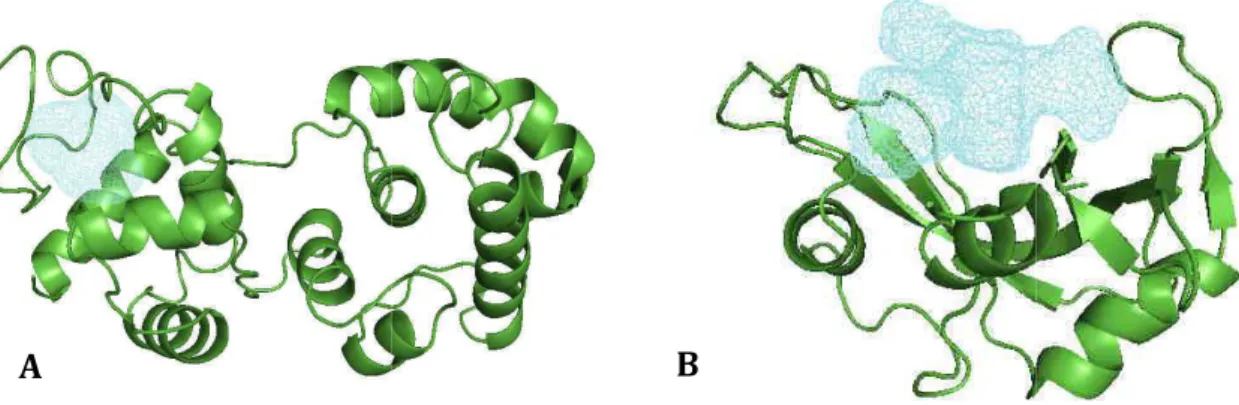
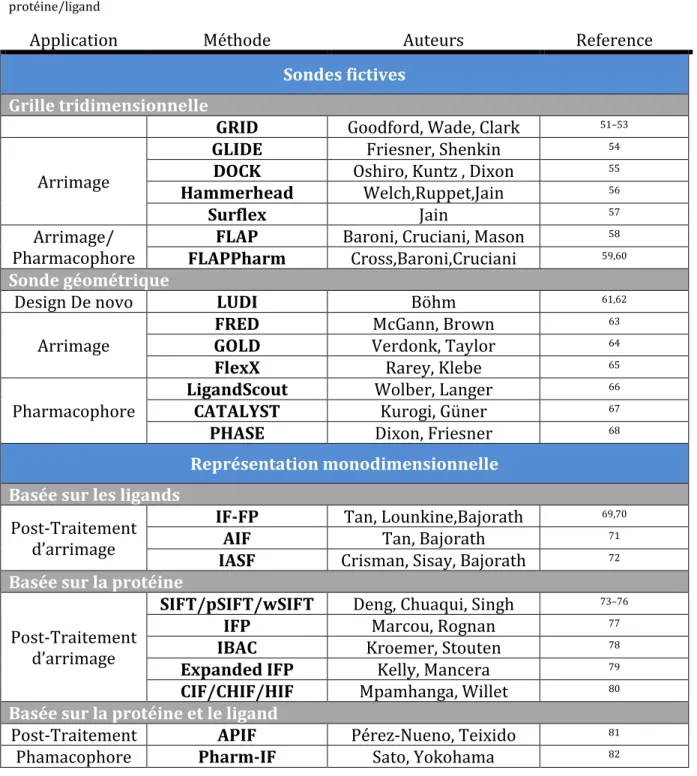
L'entrée de la molécule ou du substrat dans la protéine nécessite une poche de géométrie adéquate. Poche Cas particulier d'une cavité, très ouverte et peu enterrée, facilement accessible par une molécule sans mouvement important de la part de la protéine.
Les sites de liaisons : interactions non covalentes et représentations. 15
Anatomie
La longueur de la chaîne dépend du nombre d'atomes dans le site de liaison qui peuvent former des liaisons hydrogène avec le ligand. Comparer ses modes d'interaction de manière relativement indépendante de la structure initiale des fragments permet la recherche.

