A ferrocén és a hipovalens vegyületek együttes alkalmazása felveti annak lehetőségét, hogy a hipovalens rendszerek katalitikus aktivitása a ferrocén fotokémiai vagy redox kapcsolóként történő alkalmazásával szabályozható. E vizsgálatok és eredmények ismertetésére először külön-külön áttekintem a hipovalens vegyületekre és ferrocénekre vonatkozó szakirodalom legfontosabb szempontjait, majd ismertetem a már ismert ferrocenofánok tulajdonságait.
Irodalmi áttekintés
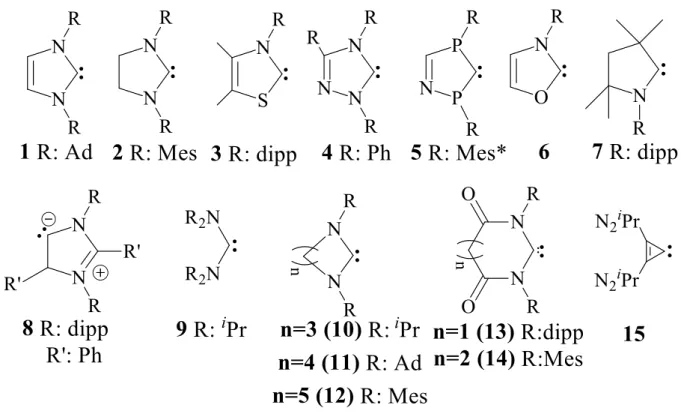
Karbének és nehezebb analógjaik
Ferrocén
A ferrocén négy legalacsonyabb energiájú pályája közül kettő-két degenerált (I és J, valamint K és L pálya a 12. ábrán), dxy (I) és dx2-y2 pályája (J) a vasból és a1 pályája Cp gyűrűkből (K) ) átfedés nem, míg a vas dz2 és a1 pályái között minimális átfedés figyelhető meg (L).

Ferrocenofánok
Ennek a torzításnak a mértéke nyilvánvalóan függ az áthidaló atomok számától (általában a legnagyobb az [1]ferrocenofánok esetében), valamint az atomok minőségétől.

Hipovalens [3]ferrocenofánok
A karbének mellett a [3]ferrocenofánok köre más hipovalens központi atomokkal jelentősen bővült az elmúlt években.
![18. ábra: Az elmúlt években előállított nitrogén, vagy foszforatomok által stabilizált, hipovalens központi atomot tartalmazó [3]ferrocenofánok](https://thumb-eu.123doks.com/thumbv2/9dokorg/2498596.294543/26.892.116.788.427.898/előállított-nitrogén-foszforatomok-stabilizált-hipovalens-központi-tartalmazó-ferrocenofánok.webp)
Ferrocén alapú biszfoszfán ligandumok és komplexeik
Megállapítást nyert, hogy a ligandum terc-butil szubsztituensekkel rendelkező Pd-kelát komplexe hatékonyabb a ketonok arilezésében és a Suzuki-reakcióban, mint a hagyományos dppf Pd-kelát komplexe.200,201 Kiterjedt kísérleti és elméleti munkák is készültek a a 202-207 ligandumon lévő szubsztituensek változásából adódó donor tulajdonságok vizsgálata Komplexek esetén a kísérleti vizsgálathoz a Tolman paramétert használjuk, amely a mérhető C≡O vegyértékrezgés hullámszáma. a NiL(CO)3 komplex, ahol L a vizsgált ligandum a donor jellemzőihez képest.202 Minél jobb a donor a vizsgált ligandum, az elektronok nagyobb mértékben adakoznak a központi atom irányába, így a központi atom nagyobb mértékben tud elektronokat küldeni a karbonilcsoportok π* relaxációs pályájára visszaadományozás útján. Látható, hogy még akkor is, ha a szubsztituenseket megváltoztatjuk, a központi atom körüli kötési szög kissé megváltozik, lehetővé téve a reaktivitás finomhangolását.

