Les canaux ont évolué sous l'influence d'une pression de sélection qui a favorisé l'apparition des principales fonctions des canaux Nav1.X : (1) sélectivité ionique, (2) activation dépendante du potentiel, (3) inactivation rapide. Ce travail de thèse vise à mieux comprendre le rôle de la partie N-terminale du canal sodique cardiaque Nav1.5, et les conséquences de certaines mutations dans cette région conduisant à des troubles de la conduction auriculo-ventriculaire et au syndrome de Brugada. .

Le cœur
Ou encore de moduler la vitesse de propagation du potentiel d'action en fonction de la région anatomique à dépolariser. La vitesse de propagation du potentiel d'action joue un rôle clé dans le syndrome de Brugada.

Le syndrome de Brugada (SBr)
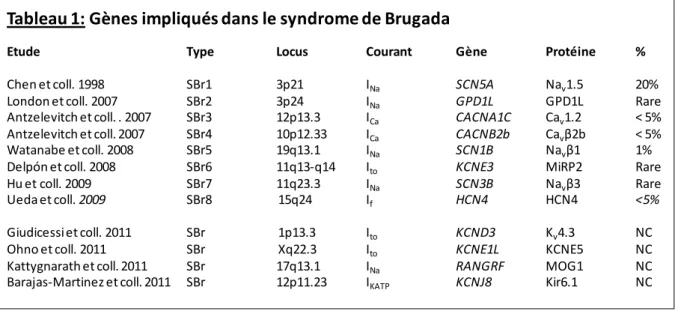
Selon les auteurs, ce syndrome pourrait donc être provoqué par une anomalie électrique ou pourrait être une forme subclinique de cardiomyopathie héréditaire diffuse. L'implication de SCN5A dans SBr a été proposée pour la première fois suite à une approche de gène candidat.
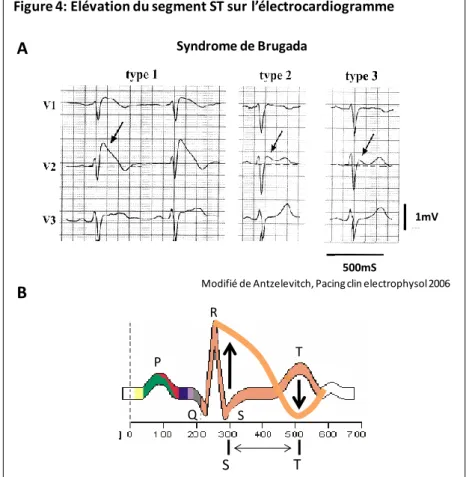
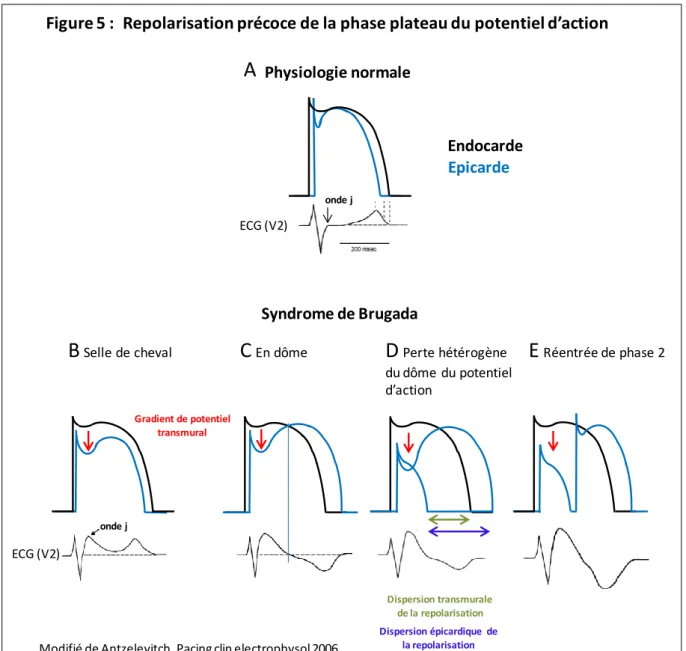
Hypothèse de la repolarisation précoce
Si la perte de la phase plateau du potentiel d'action des cellules endocardiques est excessive, le gradient de potentiel représenté en rouge peut être à l'origine de l'élévation du segment ST sur l'électrocardiogramme (B à E). La dispersion épicardique de repolarisation (flèche bleue) peut créer un gradient transmural de repolarisation (flèche verte) (D), pouvant conduire au phénomène de réentrée de phase 2 (E).