Flexibilité
Interactions non-covalentes
- Interactions clés
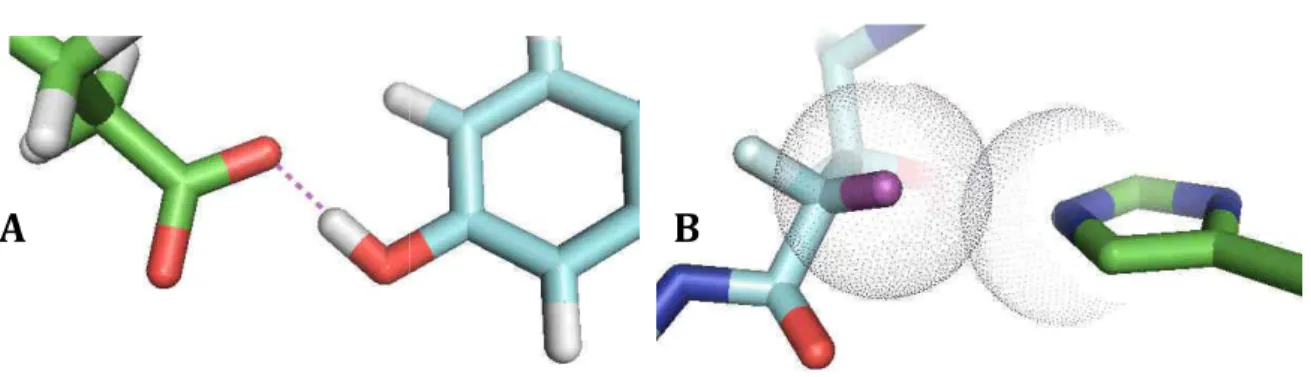
- Liaison hydrogène
- Interaction ionique
- Coordination de metaux
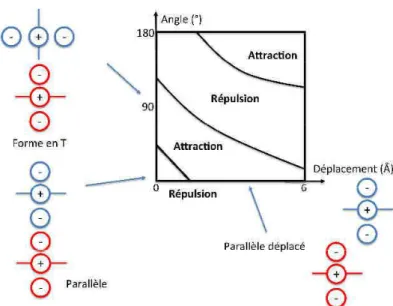
- Interaction aromatique
- Autres interactions non-covalentes
- Interaction π/Cation
- Interaction C-H/π
- Liaison halogène
- Liaison hydrogène faible
- Phénomènes de désolvatation
- Conclusion
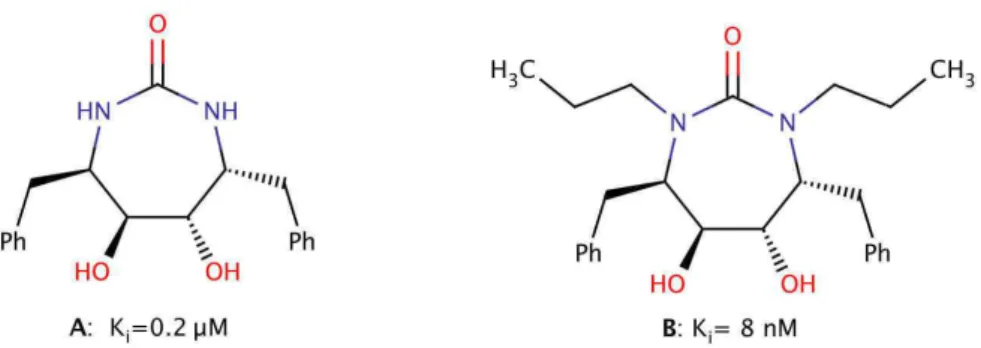
L'arrangement et la composition en acides aminés du site de liaison caractérisent la fonction de la protéine et lui permettent d'accepter des ligands. Ainsi, lorsque l’on choisit le fragment protéine-tyrosine phosphatase (code PDB : 2nt7) comme référence, on retrouve un acide carboxylique parfaitement aligné (code PDB : 2qbp) comme meilleur bioisoster.