Célkitűzések
Alkalmazott számításos kémiai módszerek
Mivel az oxidáció köztudottan reverzibilis a ferrocén rendszeren120, a spinsűrűség elhelyezkedése a ferrocénen bizonyítékul szolgál az oxidáció reverzibilis természetére. Meg kell azonban említeni, hogy a spinsűrűség meghatározása mellett fontos az oxidáció során lezajló esetleges bomlási folyamatok vizsgálata is. Legalább annyi függvényre van szükségünk, ahány elektron, de az is fontos, hogy az atom gömbszimmetriája megmaradjon.
A 31+G*)215-öt, a Dunning-féle korrelációkonzisztens (pl. cc-pVTZ)216-ot és az Ahlrichs-féle hasított vegyértéket (pl. def2SVP)217 bázisokat használtam, amelyek nagyobb sorozatszámú rendszerekre lettek optimalizálva. Ha feltételezünk egy olyan hipotetikus rendszert, amelyben az egyes elektronok között nincs kölcsönhatás, de a kapott rendszer elektronsűrűsége megegyezik a vizsgált rendszerével, könnyen megkaphatjuk a rendszer kinetikus energiáját (Kohn-Sham elmélet). ).218 Bár ezek az elvek elméletben lehetővé teszik a pontos leírást, az energiát leíró egyenletnek mégis van egy paramétere (az ún. csere-korrelációs függvény), amely csak közelítésekkel írható le. Ezek a validációk kísérleti eredményeken (pl. kristályszerkezet) vagy magas szintű csatolt klaszter (CC)225 számításokon alapulnak.
Bader elemzése azon alapul, hogy az energia meghatározásán túl az elektronsűrűség vizsgálata is rengeteg további információt ad a rendszer elektronszerkezetéről. Ha a vizsgált folyamatok a molekulaszám változása nélkül alakultak ki, akkor a termodinamikai profil leírására az energiaértékben, egyébként az 1 atm nyomáson és 298,15 K hőmérsékleten számított szabadentalpia értékében mutattam ki változást. és mindegyik kinetikáját. a reakciót. A Molden 4.0238 programmal megszerkesztettem a szükséges kezdeti geometriákat és kiértékeltem a programcsomagokból számolt adatokat (pl. frekvenciák, atomi távolságok), míg a pályákat és szerkezeteket az IQmol239 programmal írtam le.
Eredmények ismertetése és azok értékelése
Hipovalens központi atomot tartalmazó [3]ferrocenofánok vizsgálata
Ebben az esetben az I-jelölésű rendszerek több hasonlóságot mutattak a hattagú (IV) referenciaszerkezetekkel (ahol a megfelelő rendszerek között kötési szögkülönbség volt), mint az öttagú rendszerekkel (ahol a különbséget a ferrocenofánokhoz hasonlították). ). Mivel korábban megfigyelték hipovalens rendszerekben, hogy a központi hipovalens atom körüli kötésszög jelentős hatással van a szingulett-triplet állapot közötti energiakülönbségre (lásd III. fejezet),1 ezért az I. rendszerek stabilitása és kémiai viselkedése várhatóan hasonló lesz a hattagú referenciavegyületekhez. A vizsgált rendszerek stabilitását a szingulett-triplet energiakülönbség és a karbéneknél bemutatott izodézmikus reakció határozza meg,46 de hasonló módon alkalmazható szililénekre és germilénekre is (lásd III.
Fontos megjegyezni, hogy a szingulett-triplet energiakülönbségek számítása során minden szerkezeten geometriai optimalizálást végeztem, így a kapott eredmények az adiabatikus gerjesztéshez tartoznak. Először is, a karbén rendszerek vizsgálatával megállapítottam, hogy a szingulett-triplet energiakülönbségek a ferrocenofán rendszerek esetében szűk tartományban vannak (kcal/mol), míg a referencia szerkezeteknél ez a paraméter sokkal nagyobb eltérést mutat (kcal/mol). . , a korábban megfigyelt X: NMe >O>S>PMe trend szerint.28 Megfigyelhető továbbá, hogy a referenciák szingulett-triplet energiakülönbségei lényegesen nagyobbak, mint az analóg ferrocenofán rendszerek esetében. Ez alól kivételt képeznek az X=PMe szerkezetek, ahol az I. szerkezet értéke 17,7 kcal/mol, míg a referencia szerkezetek energiakülönbsége 15,7 és 25,0 kcal/mol között van.
Bár ebben az esetben ez az energiaérték nagy eltérést mutat a II-IV referenciarendszerhez képest, az izodézmikus reakció energiája (92,8 kcal/mol) majdnem egybeesik a hattagú gyűrűs vegyülettel (94,0 kcal/mol ) (IV ). , amely alapján feltételezhető, hogy ez a stabilitási paraméter jobban leírja ennek a karbénnek a viselkedését. Továbbá azt is fontos megemlíteni, hogy a B3LYP függvény jelentősen alábecsüli a vastartalmú rendszerekre számított szingulett-triplet energiakülönbséget244, ezért a ferrocén rendszer ∆Es-t értékeinek kiszámításához különböző módszereket alkalmaztam. Hasonló eredmények születtek a szililén, germilén és foszfén kationok analóg rendszereinek vizsgálata során is: míg a ferrocenofán rendszerek esetében szignifikáns különbség figyelhető meg a szingulett-triplet energiakülönbségben, addig az izodezmikus reakcióenergiák hasonló értékeket mutatnak a referenciavegyületek.