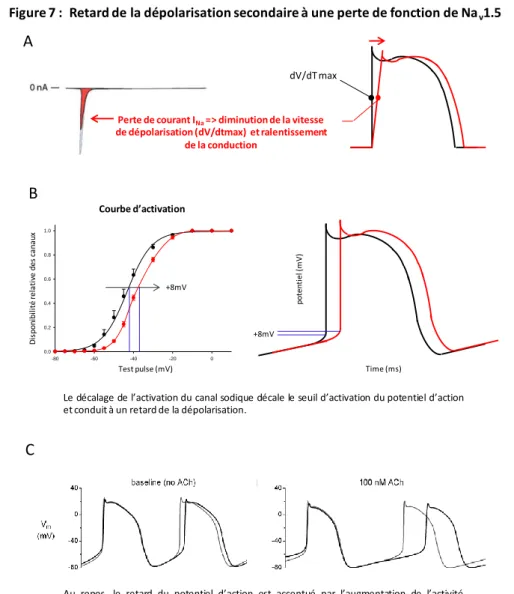
Hypothèse de la dépolarisation retardée
La sous-unité du canal sodique cardiaque Na v 1.5
Les canaux sodiques voltage-dépendants (Nav) ont été les premiers de la superfamille des canaux ioniques à être découverts. Cependant, on pense que la famille des canaux sodiques potentiellement dépendants (Nav) est apparue plus récemment au cours de l'évolution.

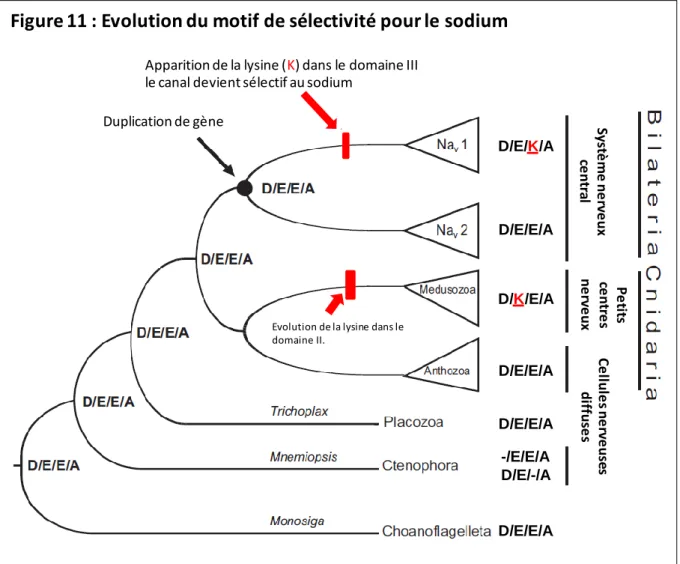
Evolution de la sélectivité du pore
Cela conforte l'idée selon laquelle les canaux Nav dérivent des canaux Cav par l'apparition de mutations de novo. L'apparition de lysine dans le filtre ionique aurait permis une plus grande sélectivité pour le sodium et l'apparition de systèmes complexes à proximité du système nerveux central chez les bilatériens et les méduzoaires.