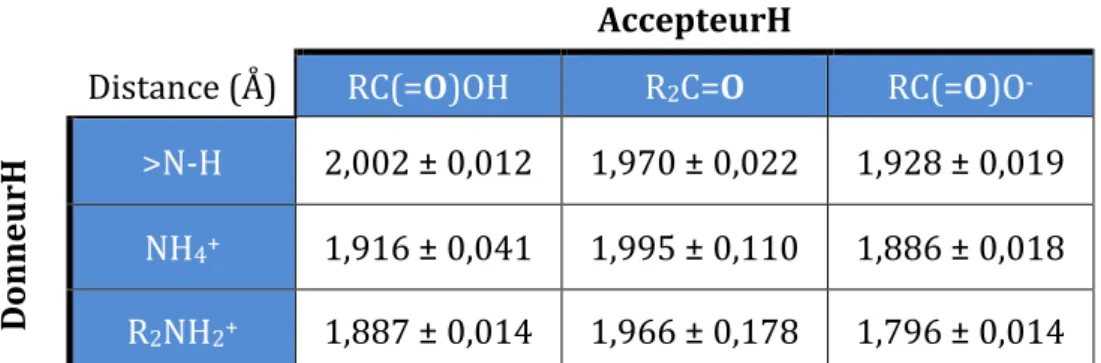
Représentation des interactions protéine/ligand
- Principales méthodes existantes
- Arrimage moléculaire
- Les pharmacophores
- Les empreintes
- Sondes fictives
- Grille tridimensionnelle
- Sondes géométriques
- Conclusion
- Représentation monodimensionnelle
- Basée sur le ligand
- Basée sur la protéine
- Basée sur la protéine et le ligand
Conclusion
La gamme d’outils qui codent ces interactions est impressionnante et couvre un large domaine méthodologique. De l’utilisation de descripteurs tridimensionnels géométriques ou énergétiques à la conversion en empreintes digitales, chacune de ces méthodes propose une vision différente de ces interactions.
Bibliographie
Description et comparaison de cavités protéiques
Contexte
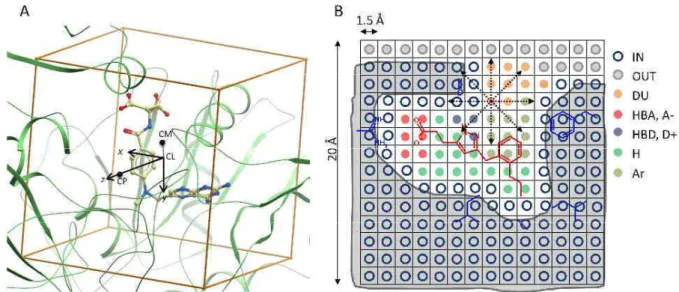
La comparaison de ces sites permet d'attribuer une fonction à une protéine orpheline et d'observer une conservation locale ou globale de structure et de propriétés physicochimiques au sein d'une famille de protéines ou entre différentes familles. Cela implique une conservation structurale, plus ou moins locale, permettant ainsi de les distinguer et de caractériser la fonction de chacun. VolSite est une méthode d'analyse et de caractérisation des cavités d'une protéine, basée sur la discrétisation de la cavité sous forme d'un réseau tridimensionnel.
Introduction
The method is simple, rapid and particularly effective for detecting binding site similarity in the absence of sequence and fold conservation. It can be used for three main applications: (i) infer the function of a protein by measuring the similarity of its known or potential ligand binding pockets to a collection of functionally annotated binding sites, (ii) classify targets according to the similarity of their binding sites, (iii) predict the structural drugability (ligandability) of a binding site from the properties of its pharmacophore form.
Methods
- Pharmacophoric annotation of cavity grid points (VolSite)
- Druggability prediction
- Binding site alignment (Shaper)
- Methods
- Similarity metrics and statistical evaluations
- Parameters selection and optimization
- Virtual screening of the sc-PDB database
- Dataset of promiscuous protein-ligand complexes
- All-against-all comparison of sc-PDB druggable binding sites
- Classification of GPCR X-ray structures
Binary classification of pairs (similar/dissimilar) allowed calculation of the area under the ROC curve for all lists. The standard Shaper parameters were thus defined as those that maximized the value of the area under the ROC curve. All entries were ranked by decreasing RefTversky similarity score and the ranking list used to calculate the area under the ROC curve for a binary classification model by repeatedly considering each of the set as positive cases.
