Binding of CBD at this site decreases the receptor's sensitivity to its orthosteric agonists (negative allosteric modulation). Then I added the rest of the CB1R N-terminal domain (residues 1-95) and modeled the entire N-terminus using the replica exchange with solute quenching (REST2) approach (16 replicas, 6 simulations, more than 1000 ns each, over 115 μs total).
Co to są receptory GPCR?
Sygnałami rozpoznawanymi przez GPCR są zewnątrzkomórkowe bodźce chemiczne, takie jak neuroprzekaźniki czy hormony, a także bodźce świetlne (w przypadku rodopsyny), które powodują zmianę konformacji receptora z nieaktywnej na aktywną. Poprzez receptory GPCR sygnał przechodzi przez błonę komórkową, wiążąc i aktywując białka efektorowe (białko G lub arestynę) od wewnątrz.
Budowa receptorów GPCR
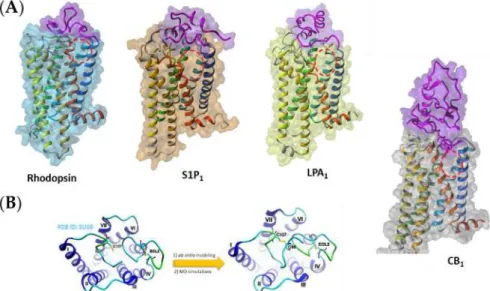
Ponadto jony wapnia uwalniane z retikulum endoplazmatycznego (ER) jako wtórny efekt interakcji aktywowanego receptora GPCR z białkiem Gq mogą również wpływać na polaryzację błony poprzez otwieranie zależnych od Ca2+ kanałów chlorkowych, powodując wypływ jonów Cl z komórek , powodując depolaryzację błony, generując sygnał elektryczny [1]. Cechy strukturalne charakterystyczne dla niektórych klas receptorów GPCR; .. Klasa A) Receptor β1-adrenergiczny powiązany z cyjanopindololem (identyfikator PDB: 4BVN), klasa B) Receptor glukagonu powiązany z analogiem glukagonu (identyfikator PDB: 5YQZ), klasa F) Wygładzony receptor związany z Anta XV (identyfikator PDB: 4QIM) , klasa C) homodimer receptora glutaminianu mGlu1 w postaci APO (ID PDB: 7DGD);
Aktywacja receptorów GPCR
Mechanizm aktywacji receptorów GPCR
Białko G jest aktywowane poprzez interakcję z receptorem, co z kolei prowadzi do wymiany cząsteczki GDP na GTP. W efekcie dochodzi do przegrupowania sieci wiązań wodorowych w obrębie receptora, co z kolei prowadzi do zmian w konformacji receptora (przesunięcia helis transbłonowych względem siebie i zmiany geometrii niektórych z nich, zwłaszcza TM6). ).
Główne przełączniki molekularne występujące w receptorach GPCR klasy A
Mostek solny łączący helisy TM3 i TM6 („blokada jonowa”), zlokalizowany po wewnątrzkomórkowej stronie receptora w obrębie motywu D/ERY na TM3 (często między resztami R3.50 i E6.30), zostaje przerwany podczas aktywacji receptora . Na przykład najbardziej konserwatywna reszta helisy TM3 będzie miała numer 3,50, podczas gdy reszta znajdująca się trzy pozycje przed nią będzie miała numer 3,47, podczas gdy reszta dziesięć pozycji po reszcie 3,50 będzie miała numer 3,60.
Ligandy receptorów GPCR oraz ich klasyfikacja
Podział ze względu na wpływ na aktywność receptora
W wyniku wiązania ligandu zmienia się konformacja receptora, sprzyjając lub uniemożliwiając wiązanie białek efektorowych z wewnątrzkomórkową częścią receptora. Ligandy, które zmniejszają poziom aktywacji receptora (tj. zapobiegają wiązaniu i aktywacji białek efektorowych) nazywane są odwrotnymi agonistami.