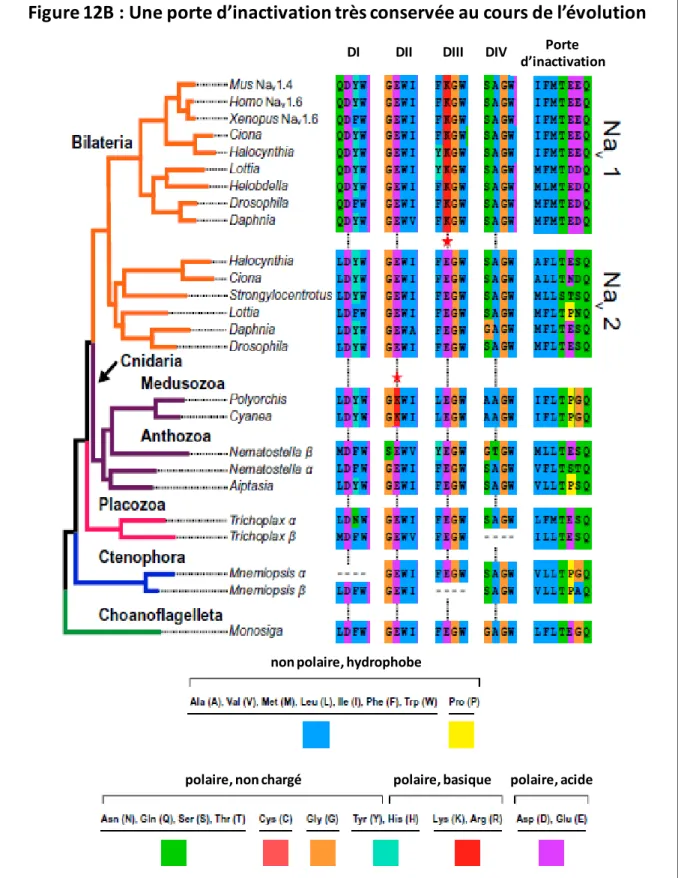
Evolution de l’inactivation rapide
Comprendre la fonction de ces canaux permettra non seulement de comprendre l'histoire des protéines nécessaires à l'adaptation de la motilité, mais aussi celle de l'évolution du système nerveux et des cellules excitatrices [36]. De plus, l'étude de l'évolution de ces systèmes plus primitifs pourrait permettre de mieux comprendre les systèmes complexes de cellules excitables chez les mammifères (36).
La famille des canaux sodiques Na v 1.X chez les mammifères
Dans le cas du canal sodique cardiaque Nav1.5, une seule substitution de la cystéine 373 à la phénylalanine (C373F) réduit de deux cents fois sa résistance au TTX, et le canal devient aussi sensible que les canaux sodiques codés par les chromosomes. 2 [64]. Dans les canaux sodiques Nav1.8 et Nav1.9, le résidu à la même position est la sérine, ce qui leur confère une plus grande résistance à la toxine [65].

Les isoformes du canal sodique Na v 1.5
Structure tridimensionnelle des canaux Nav1.X résolue par cryo-microscopie électronique à partir d'images de Sato et al. Architecture du canal NavAb : les acides glutamiques sont violets et le volume du pore est représenté en gris.