Results and discussion
- Binding site description
- Prediction of structural druggability
- Determining a robust similarity threshold for pairwise comparison of binding sites. 88
- Virtual screening for similarity to a known cavity
- Binding site similarity detection as a function of target sequence and structure
- Network of binding sites for a protein family
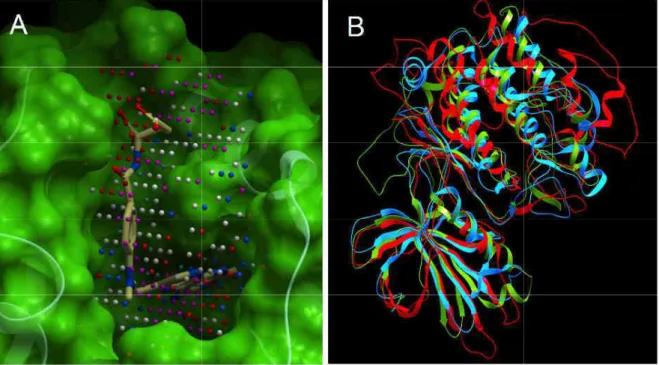
Description de cavités proteiques Significance level p of detecting the same or higher similarity score. Of the three tools, Shaper clearly has the highest proportion of similar binding sites (69%). We paid particular attention to (i) comparing different entries of the same receptor co-crystallized with ligands exhibiting different functional effects (agonist, inverse agonist, antagonist);. ii) the influence of the binding site determination (immediate proximity of the ligand, full cavity) on the obtained networks.
Conclusion
Commentaires
- Modifications post-publication
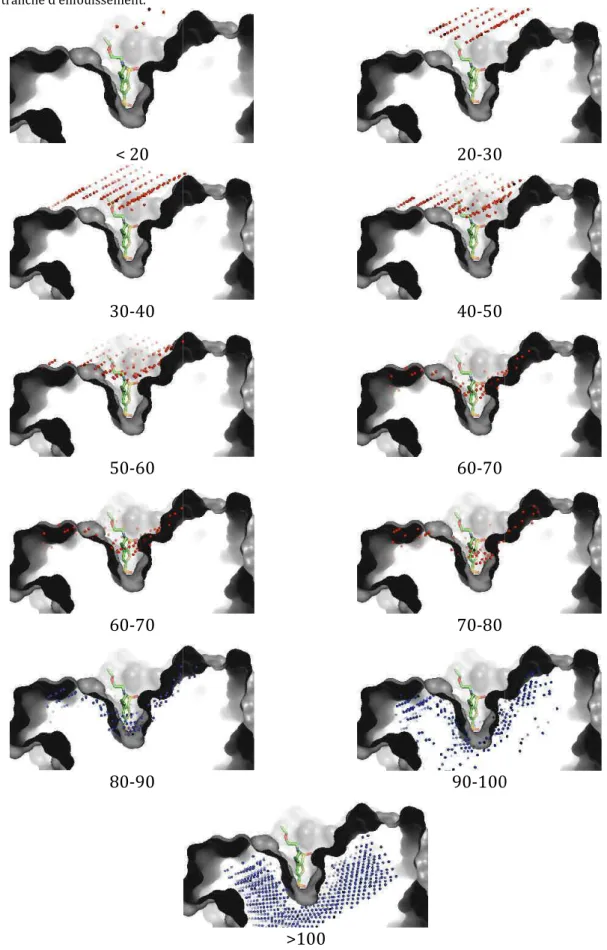
- Valeurs d’enfouissement
- Agrégation
- Detection des cavités d’une protéine
- Assignation de propriétés physico-chimiques aux cubes
La surface de la protéine est en gris, l’extérieur de la protéine en vert et l’intérieur de la protéine en bleu. Un point d'amélioration potentiel pour VolSite concerne l'attribution des propriétés physico-chimiques des cubes creux. Les propriétés de la cavité ne seraient pas des propriétés à n pour cent, mais un rapport de propriétés.
Annexes
Bibliographie
Comparaison de modes d’interaction protéine/ligand
Datasets of protein-ligand complexes
- Set 1:900 similar and 900 dissimilar protein-ligand complexes
- Set 2: sc-PDB Fragments
- Set 3 : CCDC/Astex subset of protein-ligand complexes
- Set 4 : DUD-E target and ligand sets
All protein-ligand complexes were retrieved from the sc-PDB dataset23, which archives 9877 high-resolution X-ray structures of medicinal protein-ligand complexes. Pairs of protein–ligand complexes were considered similar if: (i) their pairwise binding site similarity (expressed by Shaper similarity score29) was higher than 0.44 and ii) their pairwise ligand similarity (expressed by a Tanimoto coefficient on ECFP4 fingerprint) was between 0.55 and 0.75. The same number of pairs of different protein-ligand complexes were retrieved assuming that their pairwise binding site similarity and their pairwise ligand similarity were lower than 0.20.