Podział ze względu na miejsce wiązania liganda
Pozytywne modulatory allosteryczne (PAM) to ligandy, które wiążąc się z miejscem allosterycznym, wzmacniają sygnalizację agonistów ortosterycznych (niezależnie od ich wpływu na powinowactwo wiązania ligandu ortosterycznego, które może być dodatnie, ujemne lub zerowe). Oprócz modulatorów istnieją inne typy ligandów allosterycznych, takie jak: agonista allosteryczny, który może aktywować receptor nawet przy braku agonisty ortosterycznego, antagonista allosteryczny, który może hamować aktywację receptora indukowaną przez agonistów ortosterycznych (w sposób kompetycyjny), a wreszcie neutralne ligandy allosteryczne (NAL) wiążące je z miejscem allosterycznym receptora bez wpływu na aktywność receptora lub modulowania jego odpowiedzi na ligandy ortosteryczne [19].
Zalety modulatorów allosterycznych
Receptory GPCR wiążące ligandy hydrofobowe
- Droga wejścia ligandów ortosterycznych do receptora
- Receptor fosforanu-1-sfingozyny S1P 1
- Receptor kwasu lizofosfatydowego LPA 1
- Receptor kannabinoidowy CB 1
- Przykłady allosterycznych modulatorów receptora CB 1
- Kannabidiol – jego właściwości i miejsce wiązania
- Wpływ N-końca na wiązanie ligandów allosterycznych oraz ortosterycznych do CB 1 R
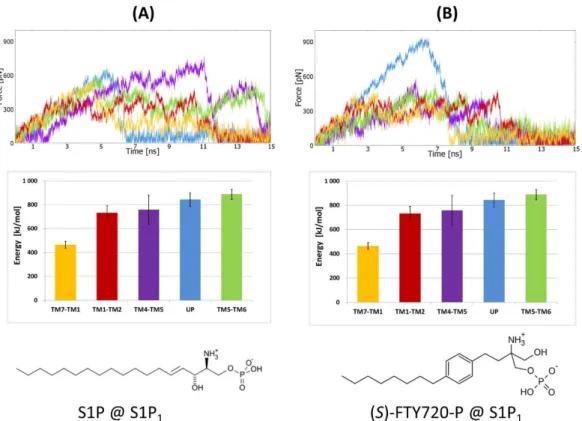
Agonista S1P1, który reguluje w dół ten receptor (FTY720-P, fosforan fingolimodu) (ryc. 10B) był używany w leczeniu stwardnienia rozsianego [29,30]. Ta właściwość ligandów S1PR1, a także fakt, że N-koniec tego receptora tworzy strukturę α-helikalną, która blokuje dostęp do miejsca wiązania od strony zewnątrzkomórkowej (ryc. 9, fioletowy), wskazuje, że te ligandy receptora S1PR1 bezpośrednio z błony komórkowej.
Hipoteza badawcza
Cel naukowy rozprawy
Przygotowanie struktur do symulacji dynamiki molekularnej
Dokowanie ligandów
Za pomocą tego programu wyznaczono potencjał elektrostatyczny wokół ligandu metodą kwantowo-chemiczną DFT (Density Functional Theory), potencjał hybrydowy B3LYP oraz bazę 6-311G**, a następnie obliczono ładunki cząstkowe na atomach ligandu w procedury optymalizacyjnej, tak aby wyznaczony potencjał elektrostatyczny został jak najlepiej zrekonstruowany. Allosteryczne ligandy (CBD i LXA4) zadokowano do allosterycznego miejsca 1 (ryc. 14) CB1R przy użyciu GOLD (Genetic Optimization for Ligand Docking) v.
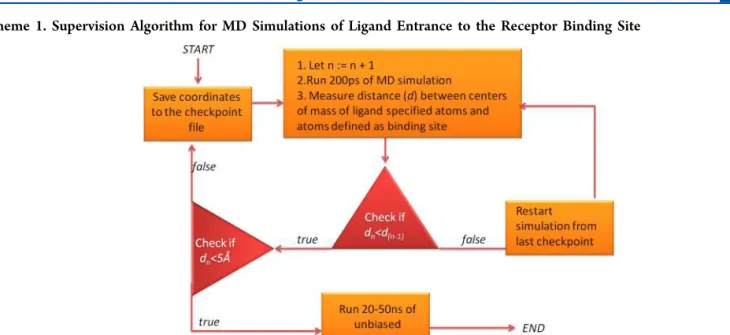
Pełnoatomowe symulacje dynamiki molekularnej (MD, SMD i SuMD)
W symulacjach SMD ligand był odciągany od miejsca wiązania receptora ze stałą prędkością (dla atomu wirtualnego v = 0,31 m/s) (Schemat 1). Wirtualny atom porusza się ze stałą prędkością (v), a wirtualna sprężyna wywiera siłę na narysowany atom (atom SMD), zmienną co do wielkości, ale stałą w kierunku.