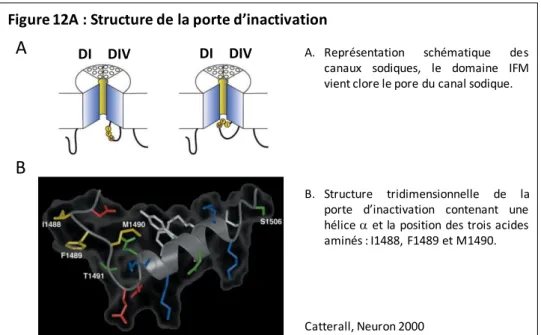
Structure du canal sodique Na v 1.5
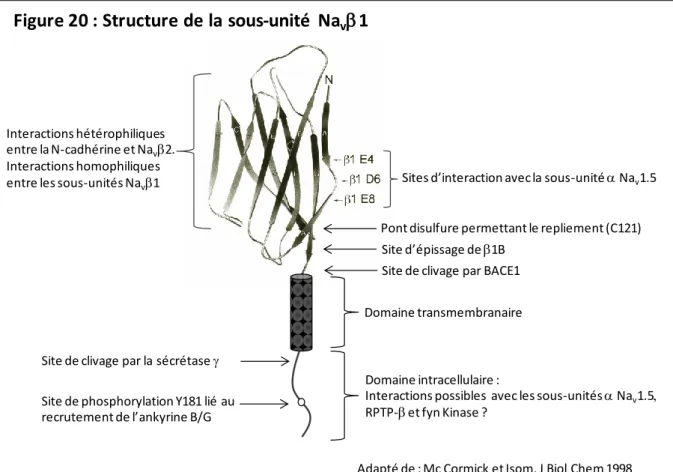
Les sous-unités Na v structure et fonctions
Cela suggère que cette région extracellulaire des sous-unités est importante dans l'interaction et la régulation de la sous-unité , Nav1.5. L’effet de la sous-unité 2 sur le courant sodique cardiaque est également très controversé. Décalage positif de la courbe d'activation Augmentation du courant tardif INaL[156] R85H N-terminal FA Diminution du courant de crête [160].
Les partenaires du canal sodique cardiaque Na v 1.5
L’ankyrine-G / syndrome de Brugada de type 8
La syntrophine 1 / SQTL de type 12
De plus, la co-expression dans les cellules HEK293 de Nav1.5, NOS, PMC4Ab et du mutant A390V augmente le courant INa persistant et déplace l'inactivation vers des potentiels positifs, conduisant à un élargissement du courant de fenêtre entrant par rapport à la co-expression avec la syntrophine sauvage. Ces données indiquent que NOS, PMCA4b et SNTA1 jouent un rôle clé dans la régulation de la nitrosylation de Nav1.5 et de l'amplitude du courant persistant INa. Fait intéressant, la coexpression dans les cellules HEK293 de Nav1.5 avec les mutants G54R et P56S ne semble pas modifier le courant sodique par rapport à la coexpression avec la syntrophine de type sauvage.
La SAP97
Il a également été récemment démontré que le complexe Nav1.5-syntrophine-dystrophine était localisé au niveau de la membrane latérale des cardiomyocytes, contrairement au complexe Nav1.5-SAP97 qui est localisé au niveau des disques intercalaires, suggérant l'existence de deux ensembles de canaux. [203].
MOG1 / syndrome de Brugada de type 11
Cette dernière étude montre une perte encore plus importante de courant sodique chez la souris en raison de la double invalidation du gène de la dystrophine et de l'utrophine. Ce mutant est capable d'exercer un effet négatif dominant sur MOG1 de type sauvage et altère le transport du canal Nav1.5 vers la membrane.
L’alpha-actinine-2
L’ubiquitine ligase Nedd4
Ce mutant est capable d'un effet négatif dominant sur le MOG1 de type sauvage et altère le transport du canal Nav1.5 vers la membrane [ 206 , 207 ]. Il a été démontré que Nedd4-2 se lie directement au site consensus PY de Nav1.5 et ubiquitine le canal dans les cellules de mammifères (Figure 22) [131]. De plus, une autre étude montre que l’augmentation de l’expression du canal Nav1.5 inhibe le protéasome dans les cardiomyocytes néonatals du rat (214).
La CAMKII
Enfin, une fraction des canaux Nav1.5 ubiquitinés est présente dans les tissus cardiaques, suggérant que le recyclage et la stabilité de ce canal in vivo sont régulés par l'ubiquitination. A noter que SAP97 interagit avec les canaux sodiques des disques intercalés, tandis que le complexe syntrophine-dystrophine interagit avec les canaux sodiques présents au niveau de la membrane latérale.