Detection of protein-ligand interactions
Only the sho ingle ligand atom and many protein atoms are kept (interaction 2 is not followed 4) are then grouped to yield IPAs 3 and 5. C) Case of aromatic interactions ms (yellow balls). For aromatic interaction atoms, which do not respect the aromatic interaction, a hydrophobic interaction (green dashed line) and IPA (IPA 2) is .. step avoids property redundancy on ligand atoms by keeping the type of interaction per ligand atom. Residual romantic interactions between protein that rules the aromatic interaction - distance between both ng the aromatic interaction rules but e) and IPA (IPA 2) is defined between.
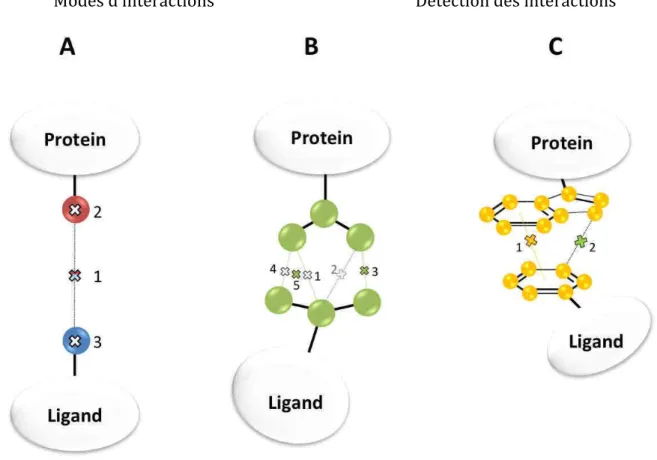
Fingerprinting triplets of interaction pseudoatoms
Finally, the geometric validity of the pharmacophoric triad is checked by applying the triangle inequality rule which states that one distance cannot be longer than the sum of the other two. Full-length fingerprints were calculated for the 9877 protein-ligand complexes of the sc-PDB dataset (2011 release) and the counting status of each triplet was calculated. Second, 361 triplets (226 bins) with a frequency between 10 and 20 were merged into 185 triplets (46 bins) of the same composition but without distance information.
Shape matching of IPAs (IShape)
To speed up the calculations, the 'Grid' volume overlay method was chosen to represent the volume of the target IPAs, and all atom radii were set to that of carbon (1.7 Å). In other words, the alignment proposed by simple shape matching is evaluated to account for the overlap of pharmacophoric features. The best similarity threshold is found from the maximum of the F-measure curve when the threshold varied from 0 to 1 with an increment of 0.01.
Graph matching of IPAs (Grim)
0.0720 RMSD root-mean-square deviation of the matched clique -0.0003 DiffI absolute value of the difference in the number of e.
Docking
Fingerprinting interaction patterns in sc-PDB complexes
A reliable similarity metric to compare protein-ligand interaction patterns
Between them, the IShape similarity score varies between 0.41 at the F-measure optimum and 0.63 at the precision optimum (Figure 4C). Therefore, our clique detection method uses a weight on pharmacophoric features that is inversely proportional to their abundance in the sc-PDB (hydrophobes are less important than polar features in the clique ranking). True positives are pairs of similar protein-ligand complexes that are predicted to be similar, while false positives are pairs of dissimilar complexes that are predicted to be similar.