A [3]ferrocenofán alapú karbének organokatalitikus aktivitásának vizsgálata
Ez alapján megállapítható, hogy az ωB97X-D mind a kisebb 6-31+G* bázissal, mind a nagyobb cc-pVTZ bázissal, valamint a magasabb szintű CC lokális módszerekkel (LNO-CCSD(T) /def2TZVP ) hasonló eredményt adnak (a különbség a legtöbb esetben 5 kcal/mol alatti, esetenként 5-6 kcal/mol körüli). Ha a karbén és aldehid van der Waals adduktját használtam referenciaként, akkor a diszperziós kölcsönhatás eltérő leírásában nagy különbség volt a különböző DFT módszerek között. A hasonló szerkezetek jelenlétét más karbének esetében NMR spektroszkópiával igazoltuk,42247 és egyéb elméleti munkákat is figyelembe vettünk a benzoin kondenzációjának vizsgálatakor.248249 Ez a szerkezet.
A termodinamikai tulajdonságok mellett fontos az egyes intermedierekre való átmenet aktivációs gátjának vizsgálata is, csak monomolekuláris folyamatot feltételezve. Mivel a Breslow intermedier protikus körülmények között is képződhet (akkor is, ha a katalizátor kis koncentrációban van jelen), érdemes a katalitikus ciklus többi lépését is megvizsgálni. A növekedés oka részben az, hogy a ferrocenofán alapú karbénből és az aldehid van der Waals komplexéből képződött Breslow intermedier 5,4 kcal/mol-al stabilabb, mint az analóg.
Az optimalizált struktúrákat vizsgálva nem találtunk nyilvánvaló okot a megnövekedett stabilitásra (sztérikus közelség, megnövekedett kötéshossz, kötési szög) és a magasabb energiaszinten működő reakcióprofilra. Érdekes módon a hattagú gyűrűrendszer energetikája jobban hasonlít a másik referenciaszerkezethez, mint a ferrocenofánokhoz, annak ellenére, hogy a karbén atom körüli hasonló kötésszög hasonló organokatalitikus aktivitást jelentene. Ezek az eredmények azonban azt mutatják, hogy a ferrocenofán alapú karbén nem célszerű organokatalizátorként használni benzoinkondenzáció esetén, mert annak ellenére, hogy a nukleofilitás növekedése nagyobb reakciókészséget jelez, az elpusztult katalizátort képező oxo tautomer stabilitása. . végül a Breslow intermedierhez képest növekszik, ami gátolja a benzoin termék képződését.