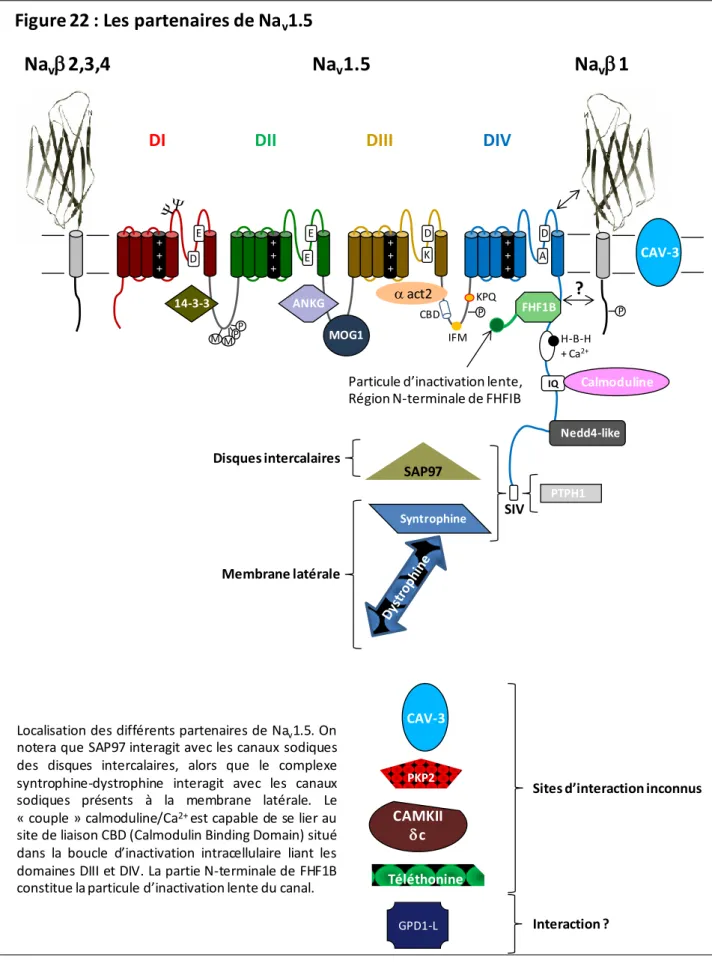
La protéine Tyrosine Phosphatase PTPH1
La protéine 14-3-3
La cavéoline-3 / SQTL type 9
De nombreuses isoformes du 14-3-3 sont exprimées dans le cœur [227] ; leurs rôles physiologiques, associés ou non à la régulation de Nav1.5, et leur éventuelle implication dans la pathologie humaine nécessiteront des investigations plus approfondies. Ces résultats sont cohérents avec ceux des mutants Nav1.5 impliqués dans le LQTS de type 3 (169). Semblable aux mutations trouvées dans LQTS, la coexpression de ces trois mutants avec Nav1.5 augmente le courant INa soutenu.
FHF1B ou FGF12-1b
Dans les cellules HEK293, la co-expression de FHF1B avec Nav1.5 déplace la courbe d'inactivation vers des valeurs hyperpolarisées sans affecter les autres paramètres étudiés [238]. De nombreuses mutations du canal Nav1.5 associées à LQTS ou SBr sont localisées dans cette région. La particule d'inactivation FHFIB est plus grosse que la particule Nav1.5 et aurait une plus grande surface d'interaction avec la partie intracellulaire de Nav1.5, lui conférant une meilleure stabilité.
La calmoduline
Récemment, la partie N-terminale du FHFIB a été décrite comme la particule à inactivation lente des canaux sodiques (Figure 23) [108]. La particule d'inactivation de canal « IFM » et celle composée de la partie N-terminale de FHF seront en compétition pour l'accès au site d'inactivation de canal. La partie proximale de la partie C-terminale de Nav1.5 possède également une région similaire à un motif « EF-hand » connu pour la liaison du calcium [ 99 , 248 ].