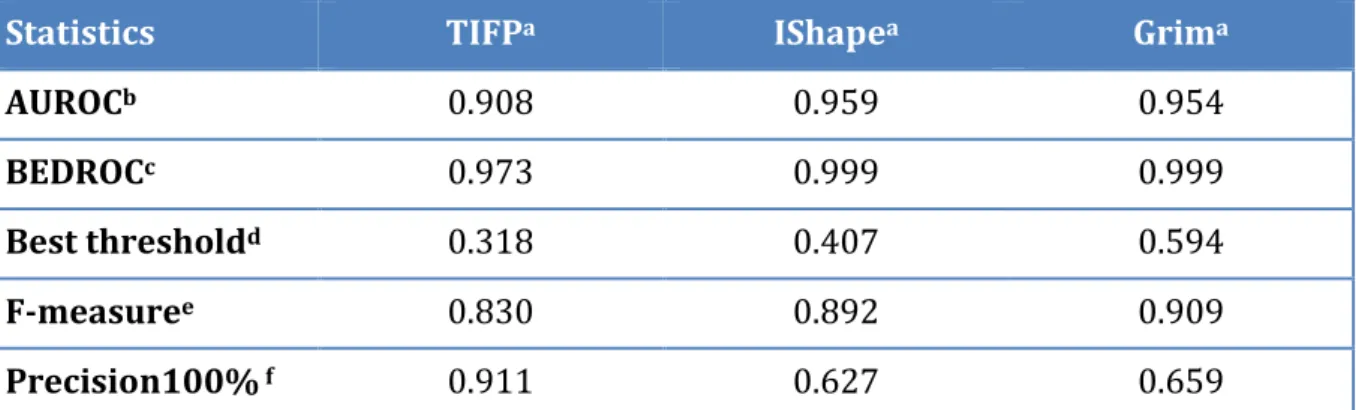
Interaction pattern similarity depends tightly on binding site similarity
137 Résultats et discussi d’interactions Mode d’interaction et site d some key residues must be fulfilled to achieve the significance of the ligand structures, which revealed that different ligands have commonality and interaction pattern usually in common. Binding site similarity (Shaper29 similarity score) versus int D) Ligand similarity (Tanimoto coefficient of ECFP4 fingerprint ilarity score). Results et discussion d’interaction et site de liaison to achieve significant binding. similar ligands that share a common substructure trypsin inhibitor binding to .. interaction pattern similarity for 9877 nt) vs. interaction pattern similarity S public keys) vs. interaction pattern ilarity score) vs. interaction pattern n fingerprints ECFP4) vs.
Some applications of interaction pattern fingerprints and graphs
- Interaction-based alignment of protein-ligand complexes
- Post-processing docking poses
- Scaffold hopping with interaction pattern conservation
Grim rescoring significantly improves the performance of rescoring (70% success, Figure 9A), especially for high-precision positions (< 1 Å rmsd). Reference sc-PDB entries were selected according to the recommended Uniprot name for the target of interest and provided for each ligand a variable number of references (from none to 115). For 7 out of the 10 targets, the benefit of Grim rescoring was related to the number of protein-ligand X-ray reference structures.
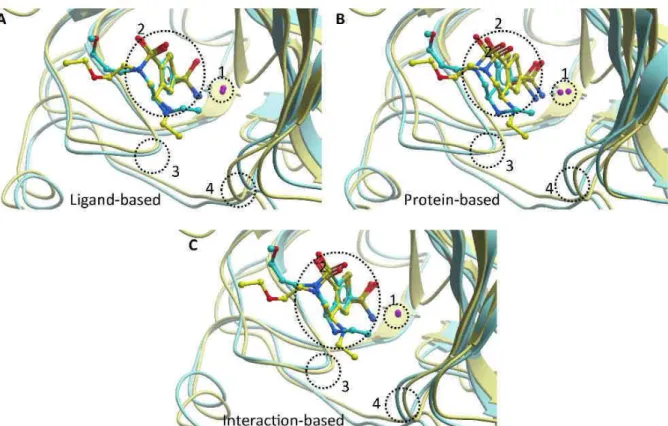
Acknowledgment
We hereby propose a generic fingerprint (TIFP) of protein-ligand interaction patterns, as well as two computational methods (IShape, Grim) to efficiently compare and align protein-ligand complexes. It allows ultrafast comparison of protein–ligand complexes but suffers from the predominance of hydrophobic contacts in most PDB–ligand X-ray structures. Finally, the proposed alignment tools (particularly the Grim method) allow to match protein-ligand complexes directly from the corresponding interaction patterns, without having to choose between a ligand-based or a target-based view.
Commentaires
- Détection des interactions
- Grim vs IShape
- Comparaisons
- Similarité
- Alignement de complexes protéine/ligand
- Post-traitement d’arrimage moléculaire
- Conclusion générale
Le cas de Grim est différent, car il deviendra plus exigeant à mesure que le nombre de points d’interaction augmentera. Mode d'interaction Post-traitement des positions d'accueil La sélection uniquement des résidus de sites de liaison améliore l'alignement par rapport à la protéine entière. L’un des changements importants serait donc de connaître le nombre de points d’interaction polaire correspondants pour compléter le score du graphique.