Karbének egyszerű foszforvegyületekkel képzett adduktjainak vizsgálata
A kettős kötés karakterének csökkenése a HOMO-pálya polarizációját eredményezi, amiből az következik, hogy az instabil karbénekkel képződő adduktok esetében alig van polarizáció (lásd a 30. a. ábrát), míg az ún. stabilizált karbének, a kötés jelentősen polarizált (lásd 30. A foszfinid (K ) adduktálódik, a disszociációs energia nem változik, ha a karbén izodezmikus energiája 90 kcal/mol felett van, így az ábrán látható bekarikázott értékek nem A korrelációban szereplők a karakter súlyára vonatkoznak, másrészt ezeknek az adduktoknak a stabilitását is jól korrelálják a karbén stabilitásával (lásd 29. ábra. .. piros, zöld, lila vonal).
A foszfinidénekből és stabil karbénekből képződő adduktok esetében megfigyelhető stagnáló kötésszilárdság ebben az esetben nem figyelhető meg, mivel a legstabilabb karbénekből (32-35) származó L-típusú adduktot nem sikerült optimalizálni. A P-C kötés távolsága ebben az esetben az egyszeres kötés hosszának tartományába esik Å), és az addukt a PF4 anionhoz hasonló "libikóka" geometriát vesz fel,255 ahol a PCl3 szinte teljesen síkré válik és T alakba rendeződik. . Az adduktok stabilitása a legstabilabb esetben is 20 kcal/mol, ami azonban jóval alacsonyabb, mint a P-C kötés irodalmi energiája (kb. 63 kcal/mol).51 Mivel a PCl3 expanziója nagy beruházást igényel. energia (számítási szinten 52,0 kcal/mol M06-2X /cc-pVTZ) szükséges, így összességében 10-20 kcal/mol nagyságrendű disszociációmentes entalpiára kapunk magyarázatot.
Fontos megjegyezni, hogy a karbén 31 is a nagy kötési szögű karbének közé tartozik, de az alacsony HOMO energia miatt csak gyenge adduktot képez a foszfánnal, így a sztérikus hatás ebben az esetben kevésbé fontos. Bár egyes esetekben a két szerkezet közötti P-C kötés távolság szignifikáns volt, a két szerkezet stabilitása közötti különbség egyetlen esetben sem haladta meg a 2 kcal/mol értéket. Ezek az akadályok azonban nem túl magasak a kcal/mol M06-2X/cc-pVTZ számítási szintjén, mert nagy különbség van az egyes szerkezetek termodinamikai stabilitásai között (10,9 kcal/mol az M06-2X/cc-pVTZ-nél). számítási szint), így valószínűleg nem lehet szobahőmérsékleten mindhárom szerkezetet egymás mellett megfigyelni.

Szubsztituens hatás vizsgálata 1,1’-diaminoferrocének és trihalofoszfánok
Az egyes termékekre jellemző szelektivitási jellemzők megértése érdekében különböző szubsztituensek esetén DFT számításokat végeztem, amelyek során a vizsgált reakciók 36. A trialkil-amin bázis, különösen a keletkező trialkil-ammónium só jelenléte, amely éterben rosszul oldódik még jobban eltolja az egyensúlyt a termék irányába. A termodinamikai eredmények még nem elegendőek a folyamat teljes megértéséhez, ezért minden folyamathoz megvizsgáltuk a kinetikai akadályokat, amelyeket az 1. ábra mutat be. 15.
Trietil-amin bázis esetén R=SiMe3 esetén a ciklokondenzáció aktivációs gátja 14,9 (trimetil-amin esetén 6,9) kcal/mol-al alacsonyabb, mint a PC13 addíció aktivációs gátja. Érdekes módon az R=SiMe2tBu szubsztituens esetében a sorrend a katalizátorként használt aminbázis minőségétől függ, mivel a PCl3 hozzáadásának aktiválási gátja 2,8 kcal/mol-al alacsonyabb, mint a ciklokondenzációs gát trietil-amin bázis esetén. de 6,9 kcal/mol magasabb, ha trimetil-amin bázist használunk. Az Arrhenius-egyenlet szerint közel 50-szeres reagensfelesleg alkalmazása a kísérlet során 2,2-2,3 kcal/mol-al csökkenti a ΔG0#(3) értékét, miközben a ΔG0#(2) értéke változatlan marad.
Mindezek alapján megállapítható, hogy az egyes termékek képződése szempontjából fontos (2) és (3) reakciók eltérő relatív kinetikája egyes szubsztituensek esetében összhangban van a kísérleti megfigyelésekkel, ill. ezen eredmények kiterjesztésével, a szubsztituensek minőségének hangolásával elősegíthető a folyamat különböző végtermékekre való áttolása. A zárójelben a PCM implicit oldószer modellje nélkül végzett számítások eredményei láthatók (kcal/mol egységben A ferrocenofán alapú diaminofoszfénium kation termelési kapacitását egyrészt a TD-DFT IV igazolja. ) a HOMO-3 → A LUMO 515 nm-en, míg a HOMO-3 → LUMO 347 nm-en nagyobb intenzitású (oszcillátor erőssége 0,0107).