La protéine GPD1L / syndrome de Brugada de type 2
Plus récemment, il a été suggéré que le site de liaison connu de CaM pourrait ne pas être la seule région de régulation du calcium dans Nav1.5. Le calcium intracellulaire est un élément important dans la régulation de l’activité des canaux, mais des études supplémentaires seront nécessaires pour comprendre les mécanismes exacts. Cependant, ils n’ont pas déterminé le site exact de l’interaction ni si cette interaction était médiée directement ou indirectement par des protéines capables d’interagir avec le canal (Fig.
La téléthonine
La plakophiline-2
Objectifs du travail de thèse
Peu de mutations dans cette région ont été caractérisées, mais elles semblent avoir un effet important sur la fonction des canaux. De plus, trois études montrent que cette région du canal pourrait être impliquée dans l'activation potentiellement dépendante des canaux Nav1.X. Comment cette mutation dans la région N-terminale du canal sodique cardiaque peut-elle induire une perte de fonction conduisant à une pathologie.
Etude fonctionnelle de mutations de la partie N-terminale du canal sodique Na v 1.5
Introduction
Résultats
Nous avons également observé un déplacement de +8,5 mV de la courbe d'activation vers des potentiels positifs. Cependant, ce phénomène n’explique pas l’effet négatif dominant observé ainsi que le déplacement de la courbe d’activation vers des valeurs positives. Ces résultats suggèrent que les canaux mutants retenus dans la cellule pourraient avoir la capacité d'inhiber le transport du canal de type sauvage vers la membrane.
Conclusion
Enfin, nous avons émis l'hypothèse que si un canal mutant était capable d'inhiber le transport vers la membrane du canal sauvage, ce dernier pourrait permettre le transport d'une petite fraction de ces mutants vers la surface de la cellule. De plus, les courants récupérés présentent un décalage en V1/2 de la courbe d'activation presque deux fois plus important que ceux mesurés dans le cas de coexpression avec le canal sauvage. Ceci suggère que les canaux mutants restaurés présents au niveau de la membrane seraient capables de modifier l'activation potentiellement dépendante des canaux de type sauvage.
Article
Etude fonctionnelle d’une mutation induisant la formation d’un codon stop prématuré dans
En revanche, la génération du codon stop dans la partie proximale, S1885X, qui entraîne la perte de la dernière hélice , réduit le courant sodique et affecte la cinétique d'inactivation des canaux [98]. L'expression de ce canal dans les cellules HEK293 a montré une réduction de la densité maximale de courant de sodium de 70 % par rapport à l'expression du canal de type sauvage (Figure 27A). Nous avons également observé un ralentissement du taux d'inactivation du canal et le temps de demi-inactivation (t1/2) doublé (Figure 28 et Tableau 5).

Etude fonctionnelle de la partie N-terminale du canal sodique Na v 1.5
Ce mécanisme pourrait impliquer des partenaires de la partie N-terminale du canal sodium encore inconnus. Ces résultats suggèrent que la partie N-terminale joue un rôle important dans l'ouverture et l'activation potentielle-dépendante du canal. Ces résultats suggèrent que la présence des mutants modifie le seuil d'activation potentiellement dépendant du canal de type sauvage.
Discussion préliminaire sur l’étude fonctionnelle du canal R1860GfsX12-Na v 1.5
Tan, B.H., et al., Sudden infant death syndrome-associated mutations in sodium channel beta subunits. Chauhan, V.S., et al., Abnormal cardiac Na(+) channel properties and heart rate QT adaptation in neonatal ankyrin (B) knockout mice. Lu, T., et al., Modulation of the rat cardiac sodium channel by the alpha subunit of the stimulatory G protein.