangements and there exists no binding site for a ligand. Residues 788