Symulacje REST2
Analiza wyników
Wyniki dotyczące publikacji własnej 1 (Jakowiecki, Filipek; Journal of Chemical Information
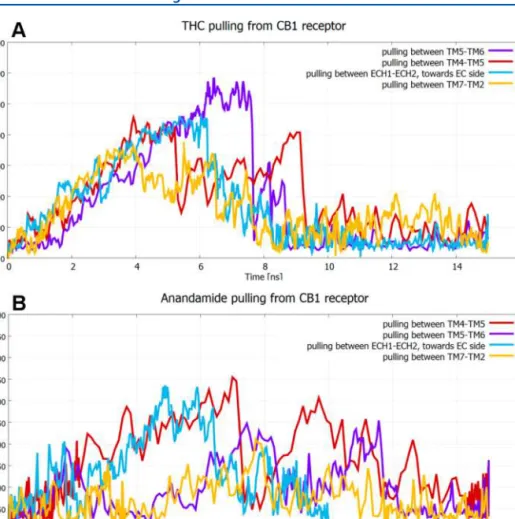
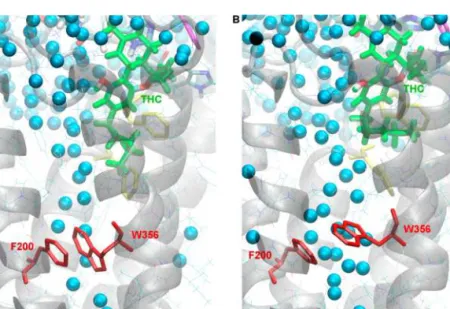
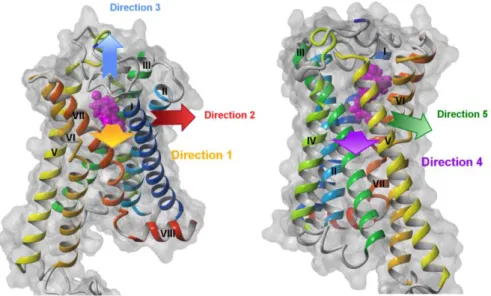
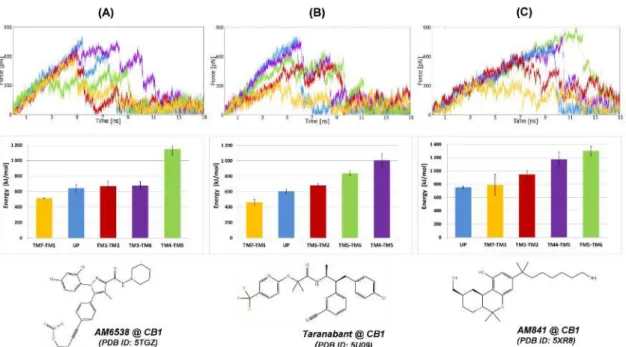
1 miało potwierdzić hipotezę o wejściu ligandów do receptora CB1 ze środowiska błony komórkowej), a drugim dowiedzieć się, w której części błony stężenie THC będzie największe. Wyniki te wskazują, że stężenie THC w błonie komórkowej jest co najmniej o jeden rząd wielkości wyższe niż w wodzie, co czyni znacznie bardziej prawdopodobnym, że THC dostanie się do miejsca wiązania receptora CB1 z błony komórkowej niż z roztworu pozakomórkowego. Przeprowadzono symulacje SMD ekstrakcji THC i anandamidu z receptora CB1 (model homologiczny) w czterech różnych kierunkach, aby zidentyfikować najbardziej prawdopodobną ścieżkę wyjścia/wejścia ligandu dla CB1R.