Nagy térkitöltésű csoportokkal rendelkező 1,1’-biszfoszfánoferrocének és Cu(I)
A geometriai optimalizálás során a szerkezetek a transzoid szerkezet felé fordultak, ezért az 1 és 1' szénatomok és a foszforatomok által alkotott diéderszöget rögzítettük, hogy a szubsztituensek méretéből adódó sztérikus hatás összehasonlítható legyen az egyes rendszerekre. . A különbség abban is megmutatkozik, hogy míg a dppf és a 42a szerkezet esetében a HOMO-1 és HOMO energia közel azonos (a különbség 0,06 illetve 0,08 eV), addig a A 42b. ábra szerint ez a különbség 0,48 eV-tal növekszik -re, mivel a HOMO a mezitilcsoport közelében van, míg a HOMO-1 a foszforon, a fenilcsoport közelében elhelyezkedő magányos elektronpár. A vizsgált rendszereken NBO számításokat is végeztünk, amelyekből jól látható a ligandumok p-karakterének növekedése (dppf esetén 53%, 42a és 42b esetén 55-56%), ami a foszfor körüli kötésszög növekedésével kapcsolatos.
A fenil-acetilén katalitikus szén-dioxid hozzáadásának hozama a megfelelő komplex katalizátorként (41% CuBr-katalizátorral, 58% CuI-vel, 29% Cu(MeCN)4(BF4)-vel), míg a dppf-. kísérleti adatokkal látható. általában azt mondják, hogy a nagy térkitöltő csoportokat tartalmazó ligandum kedvezően hat a kívánt végtermék képződésére (29% csupasz Cu(MeCN)4(BF4), 33% dppf ligandum és 76% ligandum 42a). , hogy bár a nagyobb térkitöltésű mezitil szubsztituens növeli a donor kapacitást, négy mezitilcsoport alkalmazása már csökkenti a katalitikus aktivitást a Cu(I) komplexekkel végzett katalitikus karboxilezési reakció során a túlzott sztérikus gátlás miatt.

Összefoglalás
Függelék
Irodalomjegyzék
Közlemények




![16. ábra: Az első előállított [3]ferrocenofán szerkezete az atomok közti távolságokkal jelölve](https://thumb-eu.123doks.com/thumbv2/9dokorg/2498596.294543/23.892.205.694.197.618/ábra-előállított-ferrocenofán-szerkezete-atomok-közti-távolságokkal-jelölve.webp)
![1. táblázat: Előállított [n]ferrocenofánok](https://thumb-eu.123doks.com/thumbv2/9dokorg/2498596.294543/24.892.107.795.159.945/táblázat-előállított-n-ferrocenofánok.webp)
![17. ábra: Kisméretű molekulák addíciója [3]ferrocenofán alapú karbénekre (R=Np)](https://thumb-eu.123doks.com/thumbv2/9dokorg/2498596.294543/25.892.109.787.626.1079/ábra-kisméretű-molekulák-addíciója-ferrocenofán-alapú-karbénekre-np.webp)