Recherche de fragments bioisostériques
Introduction
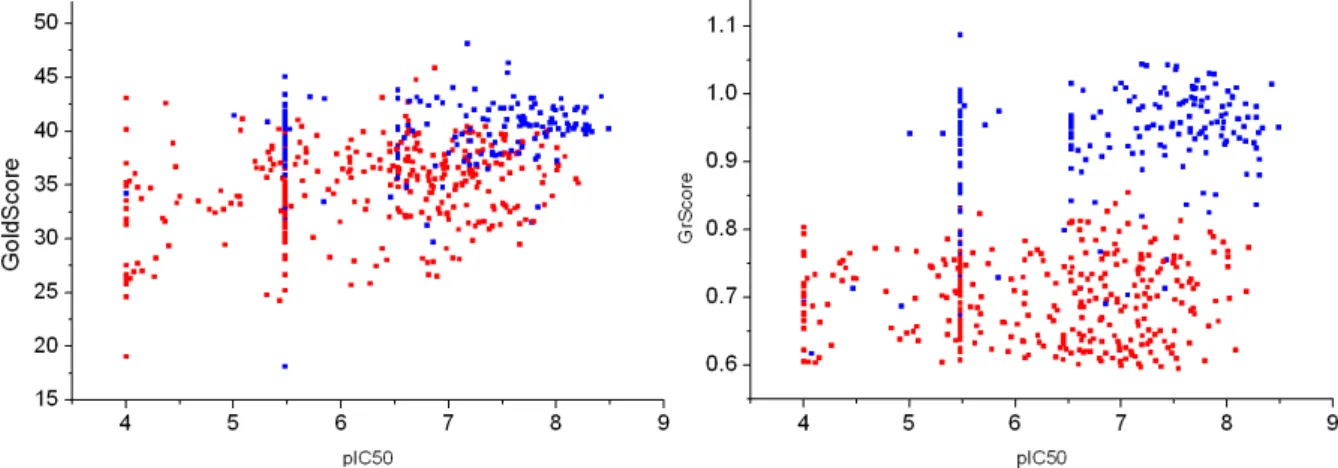
143 Résultats et discussion Modes d'interaction Cas pratiques - Des triplets contenant une interaction post-dock dominant le calcul de similarité. 145 Résultats et discussion Modes d'interaction Cas pratiques - Post docking Sur le deuxième ensemble de données de 36 complexes CCDC/Astex, des tendances assez similaires ont été observées (Figure 9B). 146 Résultats et discussion Modes d'interaction Cas pratiques - Cas post-doctoraux où la valeur EF1 est prise en compte (Tableau 3, Figure 9B).
La sélection est donc toujours dépendante des modes d'interaction du ou des ligands de référence. Il donnerait des paires de modes d'interaction et un accord de paires de sites actifs.
Matériel et méthodes
- Construction de la base de données de fragments
- Préparation des complexes proteine/ligand
- Fragmentation
- Attribution des modes d’interactions
- Conversion des fragments en 2-Dimensions
- Mesure de similarité
- Similarité structurale
- Similarité de modes d’interaction
- Interface scPDB-Frag
- Base de données
- Technologie employée
- Description de l’interface
Les empreintes digitales entières TIFP sont également générées avec l'outil fgps de la suite IChem en utilisant les paramètres par défaut. La troisième étape consiste en des critères de sélection des bioisostères potentiels : similarité structurelle (ECFP4), modes d'interaction globaux (GrScore/TIFP) ou polaire (TIFFPol) et masse moléculaire. En effet, ne représentez pas un atome existant au sein de la molécule complète, n'envisagez pas de le remplacer par un autre atome.
![Figure 2 - Exemple de fragment methylamino]-1,5-dioxo-pentan-2-yl]amino]](https://thumb-eu.123doks.com/thumbv2/1bibliocom/463676.69400/176.892.175.711.638.1076/figure-exemple-fragment-methylamino-dioxo-pentan-yl-amino.webp)
Résultats et discussions
- Fragmentation
- Difficultés lies aux points d’ancrages
- Exemple de recherche
- Recherche focalisée
- Recherche exhaustive
- Indépendance de la masse du fragment
- Perspectives
En fonction des propriétés, il peut définir des règles de similarité structurale, de modes d'interaction et de masse moléculaire (E) pour sélectionner les bioisostères. L'utilisateur a le choix de la référence parmi 28 fragments présentés, ici des fragments de molécules ciblant la protéine-tyrosine phosphatase de type I et la collagénase 3. La mise en œuvre d'une interface Web permet une interrogation facile de la base de données, et permet la visualisation ainsi que la sortie de fragments alignés.