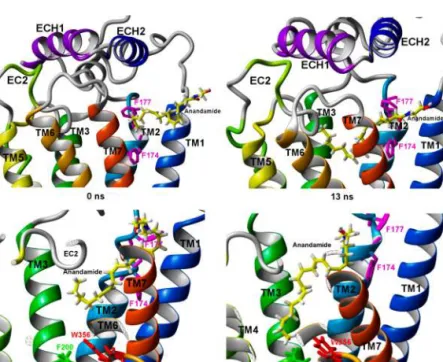
Przeprowadzono symulacje SuMD wejścia anandamidu i THC do receptora CB1 (model homologiczny) między TM7-TM1, aby sprawdzić, czy symulacja równowagi może obserwować wejście ligandu przez ten szlak i zobaczyć, która orientacja wejścia liganda jest najbardziej możliwa. Czas wejścia ligandu do miejsca wiązania wynosił 25 ns dla anandamidu i 40 ns dla THC dla wejścia przed łańcuchem alkilowym. W przypadku innych orientacji czasy wejścia były znacznie dłuższe i obserwowano tylko częściowe lub żadne wejście ligandu.
Próby symulacji wstawienia ligandu między TM4 i TM5 oraz między TM5 i TM6 okazały się bezowocne, mimo że zastosowaliśmy różne początkowe orientacje ligandu.
Wyniki dotyczące publikacji własnej 2 (Jakowiecki et. al.; Molecules 2020) [93]
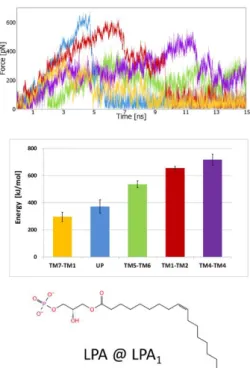
Symulacje SMD dwóch ortosterycznych agonistów pobranych z tej samej struktury krystalicznej receptora S1P1 (PDB ID: 3V2Y); (A) sfingozyno-1-fosforan (S1P); (B)(S)-FTY720-P. Przeprowadzono symulacje SMD ekstrakcji kwasu lizofosfatydowego z receptora LPA1 (struktura krystaliczna, PDB ID: 4Z34) w 5 różnych kierunkach, aby zidentyfikować najbardziej prawdopodobną ścieżkę wyjścia ligandu z tego receptora. W dwóch z nich orientację wejścia ligandu ustawiono na „przedni łańcuch alkilowy” iw obu tych symulacjach cała cząsteczka liganda weszła do receptora, chociaż dwie symulacje dały nieco inne pozycje wiązania ligandu.
W pozycji liganda otrzymanej z symulacji 1 (143 ns SuMD + 41 ns MD) grupa fosforanowa LPA tworzyła oddziaływania jonowe z resztami K39NT i R1243.28 (a w niektórych ramkach wiązanie wodorowe z Y34NT), podczas gdy drugi koniec cząsteczka wchodzi w interakcję z resztą W2716.48 (należącą do przełącznika molekularnego „przełącznika transferu”). W pozycji otrzymanej w symulacji 2 (175 ns SuMD + 33 ns MD) grupa fosforanowa liganda tworzyła wiązanie jonowe z resztą K39NT, grupa karbonylowa liganda tworzyła wiązanie wodorowe z R1243.28, a odwrotnie koniec cząsteczki tworzył oddziaływania hydrofobowe z resztą W2716. 48. W symulacjach 3 i 4 zbadano możliwość wejścia „biegunowego do przodu”, ale w obu przypadkach ligand wszedł tylko częściowo.
Symulacje SMD ligandu ortosterycznego, kwasu lizofosfatydowego ((S)-LPA), wyekstrahowanego ze struktury krystalicznej receptora LPA1 (identyfikator PDB: 4Z34).
Wyniki dotyczące publikacji własnej 3 (Jakowiecki et. al.; Molecules 2021) [94]
Sprzęganie allosteryczno-ortosteryczne w receptorze CB1 testowano w następujący sposób: Przygotowano dwa układy, oba zawierające identyczną strukturę receptora CB1 osadzoną w błonie (POPC:cholesterol, 5:1 n/n) otoczonej z obu stron wodą. Jedyna różnica pomiędzy systemami dotyczyła ligandów związanych z receptorem: i) System I zawierał CBD w miejscu allosterycznym i THC w miejscu ortosterycznym (ryc. 22). ii) System II zawierał tylko THC związane z miejscem ortosterycznym. Na podstawie analizy szlaków (przy użyciu pakietu getContacts i Pythona) zidentyfikowano kluczowe interakcje białko-białko (w obrębie receptora) oraz interakcje białko-ligand.
Obliczono częstości występowania poszczególnych oddziaływań, a następnie porównano sieci oddziaływań w obrębie receptora uzyskane dla układów I i II w symulacjach MD. Obserwowane różnice w częstości interakcji między kompleksami CB1R z CBD i THC oraz CB1R z samym THC; (A) wiązania wodorowe; (B) Wiązanie wodorowe za pośrednictwem wody; (C) oddziaływania van der Waalsa. Przedstawione interakcje najprawdopodobniej uczestniczą w allosterycznej-ortosterycznej sieci komunikacyjnej, ponieważ ich częstotliwości różnią się znacznie między symulacjami z ligandem allosterycznym (CBD) i bez niego.
Zauważyłem, że obecność CBD w miejscu allosterycznym zmniejszyła bezwzględną energię wiązania (mniej stabilny kompleks) THC w tym miejscu.
Wiązanie ligandów ortosterycznych do receptorów lipidowych (publikacje własne 1 i 2)
Szczególnie wysokie stężenie THC w części błony blisko interfejsu sprzyja wejściu ligandu przez kanał pomiędzy TM7 i TM1, który łączy błonę komórkową z ortosterycznym miejscem wiązania receptora, ponieważ wejście do tego kanału znajduje się blisko granicy między błoną a roztworem pozakomórkowym. Wyniki analogicznych symulacji SMD dla receptorów S1P1 i LPA1 (struktury krystaliczne z zakończonymi N-końcami) również wykazały wejście/wyjście ligandu pomiędzy helisami TM1 i TM7. Ligand znajdujący się w pobliżu wejścia do kanału między TM1 a TM7 (zakładając, że jego orientacja jest prawidłowa) będzie dyfundował do niego bez konieczności przekraczania barier sterycznych.
Przeciwnie, wnikaniu ligandu sprzyjają hydrofobowe i wysoce elastyczne łańcuchy boczne reszt otaczających wejście do kanału wiążącego, takich jak: M109, L111, F381 i M384. Jednocześnie reszty F174 i F177, które wcześniej zakładano, że biorą udział w mechanizmie bramkowania, nie znajdują się w świetle ortosterycznego kanału liganda, ale w kieszeni między helisami TM1 i TM2, gdzie biorą udział w wiązaniu modulatorów allosterycznych. Przeprowadziłem w sumie dziewięć symulacji SuMD dla insercji S1P i cztery dla insercji (S)-FTY720-P do receptora S1P1 (przy użyciu ligandów protonowanych w różnym stopniu, jak również różnych orientacji insercji ligandów).
Brak pełnego wejścia tych ligandów może być spowodowany silnymi oddziaływaniami jonowymi powstającymi pomiędzy ujemnie naładowaną grupą fosforanową liganda a dodatnio naładowanymi resztami receptora K41NT i R2927.34 zlokalizowanymi w pobliżu wejścia do kanału prowadzącego do ortosterycznego miejsca wiązania liganda .
Allosteryczna modulacja receptora CB 1 – sprzężenie allosteryczno-ortosteryczne (publikacja
Allosteric modulation of the CB1 cannabinoid receptor by cannabidiol-A molecular modeling study of the N-terminal domain and the allosteric-orthosteric linkage. We believe that the long N-terminus of the CB1 receptor plays an important role in the ligand binding process. The ligand binding site of the most populous rhodopsin-like (class A) GPCRs is located at the extracellular portion of those receptors.
This strategy prevented rotation of the ligand at the binding site, which could prevent this orientation. Most of the receptor structures were inactive (crystallized with antagonists or inverse agonists) and only one structure (CB1 receptor PDB ID: 5XR8) was active. For the GOR direction for (S)-FTY720-P, the ligand collided with part of the N-terminus when leaving the receptor.
We tested both ends of the ligands (hydrophilic and hydrophobic) to enter the receptor, as we have done for the same agonists, ∆9-THC and anandamide, in our simulations of a homology model of CB1[6]. SuMD simulation of ∆9-THC entry into the orthosteric binding site of the cannabinoid CB1 receptor. SuMD simulation of anandamide entry into the orthosteric binding site of the cannabinoid CB1 receptor.
Before the discovery of the allosteric binding site(s) in CB1R, the design of new ligands for this receptor was solely based on the orthosteric binding site [6]. For each of the two ligand-receptor complexes (1st complex of CB1R with CBD and THC, and 2nd complex of CB1R only with THC) five independent all-atom and unbiased MD simulations were performed (2×5×1µs).